

Abstract
Stable expression of long-term synaptic plasticity is critical for the developmental refinement of neural circuits and for some forms of learning and memory. Although structural remodeling of dendritic spines is associated with the stable expression of long-term potentiation (LTP), the relationship between structural and physiological plasticity remains unclear. To define whether these two processes are related or distinct, we simultaneously monitored EPSPs and dendritic spines, using combined patch-clamp recording and two-photon time-lapse imaging in the same CA1 pyramidal neurons in acute hippocampal slices. We found that theta burst stimulation paired with postsynaptic spiking, which reliably induced LTP, also induced a rapid and persistent expansion of dendritic spines. Like LTP, this expansion was NMDA receptor dependent. Spine expansion occurred even when LTP was inhibited by postsynaptic inhibition of exocytosis or PKA (protein kinase A); however, under these conditions, the spine expansion was unstable and collapsed spontaneously. Furthermore, similar changes in LTP and spine expansion were observed when hippocampal neurons were treated with protein synthesis inhibitors. Like LTP, spine expansion was reversed by low-frequency stimulation (LFS) via a phosphatase-dependent mechanism, but only if the LFS was applied in a critical time window after induction. These results indicate that the initial expression of LTP and spine expansion is dissociable, but there is a high degree of mechanistic overlap between the stabilization of structural plasticity and LTP.
Keywords: LTP, dendritic spines, actin, AMPA receptors, depotentiation, two-photon imaging
Introduction
Activity-induced modification of neuronal connections is essential for the development of the nervous system and may also underlie learning and memory functions of the mature brain. With the induction of long-term potentiation (LTP), there is an increase in the number and/or the activity of AMPA receptors (AMPARs) underlying synaptic transmission (Bliss and Collingridge, 1993; Malinow and Malenka, 2002), as well as an increase in the number of spines (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999) or enlargement of the spine head (Fifkova and Van Harreveld, 1977; Desmond and Levy, 1983, 1986; Matsuzaki et al., 2004). As with synaptic plasticity, changes in spine morphology are bidirectional, with stimuli that induce LTP causing spine growth (Matsuzaki et al., 2004) and stimuli that induce long-term depression (LTD) causing spine shrinkage (Okamoto et al., 2004; Zhou et al., 2004; Hsieh et al., 2006).
Changes in spine structures have long been proposed to contribute to synaptic plasticity, perhaps by providing an anatomical substrate to act as the physical scaffold for synaptic modifications (Fifkova and Morales, 1992; Harris, 1999; Yuste and Bonhoeffer, 2001; Nimchinsky et al., 2002; Kasai et al., 2003; Hayashi and Majewska, 2005; Segal, 2005) or by allowing diffusion between the dendrite and the spine (Bloodgood and Sabatini, 2005; Ashby et al., 2006; Korkotian and Segal, 2006). However, the relationship between spine morphology and synaptic plasticity remains poorly understood. Individual reports differ as to whether spine plasticity is persistent (Matsuzaki et al., 2004; Kopec et al., 2006), transient (Lang et al., 2004; Ehrlich et al., 2007), or absent altogether (Bagal et al., 2005; Stewart et al., 2005) during LTP. It has been reported that endocytosis of synaptic AMPARs is sufficient to cause spine shrinkage on its own during long-term depression (LTD) (Hsieh et al., 2006); however, a recent study found these two processes to be independent of each other, at least on a short timescale (Wang et al., 2007). One difficulty in evaluating the relationship between spine and synaptic plasticity is that the mechanisms underlying NMDAR-dependent changes in spine morphology are poorly understood.
In this study, we used simultaneous two-photon time-lapse microscopy and whole-cell patch-clamp recording to define the processes underlying NMDAR-dependent spine expansion and its subsequent stabilization or reversal, and we compared those processes to the ones underlying LTP. We identified three mechanistically distinct processes involved in the spine expansion that accompanies LTP: an initial expansion of the spine head that can occur even under conditions in which the expression of LTP is mostly blocked, a subsequent stabilization of that expansion that shares several mechanistic properties of LTP, and an activity-dependent reversal of spine expansion that is distinct from shrinkage of naive dendritic spines and closely correlated with physiological depotentiation of previously established LTP.
Materials and Methods
Slice preparation and recording procedure.
These procedures have been published previously (Zhou et al., 2004; Wang et al., 2007). Briefly, coronal sections (350 μm) were taken from postnatal day 13 (P13) to P18 rat pups (Sprague Dawley) using a Leica (Nussloch, Germany) VT1000 tissue slicer in 4°C artificial CSF (ACSF). Slices were allowed to recover for 30 min at 32°C, and recording and imaging started at least 1 h after recovery.
Slices were placed in a custom-made recording chamber on the stage of an Olympus (Tokyo, Japan) BX61W microscope and perfused at a rate of 1–2 ml/min with ACSF. All recording and imaging experiments were performed at 30–32°C. Whole-cell patch-clamp recordings were made from pyramidal neurons in CA1 of hippocampus under visual guidance. The recording pipettes were filled with the following (in mm): 128 potassium gluconate, 10 NaCl, 2 MgCl2, 10 HEPES, 0.5 EGTA, 4 Na2ATP, 0.4 NaGTP, 15 phosphocreatine, and 1 calcein, pH 7.3. Calcein is a biologically inert fluorescent dye that we used for labeling of the dendritic spines. All experiments were performed in the presence of a GABAA antagonist, picrotoxin (50 μm). Synaptic inputs were stimulated using a glass pipette with 3 μm opening positioned at ∼20–30 μm away from the imaged spines. We showed previously that this local stimulation is effective in evoking synaptic responses in the imaged spines, as revealed by Ca2+ imaging (Zhou et al., 2004).
CA1 pyramidal cells in the hippocampus were held in current-clamp mode throughout the experiments and EPSPs were recorded with Axopatch 700B amplifier and analyzed with pClamp 9.0 software (Molecular Devices, Sunnyvale, CA). The initial slope of EPSPs was used to measure synaptic responses. Stimulation at 0.05 Hz was used to establish baseline synaptic responses. The stimulation strength was set to evoke EPSPs between 5 and 8 mV. A train of theta burst stimuli (TBS) consisted of five bursts of stimuli at 5 Hz and each burst contained five pulses at 100 Hz. Each train was repeated twice with a 20 s interval. During TBS, the postsynaptic cells were depolarized through current injection to ensure that at least three spikes were generated during each burst. This induction protocol is termed theta burst paring (TBP). Low-frequency stimulation (LFS) consisted of 1 Hz stimulation for 5 min. Electrophysiological recordings were performed either with or without concurrent imaging. Because changes in synaptic responses were similar from both experiments, the data were pooled regardless of whether imaging was performed concurrently. For display purposes, EPSPs shown in the figures are averages of 5–10 consecutive EPSPs.
For most experiments, a 10-min-long baseline recording was obtained before TBS. However, it has been reported previously that postsynaptic infusion of BoTox abolished the delivery of AMPARs to synapses by blocking constitutive trafficking (Lüscher et al., 1999; Wang et al., 2007), leading to a rundown of synaptic transmission for ∼10 min after whole-cell recording. To ensure that this rundown did not contaminate our measurement of LTP, we waited 15–20 min after the start of recording before applying the TBP in these experiments. We also used a 15- to 20-min-long baseline in all experiments in which postsynaptic agents [BoTox, protein kinase A inhibitor (PKI), 8-Br-cAMP, cycloheximide (cyclo)] were introduced via the patch pipette, to allow additional time for these agents to dialyze into the cell and to reach their targets.
Image acquisition and analysis.
Time-lapse imaging was performed on a custom-made two-photon laser-scanning system modified from an Olympus Fluoview FV 300, driven by a Chameleon two-photon laser (Coherent, Santa Clara, CA) tuned to 810 nm. The intensity of laser power at the entry of the microscope was 30–40 mW and monitored continuously. Experiments were performed in the frame scan mode.
Imaging and analysis procedures were based on our previous work (Zhou et al., 2004; Wang et al., 2007). Images were usually taken every 15 min at a resolution of 512 × 512 or 256 × 256 pixels per frame, and an average of two was used in some experiments. For each time point, a stack of images covering the entire three-dimensional range of the spines were taken with a z-step size of ∼0.5 μm. In all figures, two-dimensional projections of dendritic spines were displayed with the maximal signal.
Image analysis was performed blind, with the person analyzing the images having no knowledge of the identity of the samples during the analysis. Spines were distinguished from filopodia based on our previous criteria (Zhou et al., 2004). Analysis was performed on all spines in the image field that were well resolved (i.e., protruding tangentially from the dendrite) and clearly separated from other spines. Volume analysis of individual spines was performed using a circular region of interest (ROI) positioned to cover the entire spine head, for either thin spines or mushroom spines. For stubby spines, the ROI covered the entire spine. All spines identified in the first image acquisition (typically 10 min before TBS/TBP) were analyzed in subsequent time points.
To measure spine head volume, images were first thresholded to eliminate background fluorescence. The integrated fluorescence intensity inside a spine head was measured for individual spines at different time points and normalized to the fluorescence intensity of the dendrites from the same image stack to correct for potential changes in excitation (Holtmaat et al., 2005). Fold change (volume) was determined by averaged values after TBS over the averaged values before TBS. This fluorescence intensity is expected to be proportional to the accessible spine volume (Holtmaat et al., 2005). To verify that this was the case, we also measured the diameters of spines in the TBS experiments using Neuronstudio program (Wearne et al., 2005; Rodriguez et al., 2006). Spine diameters were measured on projected two-dimensional images using two-dimensional rayburst algorithm. Images were first blurred to enhance the rayburst function. Spine volume was calculated from the spine diameters obtained using the Neuronstudio program, after calibration of the point-spread density function using fluorescence beads (Invitrogen, Carlsbad, CA). Similar results were obtained using either approach.
Statistical analysis.
The Student t test and Mann–Whitney U test were used for comparison between different conditions as appropriate, depending on whether or not the data were normally distributed. Paired statistics were used to compare effects before and after TBS/TBP for individual experimental manipulations. A minimum criterion of p < 0.05 was used to determine statistical significance in all cases. In all figures, data are presented as mean ± SEM. Reagents were obtained from Sigma-Aldrich (St. Louis, MO), except where noted.
Results
Schaffer collateral synapses undergo both synaptic potentiation and spine growth in response to intense activation of NMDARs, caused by tetanizing CA3–CA1 afferents. In the present study, we induced LTP using theta burst stimulation paired with a modest postsynaptic depolarization, so that the EPSPs during the burst elicited postsynaptic spikes. This TBP protocol is Hebbian, in the sense that presynaptic bursts are temporally correlated with postsynaptic spikes, and this activity pattern is thought to mimic the firing patterns seen in the hippocampus during behavioral learning in vivo (Otto et al., 1991).
Briefly, we recorded from CA1 pyramidal cells in whole-cell current-clamp mode using an internal pipette solution containing the fluorescent dye calcein (a biologically inert fluorescent dye) as an intracellular label, and we imaged the spines using two-photon time-lapse fluorescence imaging. EPSPs were elicited using weak stimulation applied through a theta-glass microelectrode positioned near the cell, and spines within ∼20–30 μm of the stimulating electrode were examined (to maximize the likelihood that the spines being imaged were activated by the stimulus) (Zhou et al., 2004) (see below). We then examined whether TBP could elicit changes in synaptic strength, spine morphology, or both.
Both LTP and spine expansion require NMDAR activation and postsynaptic spiking
After TBP, the volume of the spine head rapidly and persistently expanded in a portion of spines imaged (Fig. 1A, arrows), whereas other spines were unaffected (Fig. 1A, arrowheads). When all spines within the imaged regions near the stimulating electrode (Fig. 1D–F) were examined as a group, the spine volume 45 min after TBP was 141 ± 6% of the baseline level (Fig. 1B) (N = 146 spines/19 cells; p < 0.001 compared with baseline, paired t test). As is widely observed when eliciting LTP with Hebbian theta burst induction protocols (Magee and Johnston, 1997; Pike et al., 1999; van Praag et al., 1999; Golding et al., 2002; Watanabe et al., 2002), LTP immediately after TBP was small but then gradually increased to a final, stable potentiation over the next 30 min (Fig. 1C) (244 ± 17% at 45 min after TBP; N = 23; p < 0.001 compared with baseline, paired t test). The reason for the slow development of LTP under these conditions remains unknown; however, it does not involve a continual engagement of the LTP induction process by basal activity after TBP, because full LTP could still be observed 45 min after TBP even if stimulation was turned off during this period (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Figure 1.
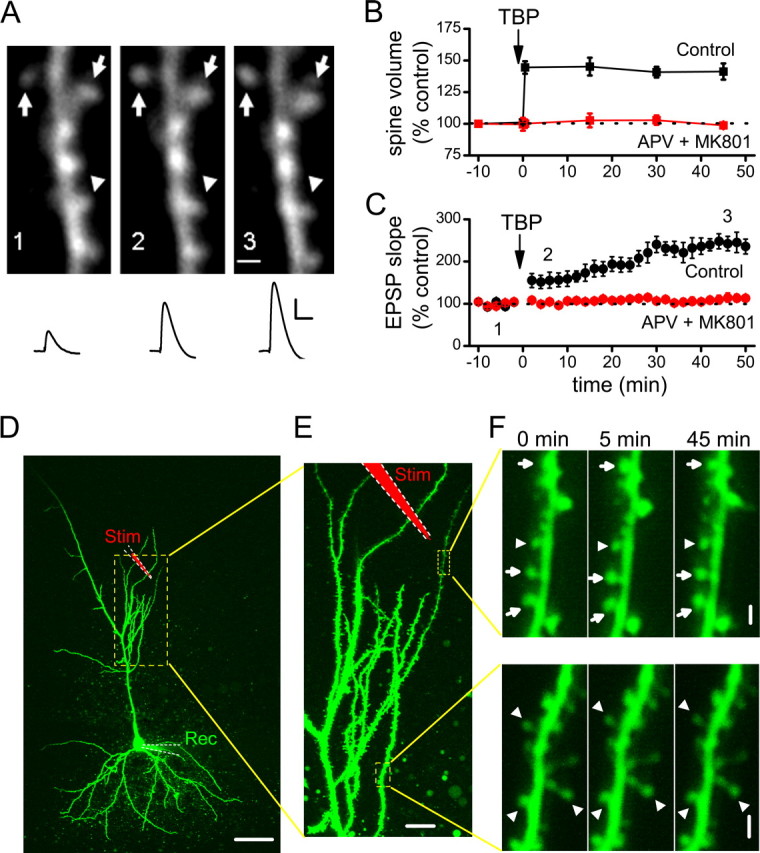
Induction of spine expansion and LTP with TBP. A, A representative experiment in which both spine size and synaptic responses (EPSPs) were monitored. TBP induces rapid expansion of spine heads in a portion of spines imaged (arrows), while leaving the rest of the spines unaffected (arrowheads). Representative EPSPs recorded at the same time points are shown under the images. Calibration: 5 mV, 50 ms. Scale bar, 1 μm. B, Population data showing the induction of spine expression associated with TBP-LTP. Spine head expansion occurred within 30 s of TBP, which was given at time “0.” This increase in spine volume was long-lasting, persisting for at least 45 min after TBP (black symbols), and was blocked by bath perfusion of NMDAR antagonists (APV and MK-801; red symbols). Error bars indicate SEM. C, The expression of LTP after TBP showed a biphasic time course, with a small (∼50%) but immediate increase in the EPSPs after TBP followed by a larger but gradual increase in the potentiation that reached a plateau ∼30 min after TBP (black symbols). LTP was also completely blocked by NMDAR antagonists (red symbols). D, An image showing a representative neuron recorded and imaged, together with the stimulating and recording electrodes. The local stimulating electrode is marked as red (Stim), whereas the recording electrode is marked green (Rec). The region inside the yellow dashed lines are shown at a higher magnification in E. Scale bar, 50 μm. E, Two regions being repetitively imaged sequentially are marked by the yellow dashed lines. Scale bar, 10 μm. F, Changes in spine size in the two imaged regions before and after TBP. A portion of spines in the region close to the stimulating electrode (top panel) showed persistent change after TBP (expanded spines are marked by arrows, whereas nonchanging ones are marked by arrowheads; the timing of image acquisition are indicated by the numbers above them. TBP was given at 0 min). Spines in the region far away (bottom panel) did not show persistent changes. These two regions were imaged subsequently within 1 min. Scale bar, 1 μm.
To determine that change in spine expansion was selectively occurring at the spines that were close to the stimulating electrode rather than a nonselective, cell-wide phenomenon, we monitored spines in two imaged regions ∼100 μm apart (Fig. 1D–F). One region was close (∼20–30 μm) (Fig. 1E, top dashed box) away from the stimulating pipette, whereas the other was far away (∼100 μm) (Fig. 1E, bottom dashed box). After giving TBP, only spines in the region close to the stimulating electrode showed persistent increases in size (Fig. 1F, top panels). This result confirmed the local nature of spine modification (Zhou et al., 2004); for the rest of the study, analysis was performed on spines within the image window that was close to the stimulating pipette, and all spines within that window were included in the analysis.
To determine whether both spine expansion and synaptic plasticity depend on NMDAR activation during TBP, we examined whether either effect occurred in experiments performed in the presence of NMDAR antagonists dl-APV (100 μm) and (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate (MK-801) (20 μm). Under these conditions, both LTP (Fig. 1C) (114 ± 9% at 45 min after TBP; N = 15; p = 0.78 compared with baseline) and spine expansion (Fig. 1B) (98 ± 2% at 45 min after TBP; N = 51 spines/5 cells; p = 0.71 compared with baseline) were completely abolished.
The involvement of NMDARs in spine expansion suggests that the induction of spine expansion may require activity in the postsynaptic cell, to remove the Mg2+ block of the NMDAR. Theta burst stimulation led to robust LTP and spine expansion when the EPSPs elicited spiking during the burst, as shown in a representative example (Figs. 1A, 2A) (for population data, see Fig. 1B,C), but not if the theta bursts were subthreshold (Fig. 2A; B,C; p < 0.001 compared with Fig. 1B,C). Thus, spine expansion in response to TBP requires activation of NMDARs and postsynaptic spiking, in common with LTP.
Figure 2.

Both spine expansion and LTP require postsynaptic spiking during theta burst stimulation. A, The expression of LTP depends on postsynaptic spiking, as shown in representative experiments. A1, Top, Sample voltage traces showing spiking activity triggered by EPSPs during TBP (top), and subthreshold EPSPs during TBS (bottom). Calibration: 25 mV, 25 ms. Bottom, Sample EPSP traces before and after TBP (left) or TBS (right). Calibration: 3 mV, 20 ms. A2, The same experiments as in A1 are shown, with EPSP slope plotted as a function of time. B, A summary of TBS experiments showed that TBS on its own did not alter synaptic transmission. C, No significant spine expansion was observed after TBS. Error bars indicate SEM.
Spine expansion after LTP is not restricted to small spines
The spine expansion induced by TBP was highly variable, doubling the volume of some spines while leaving others completely unaffected. To estimate the fraction of spines that undergo expansion during TBP, we examined the population changes in spine volume more quantitatively. To assess possible sources of noise in the measured spine volume, we normalized the volume of each spine to 100%, and then measured the volume a second time in the same conditions. Spine volume varied considerably from one measurement to the next (Fig. 3A, filled circles), but was normally distributed with no apparent drift or systematic bias.
Figure 3.
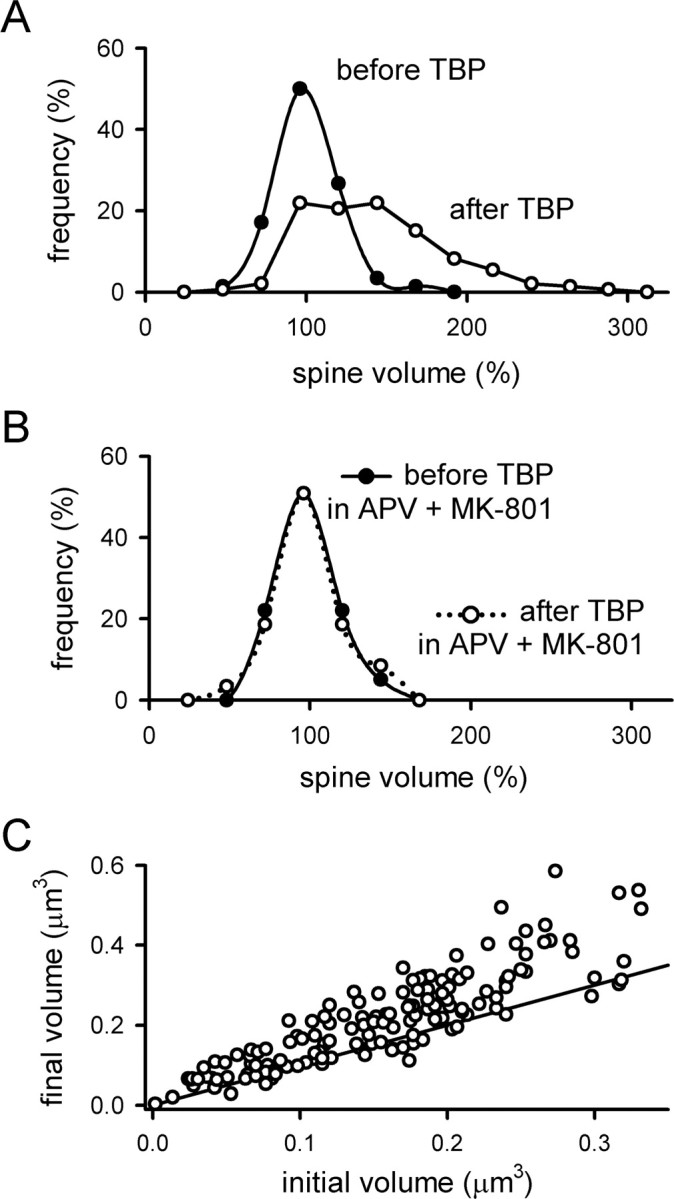
Analysis of changes in spine volume associated with TBP. A, The distribution of spine volumes was measured immediately before TBP (filled circles) and 45 min after TBP (open circles). In both cases, the volume of each spine was normalized to the values seen 10 min before TBP. Before TBP, the distribution is centered around 100%, indicating that there is no systematic drift in the measured mean spine volume between measurements. After TBP, a large number of spines have increased in volume, causing a spread in the distribution toward larger values. B, The distribution of normalized spine volumes was examined as in A, but when TBP was given in the presence of NMDAR antagonists (APV and MK-801). Neither the mean volume nor the distribution of volumes was affected by TBP in the absence of NMDAR activity. C, The increase in spine volume is not restricted to small spines. The absolute spine volumes immediately before and 45 min after TBP were plotted against each other for each spine. The line has a slope of 1, indicating no change in spine volume; points that fall above the line indicate an increase in volume. Spines showed an increase in volume over the entire range of initial sizes.
After TBP, the distribution of spine volumes shifted to the right and developed a significant skew toward larger spine volumes (Fig. 3A, open circles). The fraction of spines that did not expand with TBP was estimated by scaling the control distribution to the post-TBP distribution for spine volumes of ≤100%. Using this approach, we estimate that ∼60% of the spines expand in response to TBP, whereas the remaining 40% stay the same. As expected, when the same experiment was repeated in the presence of APV and MK-801, the distribution of spine volumes after TBP was identical to the distribution preceding TBP (Fig. 3B). The spines that did not expand might be genuinely unable to change in response to TBP, but an alternative is that these spines were postsynaptic to axons that were not adjacent to our stimulating electrode. We note that the overall fraction of spines that are competent to undergo expansion will depend on the size of the imaging window and the distance between the imaging window and stimulating electrode; however, the time course and stability of expansion in those spines that do expand should be independent of these factors.
It has been suggested that LTP induction selectively increases the size of small spines, but not large ones (Matsuzaki et al., 2004). We examined this issue by plotting the final spine volume 45 min after TBP against the initial spine volume of the same spine before TBP (Fig. 3C). Spines that do not expand should cluster around a line with a slope equal to 1, indicating that the spine volume before TBP is, on average, the same as the spine volume after TBP (Fig. 3C). Contrary to this expectation, we found that a substantial spine expansion was induced by TBP over the entire range of initial spine volumes. This conclusion is consistent with that of Kopec et al. (2006), and suggests that even large spines are competent to undergo expansion in response to TBP.
Spine expansion occurs but does not persist under conditions in which LTP is inhibited
It has long been thought that structural and synaptic plasticity are closely related, but it remains unclear how this might occur. We reasoned that if LTP and spine expansion are coupled, either by a direct interaction between the two processes or by shared signaling cascades, then manipulations that inhibit LTP should also prevent spine expansion.
As the first step to test this idea, we examined whether spine expansion was affected by selective inhibition of postsynaptic exocytosis. We did this because postsynaptic exocytosis is known to be required for LTP (Lledo et al., 1998), and for delivery of AMPARs to the spine surface (Shi et al., 1999; Park et al., 2004); thus, if spine expansion shares a common mechanism with LTP, we expect that both LTP and spine expansion should be blocked by inhibiting exocytosis. We therefore infused the light chain of botulinum toxin type B (BoTox) (0.5 μm), a neurotoxin that prevents exocytosis, into the postsynaptic cell through the patch pipette. To our surprise, the initial spine expansion after TBP occurred as usual in BoTox-treated cells (Fig. 4A,C) (peak expansion was 137 ± 3%; N = 61 spines/7 cells; p = 0.32 compared with control spine expansion), although LTP was significantly reduced (Fig. 4B) (121 ± 7% at 40 min after TBP; N = 7; p < 0.001 compared with control LTP; p = 0.12 compared with baseline) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). However, this expansion of the spine did not persist in BoTox-treated cells, and the average spine volume collapsed to control levels over the next 15–30 min (Fig. 4A,C). Thus, spine expansion does not require postsynaptic exocytosis, but the stability of this expansion does. This result suggests that LTP may share a common mechanism with the stabilization of spine expansion, rather than the initial expansion event.
Figure 4.
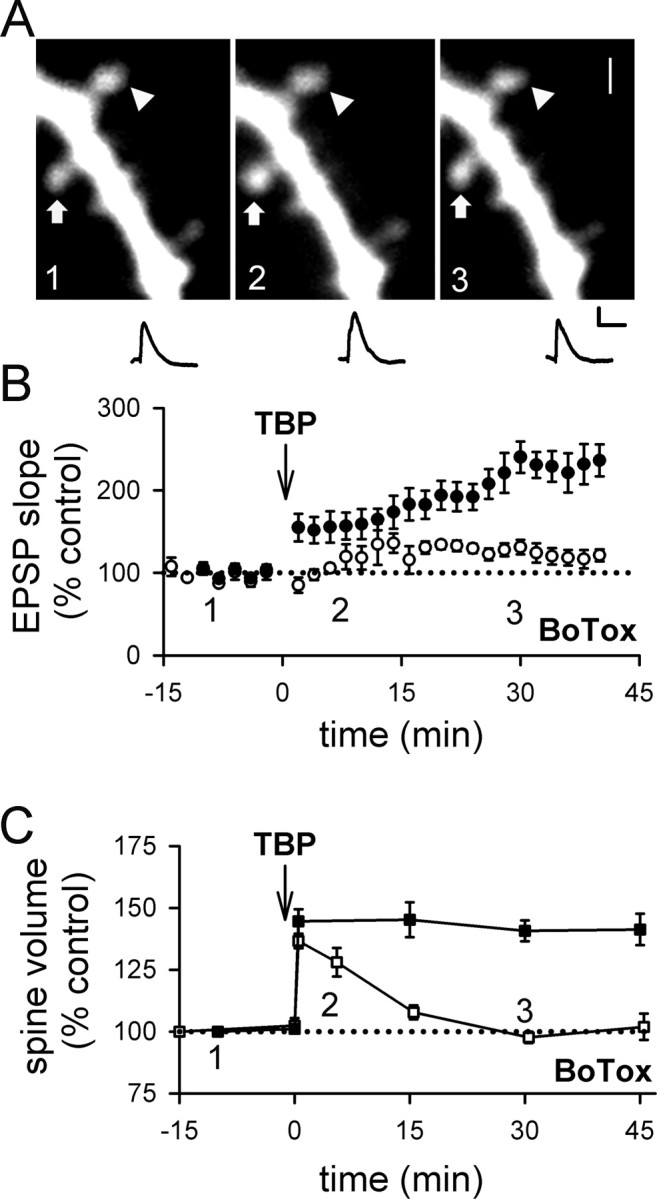
Inhibiting postsynaptic exocytosis reduces LTP and destabilizes spine expansion triggered by TBP. A, A sample experiment shows spine expansion occurred immediately after TBP, but did not persist and returned to the basal level in a neuron infused with BoTox through the patch pipette. An expanded spine is marked by an arrow, whereas an unaltered spine is marked by an arrowhead. EPSPs at the same time points in the same experiment are also shown. Calibration: 2 mV, 50 ms. Scale bar, 1 μm. B, In a population of neurons loaded with BoTox, TBP led to a gradual and small increase in EPSPs (open circles). In this and subsequent figures, responses from control neurons in Figure 1 are shown with filled circles for comparison. C, In neurons loaded with BoTox, the initial expansion occured normally in spines but the expansion was not stable and quickly returned to the baseline level (open squares). Spine expansion from control neurons are shown in filled squares for comparison. Error bars indicate SEM.
To test this idea further, we reasoned that LTP requires not only postsynaptic exocytosis, but also postsynaptic activation of protein kinases. Postsynaptic protein kinase A (PKA) is required for LTP under some conditions (Frey et al., 1993; Blitzer et al., 1995; Otmakhova et al., 2000; Duffy and Nguyen, 2003), particularly those involving postsynaptic spiking as part of the induction protocol (Brown et al., 2000; Huang et al., 2004). The specific processes affected by PKA remain unclear and may involve AMPAR trafficking and/or channel properties (Banke et al., 2000; Esteban et al., 2003; Oh et al., 2006). However, a significant body of literature suggests that PKA interacts with LTP in two ways: first, by activating a signaling cascade that involves protein synthesis and is required for some forms of LTP (for review, see Nguyen and Woo, 2003); second, by suppressing the activity of protein phosphatase 1 (PP1) to gate the induction of LTP (Blitzer et al., 1995, 1998; Brown et al., 2000). To inhibit PKA selectively in the postsynaptic neuron, we loaded the cell with the peptide inhibitor PKI (20 μm) in the internal pipette solution. Postsynaptic infusion of PKI reduced LTP (Fig. 5A) (153 ± 5%; N = 14; p < 0.05 compared with control LTP) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material) but not spine expansion (Fig. 5B) (139 ± 19%; N = 59 spines/6; p = 0.56 compared with control spine expansion), as was the case with BoTox. Also similar to the results seen with BoTox, spine expansion in the presence of PKI was unstable and quickly collapsed back to baseline values (Fig. 5B; supplemental Fig. 1C, available at www.jneurosci.org as supplemental material).
Figure 5.

Inhibiting PKA signaling reduces LTP and destabilizes spine expansion triggered by TBP. A, The gradually increasing component of LTP (as seen in control LTP in filled circles) was blocked in neurons loaded with PKI (open circles). PKI was loaded into the postsynaptic neurons with the recording patch pipette. B, Spine expansion occurred normally in PKI-loaded neurons but this expansion did not persist and quickly reversed back to pre-TBP level (open squares). Spine expansion in control neurons is shown in filled squares. C, Elevation of PKA in the postsynaptic cell is not sufficient to alter synaptic responses or spine size. In pyramidal cells infused with 8-Br-cAMP, neither EPSP slope (top; filled circles) nor spine size (bottom; filled squares) showed significant changes for 60 min after the onset of whole-cell configuration. D, Activation of postsynaptic PKA activity does not affect LTP expression. The enhancement in EPSP slopes by TBP was not different in cells loaded with 8-Br-cAMP (open circles) compared with control neurons (filled circles). E, Previous activation of postsynaptic PKA does not affect TBP-induced spine expansion. Both the initial occurrence and persistence of spine expansion was not affected in cells loaded with 8-Br-cAMP (open squares) compared with control neurons (filled squares). F, LTP was not affected by long baseline recording. At naive synapses, synaptic responses were recorded for 15–20 min before induction of LTP using TBP (open circles). This period of baseline corresponds to what was used for BoTox, PKI, or 8-Br-cAMP experiments, to allow diffusion of reagents to their synaptic targets. This 15- to 20-min-long baseline did not affect LTP induction or expression, because it was not different from LTP obtained with shorter baseline in control neurons (filled circles). Error bars indicate SEM.
Thus, PKA is required for the persistent expansion of spines in response to TBP. To address whether postsynaptic PKA is sufficient to drive spine expansion on its own at naive synapses, we loaded pyramidal cells with 8-Br-cAMP (1 mm), an activator of PKA, through the whole-cell patch pipette. Spine size was not affected in cells infused with 8-Br-cAMP (Fig. 5C) (N = 71 spines/8 cells). In addition, baseline synaptic responses were also not altered in cells infused with 8-Br-cAMP (Fig. 5C) (N = 7), consistent with previous studies (Blitzer et al., 1995; Makhinson et al., 2006). To test whether TBP-induced LTP or the associated spine expansion are affected by previous activation of postsynaptic PKA, we loaded pyramidal cells with 8-Br-cAMP for 15–20 min, and then applied TBP. No apparent changes in the magnitude or time course of LTP was seen (Fig. 5D) (224 ± 20% at 45 min after TBP; N = 8; p = 0.64 compared with control LTP), and no changes in the occurrence or stability of spine expansion was observed as well (Fig. 5E) (initial spine expansion with 142 ± 9%; N = 76 spines/6 cells; p = 0.78 compared with control initial spine expansion; and 151 ± 15% at 45 min; p = 0.56 compared with control spine expansion at 45 min). These results indicate that postsynaptic PKA activity is necessary for the spine expansion that occurs in response to TBP to persist, but it is not sufficient to elicit spine expansion in the absence of TBP.
One potential concern in the above experiments is that they required a period of 15 min to infuse these agents into the postsynaptic cell, before the induction of LTP. LTP is often reported to “wash out” during prolonged recordings, with the magnitude of LTP decreasing with time after obtaining the whole-cell configuration. To address the possibility that the selective loss of LTP was caused by washout, we performed control experiments in which normal internal solution was used but TBP was not applied until 15 min had passed (Fig. 5F). LTP under these conditions was unaffected by the prolonged baseline period (227 ± 23% at 45 min after TBP; N = 6; p = 0.59 compared with control LTP with 10 min baseline). Thus, the loss of LTP during infusion of postsynaptically loaded agents such as BoTox or PKI cannot be explained by washout.
Because PKA is required for both LTP expression and for stabilizing the spine expansion, we wondered whether these processes also depend on protein synthesis, which is thought to be a major effector of PKA. The protein synthesis-dependent phase of LTP is required to sustain the late-phase LTP (Huang et al., 1996; Pfeiffer and Huber, 2006). Recent works have indicated that protein synthesis occur very rapidly after the induction of LTP (Ouyang et al., 1999; Tsokas et al., 2005). It is unknown, however, whether protein synthesis is required for spine expansion to occur or to sustain it.
First, we incubated hippocampal slices with anisomycin (aniso) (20 μm), a peptidyl-transferase inhibitor (Rodriguez-Fonseca et al., 1995; Dinman et al., 1997), for 30 min before obtaining whole-cell recording in the pyramidal neurons. Anisomycin was also present in the perfusion ACSF throughout the entire recording period. After giving TBP in the same manner as in the previous experiments, the initial increase in EPSP slope was indistinguishable from that in control neurons; however, the slow rising phase of LTP was absent in these neurons (Fig. 6A,B) (128 ± 11%; N = 11; p < 0.001 compared with control LTP). This concentration of anisomycin did not affect basal synaptic transmission (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Interestingly, the initial spine expansion was not affected in neurons treated with anisomycin (Fig. 6A,C) (141 ± 7%; N = 80 spines/11 cells; p = 0.69 compared with control initial spine expansion), but this expansion was destabilized and returned to the baseline level spontaneously (Fig. 6A,C) (105 ± 4%; N = 80 spines/11 cells; p < 0.001 compared with control spine expansion). This behavior resembled that in neurons infused with BoTox or PKI (Figs. 4, 5). Incubation with anisomycin alone did not significantly affect the spine size at naive synapses (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
Figure 6.

Inhibiting protein synthesis reduces LTP and destabilizes spine expansion triggered by TBP. A, A representative experiment showing that EPSPs increased after TBP but decayed back toward baseline; spine expansion occurred but collapsed with time, in a neuron incubated with aniso. The sample EPSP traces and spine images were acquired at the time indicated on the graph below the images. Scale bar, 1 μm. Calibration: 5 mV, 50 ms. B, In neurons treated with aniso, TBP triggered an initial enhancement but no gradual increase in EPSPs with time (open circles). LTP in control neurons was shown in filled circles. C, The initial expansion of spines was unaffected by aniso treatment but the expansion was not sustained (open squares). Spine expansion in control neurons was shown in filled squares. D, A sample experiment showing that internal loading of cyclo led to similar changes in LTP and spine changes as in aniso-treated neurons. Scale bar, 1 μm. Calibration: 5 mV, 50 ms. E, Compared with control LTP (filled circles), the slow rising component of LTP was absent although the initial enhancement was intact (open circles) in neurons loaded with cyclo. F, Internal loading of cyclo did not affect the occurrence of spine expansion but inhibited its persistence (open squares). Spine expansion in control neurons is shown in filled squares. Error bars indicate SEM.
We also tested another protein synthesis inhibitor, cycloheximide, an inhibitor of translation (Rao and Grollman, 1967; Baliga et al., 1969; Obrig et al., 1971). In the above anisomycin experiments, the blockade could be mediated by inhibition of protein synthesis in either or both presynaptic and postsynaptic compartments. To determine whether protein synthesis in the postsynaptic neurons could affect LTP and spine expansion, we took advantage of the finding that internal loading of cycloheximide through whole-cell recording pipette selectively interfered with protein synthesis in the postsynaptic neurons (Yin et al., 2006). Cycloheximide (60 μm) was included in the patch pipette, and 15–20 min of baseline recording was allowed before the induction of LTP. The basal synaptic transmission was not affected by loading of cycloheximide (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). However, the slow rising phase of LTP was significantly reduced in the cycloheximide-filled neurons (Fig. 6D,E) (146 ± 22%; N = 7; p < 0.01 compared with control LTP), whereas the initial enhancement in spine volume was left intact (Fig. 6D,F) (134 ± 4%; N = 76 spines/7 cells; p = 0.19 compared with control initial spine expansion). Furthermore, spine expansion did not persist in these neurons (Fig. 6D,F) (107 ± 4%; N = 76 spines/7 cells; p < 0.001 compared with control spine expansion), comparable with our findings with anisomycin, PKI, and BoTox. When TBP was given in the presence of NMDAR antagonists, APV (100 μm) and MK-801 (20 μm), there was no apparent change in the size of EPSPs (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material) or spines (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material) in neurons loaded with cycloheximide. Thus, the residual LTP that was observed in the presence of protein synthesis inhibitors was NMDAR dependent. In addition, the destabilization of spine expansion was not caused by rundown of spine size that is triggered by TBP in the absence of protein synthesis.
Together, the above results unequivocally demonstrate that spine expansion can occur even when TBP-induced LTP is blocked, and is independent of postsynaptic exocytosis, PKA activity, and protein synthesis. This indicates that LTP and spine expansion are mechanistically dissociable, downstream of NMDAR activation. However, the expression of TBP-induced LTP and the consolidation of spine expansion appear to share a much stronger mechanistic overlap, because both processes are blocked by postsynaptic interference with PKA, exocytosis, and protein synthesis.
Spine stabilization and LTP consolidation are both disrupted by activity after TBP
The above results indicate that spine expansion is labile and reverses in the absence of stabilization. This observation is broadly reminiscent of LTP, which has a labile time period of ∼30 min after induction during which the reversal of LTP (or depotentiation) is easier (Larson et al., 1993; O'Dell and Kandel, 1994; Zhou et al., 2003). Within this time window, LTP can be depotentiated by low-frequency activity (LFS), through a mechanism that requires PP1 (O'Dell et al., 1994; Zhou et al., 2003). PKA can prevent depotentiation by leading to phosphorylation of inhibitor 1, which in turn inhibits PP1 (Kang-Park et al., 2003; Makhinson et al., 2006). To determine whether the consolidation of spine expansion is subject to reversal through mechanisms that are similar to those involved in depotentiation, we examined whether spine expansion is subject to activity-dependent reversal, and if so, whether the same mechanisms are involved in this reversal of spine expansion as in synaptic depotentiation.
First, we applied TBP, and then gave LFS (1 Hz for 5 min) 15 min later. LFS partially suppressed the full expression of LTP (Fig. 7A) (159 ± 14% at 40–45 min after TBP; N = 14; p < 0.05 compared with control LTP) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), and the spine volume after LFS fell to pre-TBP levels (Fig. 7B) (107 ± 5%; N = 65 spines/7 cells; p = 0.11, compared with baseline; p < 0.001, compared with spine expansion in control neurons) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Thus, spine expansion can be reversed by activity.
Figure 7.

LFS reverses spine expansion and blocks LTP. A, LFS (1 Hz for 5 min; bar) given at 15 min after TBP prevented the full expression of LTP (open circles). LTP from control neurons are shown in filled circles. B, Stable expansion of spines was disrupted by LFS given at 15 min after TBP (open squares). This reversal developed after the termination of LFS. Spine expansion in control neurons is shown in filled squares for comparison. C, Internal loading of PP1/2A inhibitor, OA, abolished the effect of LFS on LTP. Experiments were performed as in A, except that neurons were loaded with OA through the recording pipette (open circles). Control LTP is shown in filled circles. D, Internal loading of OA also prevented the LFS-induced reversal of spine expansion (open squares). Spine expansion was persistent in OA-loaded neurons, although LFS was given as in B. Spine expansion in control neurons is shown in filled squares. E, Activation of postsynaptic PKA inhibited the effect of LFS on LTP. The full expression of LTP in cells loaded with 8-Br-cAMP (open circles) was not different from that in the control neurons (filled circles). F, Elevation of postsynaptic PKA blocked LFS-induced reversal of spine expansion. Both the initial occurrence and persistence of spine expansion were not different between cells loaded with 8-Br-cAMP (open squares) and control neurons (filled squares). Error bars indicate SEM.
To identify the mechanisms underlying this effect, we examined whether spine expansion could be blocked by the PP1/2A inhibitor okadaic acid (OA). When the same experiment was repeated in cells loaded with the PP1/2A inhibitor okadaic acid, LFS had no effect on either LTP (Fig. 7C) (275 ± 43% at 40–45 min after TBP; N = 5; p = 0.95 compared with control LTP) or spine expansion (Fig. 7D) (140 ± 15%; N = 54 spines/6 cells; p = 0.95 compared with control spine expansion). It has been suggested that PP1 can be endogenously inactivated by PKA (Kang-Park et al., 2003; Makhinson et al., 2006), and so we would anticipate that persistently elevated PKA activity in the postsynaptic cell would have an effect similar to that of okadaic acid. We tested this possibility by including 8-Br-cAMP in the recording pipette solution, and we repeated the above experiments using LFS. Consistent with Kang-Park et al. (2003), LFS lost its effect on LTP (Fig. 7E) (259 ± 36%; N = 8; p = 0.69 compared with control LTP). In addition, spine expansion was not reversed by LFS (Fig. 7F) (spine expansion at 45 min with 146 ± 7%; N = 58 spines/6 cells; p = 0.64 compared with control spine expansion and p = 0.43 compared with initial spine expansion) in neurons loaded with 8-Br-cAMP.
The above results suggest that spine expansion can be reversed by LFS. However, we previously found that LFS with longer duration (15 min instead of 5 min), which induces LTD, can induce spine shrinkage even at naive synapses (Zhou et al., 2004). Thus, it is possible that the reduction in spine volume seen here after LFS is not a reversal of spine expansion, but a spine shrinkage that is independent of previous expansion. One argument against this idea is that the spine shrinkage that accompanies LTD is not blocked by okadaic acid (Zhou et al., 2004); however, to examine this issue more completely, we considered two experimental tests to distinguish between the two possibilities.
First, if the reduction in spine volume after LFS is a bona fide spine shrinkage that is independent of spine expansion, LFS should be able to reduce spine volume independent of when the spine expansion occurred. Contrary to this prediction, LFS applied 35 min after TBP had no effect on either LTP (Fig. 8A) (272 ± 48%; N = 10; p = 0.85 compared with control LTP) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material) or spine volume (Fig. 8B) (146.8 ± 4.5%; N = 52 spines/6 cells; p = 0.88 compared with control spine expansion) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), indicating that both processes are consolidated into a stable form within this time period.
Figure 8.

Reversal of spine expansion and inhibition of LTP is time dependent. A, LTP was not affected when TBP was given at 35 min after TBP (open circles). LTP from control neurons is shown in filled circles. B, Spine expansion was unaffected when LFS was given at 35 min after TBP (open squares). Expansion in control neurons is shown in filled squares. C, LFS did not alter responses at naive synapses. D, LFS also did not affect the size of naive spines. E, Summary of the effects of LFS on LTP consolidation under various conditions. All experiments except those in the last panel (LFS without TBP) received TBP before LFS. Values were taken from 40 to 45 min after TBP, or 40–45 min after LFS (for LFS without TBP). F, Summary of the effect of LFS on spine size. Results are displayed in the same manner as in E. *p < 0.05; ***p < 0.001, all compared with control TBP experiments. Error bars indicate SEM.
As a second test to differentiate the reversal of spine expansion from spine shrinkage, we applied LFS (1 Hz for 5 min) to naive synapses. LFS had no effect on EPSP slope (Fig. 8C) (107 ± 12%; N = 5; p = 0.58 compared with baseline) or spine volume (Fig. 8D) (97.1 ± 6.6%; N = 52 spines/5 cells; p = 0.47 compared with baseline) at naive synapses, providing additional evidence that the reduction in spine volume induced by LFS is a reversal of spine expansion rather than spine shrinkage. These experiments are summarized in Figure 8, E and F. Thus, the reversal of spine expansion and depotentiation of LTP have similar requirements in terms of activity dependence for induction, time to consolidation, and dependence on PP1/2A.
Discussion
In this study, we showed that spine expansion shares a common trigger with LTP (NMDAR activation), but can occur under conditions in which LTP is inhibited downstream of NMDAR activation. Surprisingly, the stability of spine expansion appears to share several mechanistic features with LTP, although the initial expansion does not. These results suggest that the initial induction events underlying physiological and morphological aspects of synaptic plasticity are mostly distinct downstream of NMDAR activation, but that the long-term stability of the two types of plasticity share similar mechanistic requirements.
Spine expansion after TBP has three distinct phases
Our results indicate that spine expansion after TBP can be separated into three mechanistically dissociable phases: (1) an initial expansion, which requires postsynaptic spiking and NMDAR activation but not postsynaptic exocytosis, PKA activity, or protein synthesis; (2) a labile phase, in which expansion is stabilized by a mechanism that requires postsynaptic exocytosis, PKA, and protein synthesis, and can be disrupted by LFS via a PP1/2A-dependent mechanism; and (3) a consolidated state, in which expansion is no longer destabilized by LFS. This third phase occurs ∼30 min after the initial expansion.
It is somewhat unexpected that we found postsynaptic exocytosis not to be required for the initial spine expansion. The surface area of the spine presumably increases during expansion, generating an expectation that additional membrane must be incorporated to accommodate this increase. Consistent with this, postsynaptic exocytosis has been reported to provide membrane for spine expansion during chemically induced LTP in cultured hippocampal neurons (Park et al., 2006) or cultured hippocampal slices (Kopec et al., 2007). Park et al. used live-cell imaging and serial section electron microscopy to demonstrate that the stimuli to induce LTP promoted the mobilization of recycling endosomes and vesicles into spines, and blockade of recycling endosomal transport abolished LTP-induced spine formation. Kopec et al. (2007) found that both LTP and spine expansion were blocked when exocytosis is inhibited by the expression of a dominant negative syntaxin-13 construct.
However, it seems unlikely that rapid insertion of new membrane is an absolute requirement for rapid spine expansion, because the surface area of a single spine (<1 μm2) (Park et al., 2006) is negligible compared with the total surface area of a pyramidal cell (>10,000 μm2) (Ambros-Ingerson and Holmes, 2005), and lipids are presumably free to diffuse within the plasma membrane from the dendrite into the spine. One possible resolution for the discrepancy between our findings and those of Park et al. and Kopec et al. is that the surface area becomes more important as a limiting factor when LTP is simultaneously induced at all spines on a neuron by chemical induction protocols (used in those studies) than when it is induced at a small fraction of spines using extracellular stimulation (our method).
The spine expansion that we have observed is presumably a consequence of cytoskeletal reorganization during LTP. Theta burst protocols are known to cause long-lasting actin polymerization in dendritic spines (Okamoto et al., 2004; Lin et al., 2005), and this effect is presumably triggered by a rapid, phosphorylation-dependent inactivation of the actin depolymerizing protein cofilin (Fukazawa et al., 2003; Chen et al., 2007). This actin polymerization is required for spine expansion to occur (Matsuzaki et al., 2004) and can be reversed by LFS in a discrete time window 30 min after delivery of the theta bursts (Kramar et al., 2006), indicating that the time and activity dependence of actin dynamics is strikingly similar to our observations regarding spine size.
Are LTP and spine expansion related?
A key question is whether structural and physiological plasticity are coordinated and, if so, how this coordination takes place. The initial spine expansion superficially resembles LTP in that it requires both postsynaptic spiking and NMDAR activation. However, this rapid expansion of the spine still occurs under several different experimental conditions in which LTP is either blocked entirely or strongly reduced, during the postsynaptic infusion of BoTox or PKI, or under protein synthesis blockade. Thus, the mechanisms underlying spine expansion and LTP are both triggered by NMDARs, but are distinct.
Does this conclusion depend on the specific mechanisms by which our manipulations interfere with LTP expression? A considerable body of evidence suggests that postsynaptic exocytosis is an integral part of the mechanisms directly underlying LTP. However, the literature regarding PKA and protein synthesis is somewhat more controversial. Some evidence suggests that these components are integral to the signaling cascade that directly underlies a component of LTP that occurs late (>1 h) after tetanization (Frey et al., 1993); it has also been suggested that this component might be preferentially induced by theta burst induction protocols (Huang and Kandel, 2005). However, some caution is warranted. Protein synthesis has been found to be permissive, rather than a direct mediator, for other forms of synaptic plasticity in the hippocampus (Nosyreva and Huber, 2006), and our present data do not rule out this alternative. We emphasize that, for the purposes of this study, our main interest is not in defining the sequence of events that is integrally involved in LTP; rather, our interest is in whether structural plasticity is subject to the same requirements as physiological plasticity or whether the two processes can be dissociated. As a result, our conclusion that spine expansion can be dissociated from LTP is robust, regardless of whether our experimental manipulations interfere directly with the mechanisms that underlie LTP, or indirectly via regulation of the competence to undergo LTP.
However, we found that LTP has a substantially stronger overlap with the spine stabilization after expansion. To summarize our different experiments and the range of effects observed on LTP and persistent spine expansion, we grouped our results according to different experimental manipulations and plotted the increase in spine volume seen after 40–45 min or more against the increase in synaptic strength seen in the same experiments during the same time window (Table 1). The data indicated that the two processes were strongly correlated, with most manipulations either abolishing or strongly reducing both processes (APV, BoTox, PKI, aniso, cyclo, LFS at 30 s after TBP, LFS at 15 min after TBP) or having little effect on either (LFS at 35 min after TBP, LFS at 15 min after TBP in the presence of okadaic acid or 8-Br-cAMP).
Table 1.
Summary of changes in EPSPs and spine volumes after TBP under all the conditions examined
| Experiments | EPSP slope (%) | Spine volume (%) |
|---|---|---|
| Control | 247.9 ± 17.9 | 141.2 ± 6.4 |
| APV | 109.9 ± 10.3*** | 98.7 ± 1.6*** |
| BoTox | 121.3 ± 7.1*** | 101.9 ± 5.3*** |
| PKI | 159.0 ± 28.1* | 100.8 ± 4.6*** |
| 8-Br-cAMP | 224.3 ± 19.8 | 151.8 ± 14.6 |
| Aniso | 128.0 ± 11.1*** | 104.8 ± 3.6*** |
| Cyclo | 146.2 ± 22.0** | 107.8 ± 3.5*** |
| LFS at 30 s | 130.2 ± 18.8*** | 99.8 ± 5.7*** |
| LFS at 15 min | 159.2 ± 13.8*** | 106.6 ± 4.6*** |
| LFS at 15 min (OA) | 275.3 ± 42.9 | 139.8 ± 14.6 |
| LFS at 15 min (8-Br) | 259.5 ± 35.8 | 146.4 ± 7.1 |
| LFS at 35 min | 271.9 ± 48.2 | 146.8 ± 4.5 |
Normalized changes are calculated for each condition, typically around 40∼45 min after TBP. The numbers are mean ± SEM.
*p < 0.05;
**p < 0.01;
***p < 0.001.
How to account for these diverse requirements (PKA, exocytosis, and protein synthesis) for both the full expression of LTP and persistent spine expansion? One possibility is that these signaling components regulate both LTP and spine expansion through parallel, but distinct, effector mechanisms. However, an alternative is that structural and synaptic plasticity are coordinated, so that one does not occur without the other. A recent study by Kopec et al. (2007) favors the latter possibility, reporting that manipulations that selectively interfere with synaptic incorporation of AMPARs also block the persistent spine expansion associated with chemical LTP. Our own data do not decisively address the idea that structural and synaptic plasticity are coordinated, although it is generally compatible with it; however, if such a coordination takes place, our data indicate that it likely occurs after initial expression of plasticity, during consolidation of the plasticity. Additional investigation of this issue will be of interest.
If LTP and the stabilization of spine expansion share overlapping mechanisms, we might expect that the two processes would be reversed in parallel as well. Consistent with this, we found that both processes are subject to activity-dependent disruption for ∼30 min. In both cases, reversal of the TBP-induced plasticity requires the activity of PP1/2A. The simplest explanation for this result is that LTP and spine stabilization are similar because of a common dependence of both processes on overlapping signaling cascades or targets; however, an alternative is that the stability of spine expansion is directly regulated by the successful expression of LTP. We note that the activity-dependent reversal of spine expansion by LFS is clearly distinct from the de novo spine shrinkage we previously described (Zhou et al., 2004): (1) it has a lower activity threshold for induction, (2) it requires elevation of PP1/2A, and (3) it can be induced only in a discrete time window after the expansion. To our knowledge, this is the first report of a physiologically relevant mechanism by which NMDAR-dependent spine plasticity can be reversed.
Our results are generally compatible with a model of sequential expression of synaptic plasticity (Edwards, 1995; Lüscher et al., 2000; Lisman and Raghavachari, 2006), in which spine expansion and initial physiological potentiation constitute a first step in plasticity, which is followed by additional subsequent physiological changes over the next 15–30 min and eventually consolidation of both processes. A recent model (Lisman and Raghavachari, 2006) appears to account for most of the events examined in this study. The early expression of LTP and spine plasticity do not require protein synthesis and most likely are mediated by phosphorylation of existing AMPARs and actin polymerization. In the next 15–30 min, LTP and spine modification enter a protein synthesis-dependent phase. Synapse growth may be likely to occur (Toni et al., 2001; Ostroff et al., 2002), accompanied with the enlargement of PSDs (postsynaptic densities) and perhaps the formation of perforated synapses. These processes are accompanied with slow increase in the AMPAR responses, likely to be mediated by incorporation of new AMPARs at the synapses. Interaction between these events leads to stabilization of both functional and structural modifications.
In summary, the stabilization and reversal of LTP and spine expansion appear to share similar mechanisms, whereas the initial events of spine expansion appear to be distinct from those underlying LTP. Our results indicate that physiological and morphological plasticity share several mechanistic requirements during the consolidation phase that takes place late in the expression of synaptic plasticity, rather than during the initial expression phase of the plasticity that takes place shortly after induction.
Footnotes
This work was supported by grants from The Whitehall Foundation and Ellison Medical Foundation (Q.Z.) and by National Institute of Neurological Disorders and Stroke Grant R01 NS045101 (M.F.). We thank P. O'Brien for the help on the two-photon setup, Drs. P. Adams, R. Blitzer, G. Huntley, R. Nicoll, and M.-m. Poo for their critical comments on a previous version of this manuscript, and Dr. D. Benson for in-depth discussion of the results.
References
- Ambros-Ingerson J, Holmes WR. Analysis and comparison of morphological reconstructions of hippocampal field CA1 pyramidal cells. Hippocampus. 2005;15:302–315. doi: 10.1002/hipo.20051. [DOI] [PubMed] [Google Scholar]
- Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagal AA, Kao JP, Tang CM, Thompson SM. Long-term potentiation of exogenous glutamate responses at single dendritic spines. Proc Natl Acad Sci USA. 2005;102:14434–14439. doi: 10.1073/pnas.0501956102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliga BS, Pronczuk AW, Munro HN. Mechanism of cycloheximide inhibition of protein synthesis in a cell-free system prepared from rat liver. J Biol Chem. 1969;244:4480–4489. [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis FF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Wong T, Nouranifar R, Iyenger R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Neuronal activity regulates diffusion across the neck of dendritic spines. Science. 2005;310:866–869. doi: 10.1126/science.1114816. [DOI] [PubMed] [Google Scholar]
- Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyebgar R, Landau EM. Long-term potentiation induced by theta frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci. 2000;20:7880–7887. doi: 10.1523/JNEUROSCI.20-21-07880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond NL, Levy WB. Synaptic correlates of associative potentiation/depression: an ultrastructural study in the hippocampus. Brain Res. 1983;265:21–30. doi: 10.1016/0006-8993(83)91329-x. [DOI] [PubMed] [Google Scholar]
- Desmond NL, Levy WB. Changes in the numerical density of synaptic contacts with long-term potentiation in the hippocampal dentate gyrus. J Comp Neurol. 1986;253:466–475. doi: 10.1002/cne.902530404. [DOI] [PubMed] [Google Scholar]
- Dinman JD, Ruiz-Echevarria MJ, Czaplinski K, Peltz SW. Peptidyl-transferase inhibitors have antiviral properties by altering programmed-1 ribosomal frameshifting efficiencies: development of model systems. Proc Natl Acad Sci USA. 1997;94:6606–6611. doi: 10.1073/pnas.94.13.6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy SN, Nguyen PV. Postsynaptic application of a peptide inhibitor of cAMP-dependent protein kinase blocks expression of long-lasting synaptic potentiation in hippocampal neurons. J Neurosci. 2003;23:1142–1150. doi: 10.1523/JNEUROSCI.23-04-01142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA. Anatomy and electrophysiology of fast central synapses lead to a structural model of long-term potentiation. Physiol Rev. 1995;75:759–787. doi: 10.1152/physrev.1995.75.4.759. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci USA. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Morales M. Actin matrix of dendritic spines, synaptic plasticity, and long-term potentiation. Int Rev Cytol. 1992;139:267–307. doi: 10.1016/s0074-7696(08)61414-x. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Van Harreveld A. Long-lasting morphological changes in dendritic spines of dentate granular cells following stimulation of the entorhinal area. J Neurocytol. 1977;6:211–230. doi: 10.1007/BF01261506. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Harris KM. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Majewska AK. Dendritic spine geometry: functional implication and regulation. Neuron. 2005;46:529–532. doi: 10.1016/j.neuron.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Theta frequency stimulation up-regulates the synaptic strength of the pathway from CA1 to subiculum region of hippocampus. Proc Natl Acad Sci USA. 2005;102:9365–9370. doi: 10.1073/pnas.0408368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Nguyen PV, Kandel ER. Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn Mem. 1996;3:74–85. doi: 10.1101/lm.3.2-3.74. [DOI] [PubMed] [Google Scholar]
- Huang YY, Pittenger C, Kandel ER. A form of long-lasting, learning-related synaptic plasticity in the hippocampus induced by heterosynaptic low-frequency pairing. Proc Natl Acad Sci USA. 2004;101:859–864. doi: 10.1073/pnas.2237201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Park MH, Sarda MA, Jones KH, Moore SD, Shenolikar S, Clark S, Wilson WA. Protein phosphatases mediate depotentiation induced by high-intensity theta-burst stimulation. J Neurophysiol. 2003;89:684–690. doi: 10.1152/jn.01041.2001. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkotian E, Segal M. Spatially confined diffusion of calcium in dendrites of hippocampal neurons revealed by flash photolysis of caged calcium. Cell Calcium. 2006;40:441–449. doi: 10.1016/j.ceca.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci USA. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang C, Barco A, Zablow L, Kandel ER, Siegelbaum SA, Zakharenko SS. Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc Natl Acad Sci USA. 2004;101:16665–16670. doi: 10.1073/pnas.0407581101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Xiao P, Lynch G. Reversal of LTP by theta frequency stimulation. Brain Res. 1993;600:97–102. doi: 10.1016/0006-8993(93)90406-d. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramar EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Raghavachari S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci STKE. 2006;356 doi: 10.1126/stke.3562006re11. re11. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Makhinson M, Opazo P, Carlisle HJ, Godsil B, Grant GN, O'Dell TJ. A novel role for cyclic guanosine 3′,5′monophosphate signaling in synaptic plasticity: a selective suppressor of protein kinase A-dependent forms of long-term potentiation. Neuroscience. 2006;140:415–431. doi: 10.1016/j.neuroscience.2006.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Obrig TG, Culp WJ, McKeehan WL, Hardesty B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem. 1971;246:174–181. [PubMed] [Google Scholar]
- O'Dell TJ, Kandel ER. Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learn Mem. 1994;1:129–139. [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–545. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE. Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2000;20:4446–4451. doi: 10.1523/JNEUROSCI.20-12-04446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Eichenbaum H, Wiener SI, Wibble CG. Learning-related patterns of CA1 spike trains parallel stimulation parameters optimal for inducing hippocampal long-term potentiation. Hippocampus. 1991;1:181–192. doi: 10.1002/hipo.450010206. [DOI] [PubMed] [Google Scholar]
- Ouyang Y, Rosenstein A, Kreiman G, Schuman EM, Kennedy MB. Tetanic stimulation leads to increased accumulation of Ca2+/calmodulin-dependent protein kinase II via dendritic protein synthesis in hipocampal neurons. J Neurosci. 1999;19:7823–7833. doi: 10.1523/JNEUROSCI.19-18-07823.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. Current advances in local protein synthesis and synaptic plasticity. J Neurosci. 2006;26:7147–7150. doi: 10.1523/JNEUROSCI.1797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike FG, Meredith RM, Olding AW, Paulsen O. Rapid report: postsynaptic bursting is essential for “Hebbian” induction of associative long-term potentiation at excitatory synapses in rat hippocampus. J Physiol (Lond) 1999;518:571–576. doi: 10.1111/j.1469-7793.1999.0571p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Grollman AP. Cycloheximide resistance in yeast: a property of the 60S ribosomal subunit. Biochem Biophys Res Commun. 1967;29:696–704. doi: 10.1016/0006-291x(67)90273-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Hof PR, Wearne SL. Rayburst sampling, an algorithm for automated three-dimensional shape analysis from laser scanning microscopy images. Nat Protoc. 2006;1:2152–2161. doi: 10.1038/nprot.2006.313. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Fonseca C, Amils R, Garrett RA. Fine structure of the peptidyl transferase center on 23 S-like rRNAs deduced from chemical probing of antibiotic-ribosome complexes. J Mol Biol. 1995;247:224–235. doi: 10.1006/jmbi.1994.0135. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Stewart MG, Medvedev NI, Popov VI, Schoepfer R, Davies HA, Murphy K, Dallerac GM, Kraev IV, Rodriguez JJ. Chemically induced long-term potentiation increases the number of perforated and complex postsynaptic densities but does not alter dendritic spine volume in CA1 of adult mouse hippocampal slices. Eur J Neurosci. 2005;21:3368–3378. doi: 10.1111/j.1460-9568.2005.04174.x. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Povilaitite P, Parisi L, Muller D. Remodeling of synaptic membranes after induction of long-term potentiation. J Neurosci. 2001;21:6245–62551. doi: 10.1523/JNEUROSCI.21-16-06245.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25:5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA. 1999;96:13427–13431. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XB, Yang Y, Zhou Q. Independent expression of synaptic and morphological plasticity associated with long-term depression. J Neurosci. 2007;27:12419–12429. doi: 10.1523/JNEUROSCI.2015-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearne SL, Rodriguez A, Ehlenberger DB, Rocher AB, Henderson SC, Hof PR. New techniques for imaging, digitization and analysis of three-dimensional neural morphology on multiple scales. Neuroscience. 2005;136:661–680. doi: 10.1016/j.neuroscience.2005.05.053. [DOI] [PubMed] [Google Scholar]
- Yin HH, Davis MI, Ronesi JA, Lovinger DM. The role of protein synthesis in striatal long-term depression. J Neurosci. 2006;26:11811–11820. doi: 10.1523/JNEUROSCI.3196-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Tao HW, Poo MM. Reversal and stabilization of synaptic modifications in a developing visual system. Science. 2003;300:1953–1957. doi: 10.1126/science.1082212. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]