

Abstract
Subunits located near the cardiolipin binding sites of bovine heart cytochrome c oxidase (CcO) were identified by photolabeling with arylazido-cardiolipin analogues and detecting labeled subunits by reversed-phase HPLC and HPLC–electrospray ionization mass spectrometry. Two arylazido-containing cardiolipin analogues were synthesized: (1) 2-SAND-gly-CL with a nitrophenylazido group attached to the polar headgroup of cardiolipin (CL) via a linker containing a cleavable disulfide; (2) 2′,2″-bis-(AzC12)-CL with two of the four fatty acid tails of cardiolipin replaced by 12-(N-4-azido-2-nitrophenyl) aminododecanoic acid. Both arylazido-CL derivatives were used to map the cardiolipin binding sites within two types of detergent-solubilized CcO: (1) intact 13-subunit CL-containing CcO (three to four molecules of endogenous CL remain bound per CcO monomer); (2) 11-subunit CL-free CcO (subunits VIa and VIb are missing because they dissociate during CL removal). Upon the basis of these photolabeling studies, we conclude that (1) subunits VIIa, VIIc, and possibly VIII are located near the two high-affinity cardiolipin binding sites, which are present in either form of CcO, and (2) subunit VIa is located adjacent to the lower affinity cardiolipin binding site, which is only present in the 13-subunit form of CcO. These data are consistent with the recent CcO crystal structure in which one cardiolipin is located near subunit VIIa and a second is located near subunit VIa (PDB ID code 1V54 referenced in Tomitake, T. et al. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15304−15309). However, we propose that a third cardiolipin is bound between subunits VIIa and VIIc near the entrance to the D-channel. Cardiolipin bound at this location could potentially function as a proton antenna to facilitate proton entry into the D-channel. If true, it would explain the CcO requirement of bound cardiolipin for full electron transport activity.
Cardiolipin (diphosphatidylglycerol, CL1) is a unique phospholipid found in membranes that couple electron transport to oxidative phosphorylation, for example, mitochondrial inner membrane and bacterial cytoplasmic membrane (2-4). In eukaryotes, CL is synthesized exclusively within the mitochondrion and is found only in the mitochondrial inner membrane. The structure of CL is quite different from other phospholipids. It contains three glycerols, two phosphates, and four highly unsaturated fatty acyl tails (Figure 1). Unlike the other major phospholipids of the inner mitochondrial membrane, CL is acidic and negatively charged. Cardiolipin is not a passive component of the inner mitochondrial membrane but is an indispensable factor for the proper function of a number of membrane-bound enzymes (5-7). Hormonal control of cardiolipin biosynthesis (8, 9) also suggests its possible role in regulation of mitochondrial function. In yeast, complete lack of CL (and PG) results in severe mitochondrial dysfunction due to the lack of translation of one of the nuclear-encoded components of the electron transport chain (10).
Figure 1.

Cardiolipin (diphosphatidyl-glycerol).
The best understood inner mitochondrial enzyme with an absolute requirement for CL is bovine heart cytochrome c oxidase (CcO; EC 1.10.2.2). Not only is cardiolipin associated with the purified complex, but three to four CL must be bound at specific sites to maintain its functional and structural integrity (5, 11-13). Without this bound CL, subunit interactions within the enzyme are destabilized and the enzyme is partially inactivated. Loss of enzymatic activity is directly related to the removal of CL, not the dissociation of subunits, since reassociation of CL at the specific binding sites restores full electron transport activity. Both the structural and functional stabilization of CcO is highly specific for CL; no other phospholipid can be substituted.
The CL bound to CcO can be divided into two classes based upon different binding affinities. One to two CL are bound less tightly and can be selectively removed by ion exchange chromatography in the presence of detergent (12). The remaining two CL are tightly associated with CcO and are difficult to remove by detergent extraction (5, 11). Coincident with removal of the loosely bound CL is the dissociation of subunits VIa and VIb2, two subunits located at the CcO dimer interface (PDB ID code 1V54 referenced in 14). The resulting 11-subunit form of CcO containing two tightly bound CL cannot dimerize due to the absence of VIa and VIb but retains full electron transport activity (12, 15). The two tightly bound CL subsequently can be removed by either phospholipase A2 digestion or extraction with a very high concentration of detergent. No other subunits dissociate from CcO upon removal of these tightly bound CL, but the resulting CL-free 11-subunit complex has only half of its original electron transport activity. Cardiolipin binding studies confirm the presence of two high-affinity and one or two lower-affinity binding sites (13). CcO, therefore, contains two types of CL binding sites: (1) two high-affinity sites that regulate electron transport and (2) one or two lower-affinity sites that stabilize subunits VIa and VIb and, therefore, CcO dimerization.
Previous attempts to locate the CL binding sites within CcO using chemical labeling approaches have led to relatively vague subunit assignments. CL has been reported to bind not only to one of the larger subunits, that is, I–IV (16), but also to one or more of the smaller subunits, that is, IV–VIII, (5, 17-19). The major difficulty encountered in each of these previous studies was that CL-modification of a CcO subunit(s) perturbed its migration on SDS–PAGE making positive identification impossible.
The most recent three-dimensional structure of bovine heart CcO reveals that two CL, four PG, three triglycerides, and four cholates are bound to each monomer within crystallized dimeric CcO (PDB ID code 1V54 referenced in 14). Whether the resolved CLs correspond to the two tightly bound CL revealed by our previous binding and functional studies is, of course, unknown. However, one of the resolved CLs is located adjacent to subunit VIa, consistent with our prediction that one or two of the lower-affinity CL bind near either subunit VIa or subunit VIb. The presence of four PG within crystallized CcO, however, is inconsistent with our biochemical analyses, which indicate phosphatidylglycerol is not present in our CcO preparations (PG/aa3 < 0.01).
To better define the binding locations of the two functional and one to two structural CL within CcO, we synthesized two new arylazido-CL derivatives. To avoid the problems encountered in previous studies, we utilized quantitative RP-HPLC subunit analysis and HPLC–ESI/MS to detect photolabeled subunits instead of SDS–PAGE. Results obtained from these approaches have allowed us to identify subunits located adjacent to the high-affinity, functionally important CL and those adjacent to the lower-affinity, structurally important CL.
EXPERIMENTAL PROCEDURES
Materials
Bovine CcO was prepared from Keilin–Hartree heart particles by the method of Fowler et al. (20) with modifications described by Mahapatro and Robinson (21). Heme content (9.4−9.9 nmol/mg), phosphorus content (15−20 P/aa3), and electron transport activity (340−360 s−1) were determined as described previously (22). Cytochrome c oxidase concentrations were calculated on the basis of ∊422 = 1.54 × 105 M−1 cm−1 (23). Individual drops of purified enzyme (20−25 mg/mL protein, 10 mg/mL sodium cholate, 100 mM phosphate buffer, pH 7.4) were quickly frozen by pipetting the solution into liquid nitrogen. Frozen 50 μL aliquots of enzyme were stored at −80 °C.
Phospholipase A2 was isolated from Crotalus atrox venom as described by Wells and Hanahan (24). Its purity was verified by SDS-PAGE. Only one Coomassie blue stained band at about 15 kDa was observed. Concentrations of phospholipase A2 were determined using = 27.2 M−1 cm−1 (24).
The C18 reversed phase column (10 μ, 4.6 mm × 250 mm, cat. no. 218YP104) was purchased from Vydac; the HiTrap Q HR column was from Pharmacia. Acetonitrile and phosphoric acid were of HPLC grade and were obtained from Fisher Scientific. HPLC grade chloroform and methanol were from EM Science. Beef heart cardiolipin and dodecylmaltoside were purchased from Avanti Polar Lipids and Antrace, Inc., respectively. All other chemicals were reagent grade.
Methods. Preparation of Cardiolipin-Free Cytochrome c Oxidase
Cardiolipin and other phospholipids were removed from purified CcO (15−20 P/aa3) using the phospholipase A2 digestion procedure of Sedlák and Robinson (12). Cytochrome c oxidase (40 μM) was incubated with PLA2 (40 μM) at room temperature in 50 mM TrisSO4, pH 7.4, containing 10 mM CaCl2 and 1 mg of dodecylmaltoside per mg of CcO. The PLA2 reaction was quenched with 50 mM EDTA after 3 h, and the resulting PL-free complex was isolated using HiTrap Q anion exchange chromatography in the presence of 0.5 mg/mL dodecylmaltoside. CL-free CcO eluted as a single elution peak at ∼0.3 M Na2SO4. The fractions containing CcO were pooled, the detergent concentration was adjusted to give a protein/detergent ratio of 1:1 (w/w), and the sample was dialyzed against buffer containing 0.1 mg/mL dodecylmaltoside to remove sodium sulfate.
Quantitation of CcO Bound Phospholipid and Cardiolipin
Total phospholipid and CL content of each form of CcO were each quantified after Bligh and Dyer extraction of lipids (25) in the presence of 2 M guanidinium chloride. Total phosphorus content (precision ± 0.4 P/aa3) was quantified by the phospho-molybdate colorometric method after wet ashing in perchloric acid (26). Total CL content (precision ± 0.1 CL/aa3) was quantified by silicic acid HPLC using Avanti Polar Lipid cardiolipin as a standard (27).
Synthesis of bis-(AzC12)-CL
Synthesis followed the procedure of Dale and Robinson (22) except the acylating carboxylic acid contained an aryl azide, that is, 12-[N-(4-azido-2-nitrophenyl)] aminododecanoic acid (ANPA) was used instead of a fatty acid (Figure 2). The synthesis involves (1) preparation of silicic acid HPLC purified dilyso-THP-CL, (2) DCC coupled acylation of the two free hydroxyl groups in dilyso-THP-CL using DMAP as a catalyst, (3) removal of the THP protecting group by exposure to 0.1 M HCl for 1 h, and (4) purification of the resulting product by normal phase silicic acid HPLC. All reactions and purification procedures were done either in the dark or under a dim red safe light. Reaction conditions were 2 μmol of silicic acid HPLC purified THP-CL, 40 μmol of ANPA, 60 μmol of DCC, and 198 μmol of DMAP reacted for 13 h at RT in 0.6 mL CH2Cl2. The aryl azide containing carboxylic acid, ANPA, was prepared as follows: (1) 4-fluoro-3-nitrophenyl azide (200 μmol in 1.3 mL ethanol) was added to 12-aminododecanoic acid (242 μmol) suspended in 1.3 mL of aqueous 1 M Na2CO3 and reacted in the dark for 24 h with stirring at 60 °C; (2) the reaction was stopped by acidification with HCl and phase-extracted three times with five volumes of ethyl ether, and the pooled ether fractions were dried under a stream of nitrogen. The orange-red product, ANPA (∊460 = 4.88 × 103 M−1 cm−1), was judged to be >99% pure by thin-layer chromatography after its purification by reversed phase HPLC.
Figure 2.

Synthesis of bis-(AzC12)-cardiolipin: (A) cardiolipin; (B) THP-CL; (C) dilyso-THP-CL; (D) 12-aminododecanoic acid; (E) 4-fluoro-3-nitrophenyl azide; (F) ANPA; (G) bis-(AzC12)-THP-CL; (H) bis-(AzC12)-CL.
Synthesis of SAND-gly-CL
The synthesis was done in three steps: (1) preparation of t-BOC-glycine anhydride; (2) synthesis of 2-glycyl-CL; (3) reaction of SAND with the free amino group in 2-gly-CL (Figure 3).
Figure 3.

Synthesis of SAND-gly-cardiolipin: (A) cardiolipin; (B) t-BOC-glycine; (C) t-BOC-glycine anhydride; (D) t-BOC-glycyl-CL; (E) 2-glycyl-CL; (F) SAND; (G) SAND-gly-CL.
(1) Synthesis of t-BOC-glycine Anhydride (Modified Method of Rammler and Khorana (28))
DCC (0.6 mmol) was reacted with 1.1 mmol of t-BOC-glycine in 3 mL of anhydrous ether at RT for 30 min. Insoluble DCC urea precipitated during the reaction. The soluble product, t-BOC-glycine anhydride, was separated from insoluble, crystalline DCC urea by filtration through a sintered glass funnel after addition of 5 mL of CHCl3 and 5 mL of CH2Cl2. A white crystalline mass containing predominantly t-BOC-glycine anhydride, together with some DCC urea, was obtained after removal of solvent from the filtrate. The remaining DCC urea was removed from t-BOC-glycine anhydride by shaking the product in 3 mL of anhydrous ether and removing the insoluble DCC urea by filtration. Approximately 0.1 g of crystalline t-BOC-glycine anhydride was collected after removal of ether.
(2) Synthesis of 2-Glycyl-CL
The disodium salt of CL (33.4 μmol) was reacted for 5 h under a stream of N2(gas) with 1.1 mmol of t-BOC-glycine in 3 mL of CHCl3 containing 57.5 μmol of 4-DMAP. After all of the CL had been acylated, as monitored by TLC on Whatman K5 silica plates using CHCl3/CH3OH/H2O (95:28:4 v/v/v) as the running solvent, excess t-BOC-glycine anhydride was hydrolyzed by addition of 1 mL of H2O and subsequent phase separation of the aqueous layer. DMAP was removed from the product, after its conversion to a salt by the addition of 50 μL of 5 N acetic acid, by two extractions with H2O. Water was removed from the CHCl3 layer containing the product t-BOC-gly-CL by addition of anhydrous Na2SO4. The t-BOC protecting group was then removed by addition of 30% trifluoroacetic acid (v/v) for 1 h (deprotection was judged complete by the generation of a phosphate and amine containing product, Rf = 0.4 after TLC on Whatman K5 silica plates with CHCl3/CH3OH/H2O (95:28:4 v/v/v) as the mobile phase. TFA was removed from the product by evaporation (N2 gas followed by vacuum). The liquid residue was dissolved in 3 mL of CH2Cl2 and washed once with 3% (w/w) aqueous solution of NaHCO3 followed by three washings with water, that is, until the pH of the aqueous layer was 7. Water was removed from the CH2Cl2 solution of 2-glycyl-CL by anhydrous Na2SO4, and the product was subsequently purified by silicic acid column chromatography (1 cm × 8 cm of 60−200 mesh) by elution with 15 mL of CH2Cl2 followed by 8 mL of CH3OH. The resulting product was judged to be pure by TLC using the running solvent CHCl3/CH3OH/H2O (95:28:4 v/v/v). The spot at Rf = 0.4 tested positive for phosphate and free amine. The yield of 2-glycyl-CL was 17.4 μmol using FW = 1551. 2-Glycyl-CL was further purified by silicic acid HPLC using cyclohexane/2-propanol/5 mM H3PO4 (50:50:2.75, v/v/v) as the elution solvent (29). 2-Glycyl-CL eluted as a single peak at 21 min compared with CL at 32 min.
(3) Synthesis of SAND-gly-CL
2-Glycyl-CL (6.45 μmol) was reacted for 24 h with SAND (13 μmol) in 1 mL of dry dimethylformamide under an atmosphere of N2(gas) in the presence of 4-DMAP (20 μmol) and triethylamine (100 μmol). The reaction was judged to be complete by the presence of a single phosphate positive but ninhydrin negative TLC spot (Rf slightly less than 2-glycyl-CL). The solvent dimethylformamide was removed by vacuum, and the residue was dissolved in 3 mL of CHCl3 (insoluble crystals were removed by filtration). The CHCl3 solution was washed six times with 3 mL of water. The yield of SAND-gly-CL (6.2 μmol) was determined using quantitative phosphate analysis (22). SAND-gly-CL was further purified by silicic acid HPLC as described above for 2-glycyl-CL. It eluted as a single peak at 17 min compared with CL at 32 min.
Photolabeling of CcO by Arylazido-CL Analogues
The CL binding sites within CL-containing 13-subunit CcO (3.5 CL per monomer) or CL-free 11-subunit CcO were photolabeled by bis-(AzC12)-CL or SAND-gly-CL as follows: (1) CcO, solubilized in 25 mM TrisSO4 buffer at pH 7.5 containing 1 mg of dodecylmaltoside per mg of protein, was incubated overnight in the dark at 4 °C with a 3−10 fold molar excess of the arylazido-CL. (2) Unbound bis-(AzC12)-CL or SAND-gly-CL was removed by anion-exchange HiTrap Q column chromatography in the dark (12). (3) The resulting azido-CL/CcO complex (4 μM) was photoactivated by a 30 min exposure to 460 nm monochromatic light in a quartz cuvette at 25 °C using a Fluorolog-3 spectrofluorometer (Jobin Yvon-Spex) equipped with a 450 W xenon lamp. Based on binding studies with 2-[14C]-acetyl-CL (13), a maximum of one to two and three to four arylazido-CL will remain bound to CL-containing CcO and CL-free CcO, respectively, after HiTrap Q chromatography. CcO oxidase treated in this way is entirely monomeric. Photolabeling of dimeric CcO was done after cholate-induced dimerization of dodecylmaltoside solubilized CcO as described by Musatov and Robinson (15). Exposure to the intense visible light did not cause a detectable perturbation in CcO, that is, the subunit content, visible spectrum, and enzymatic activity were each indistinguishable from CcO that had not been irradiated.
Subunit Labeling Quantified by RP-HPLC
Subunits photolabeled by either arylazido-CL are not eluted from a C4 or C18 reversed phase column with an acetonitrile gradient because of their greatly increased hydrophobicity. Photolabeling of a subunit, therefore, was quantified by its decreased yield from a C18 reversed phase HPLC column (30). Arylazido-CL photolabeling was quantified by comparing the percent yield of each subunit before and after photoactivation of the RP-HPLC purified, arylazido-CL-CcO complex. Subunit yields were also compared to HiTrap Q purified CcO that had not been exposed to arylazido-CL, before and after visible irradiation. All three controls gave nearly identical subunit yields, that is, within experimental error of the HPLC method. In each case, subunit yields were evaluated from the area under each HPLC elution peak after normalizing the chromatograms to make the subunit Va percent yields equal. Paired analyses were done in triplicate to determine the average percent photolabeling of each subunit. By this approach, arylazido-CL labeling of one or more of the 10 nuclear-encoded subunits, IV–VIII, were identified. Subunits I–III were not analyzed since they are not significantly labeled by arylazido-CL based upon SDS–PAGE analysis of 3[H]-ANPA-CL labeled CL-free CcO (5).
Identification of SAND-gly-CL Labeled Subunits by HPLC–ESI Mass Spectrometry
SAND-gly-CL labeled subunits were identified as 2-[m-amino-o-nitrobenzamido]-ethanethiol (ANBET)-modified subunits using HPLC–ESI/MS after cleavage of the internal disulfide and release of thiopropylglycyl-CL. Separation of CcO subunits was accomplished using a Michrom BioResources MAGIC 2002 micro HPLC. HPLC conditions were as follows: column, Michrom MAGIC MS C18 (1.0 × 150 mm, 5 μm, 200 Å); mobile phase, water/acetonitrile/n-propanol/acetic acid/trifluoroacetic acid (A, 98/1/1/0.1/0.01; B, 10/70/10/0.09/0.01); linear gradient, 2% B to 55% B in 50 min; flow rate 50 μL/min. ESI mass spectra were acquired on a Finnigan LCQ ion trap mass spectrometer using an electrospray voltage of 3 kV and a scan range of m/z 200−2000. Spectra of the CcO subunits were acquired in profile mode. Elution of ANBET-labeled subunits was detected using selected ion retrieval after calculation of expected charge states for CcO subunits having an increased mass of 239 Da.
RESULTS
Two new arylazido derivatives were synthesized from commercially available CL, bis-(AzC12)-CL (Figure 2), and SAND-gly-CL (Figure 3). Both were used to map subunits that are located near the CL binding sites of CcO. Because the photoreactive arylazido group(s) are on the end of the hydrocarbon tails within bis-(AzC12)-CL but linked to the polar headgroup in SAND-gly-CL, the two derivatives are expected to label quite different regions of CcO. The CL binding sites within two types of dodecylmaltoside solubilized CcO were investigated: (1) CL-free CcO (i.e., 11-subunit CcO that had all endogenous CL removed by PLA2 digestion with coincident removal of subunits VIa and VIb) and (2) CL-containing CcO (i.e., structurally intact 13-subunit CcO that contains three to four endogenous CL per monomer).
Covalent attachment of the very hydrophobic CL moiety to a subunit greatly increases its hydrophobicity so that it no longer elutes from a C18 RP-HPLC column with an acetonitrile gradient. No radioactivity is detected in the eluate when CcO photolabeled with [3H]-arylazido-CL is analyzed by RP-HPLC. Irreversible binding of CL-labeled subunits to a C18 RP column, therefore, was used to quantify photolabeling of 11 of 13 CcO subunits (photolabeling of subunits I and III cannot be quantified by this approach since unlabeled subunit I and III also bind irreversibly to a C18-RP column). The decreased yield of a particular subunit after arylazido-CL labeling directly assesses the extent to which it was photolabeled by either bis-(AzC12)-CL or SAND-gly-CL. The RP-HPLC subunit elution patterns of CL-free or CL-containing CcO did not significantly change when either type of CcO was incubated in the dark with bis-(AzC12)-CL and unbound ligand was removed by HiTrap Q ion exchange chromatography. However, significant decreases in the elution of several subunits occurred with both types of CcO upon photoactivation of the bound ligand (Figure 4A). With CL-free CcO the elution yield of subunits IV, VIIa, VIIb/VIIc, and VIII were consistently lower after photoactivation of bound bis-(AzC12)-CL (subunits VIIb and VIIc are not resolved; therefore, only the total yield of VIIb and VIIc can be determined). The average decreased yield of subunits VIIa, VIIb/VIIc, and VIII in three such experiments was 62% ± 15%, 44% ± 14%, and 25% ± 17%, respectively (Figure 5A). If only subunit VIIb or VIIc is photolabeled, the decreased yield of the modified subunit would be twice that observed for both subunits, that is, 88%. The yield of subunit VIII decreased in only two of three experiments; therefore, its associated error is quite large. Last, the decreased yield obtained for subunit IV (17% ± 3%) and subunit VIc (9% ± 7%), each of which contains a transmembrane helix on the surface of CcO, is indicative of the amount of nonspecific photolabeling obtained for a membrane-embedded subunit. Assuming a one to one relationship between photolabeling and loss of a subunit, a total of 1.5 mol of bis-(AzC12)-CL was incorporated, which corresponds to a photolabeling efficiency of 75% if two ligands are bound per enzyme. The fairly high labeling efficiency is probably due to the attachment of the arylazido groups at the ends of the hydrocarbon tails. This would place the bound aryl azide in an apolar environment, which is known to increase labeling efficiency due to the increased lifetime of the photogenerated nitrene (31). Bis-(AzC12)-CL photolabeling of monomeric, CL-containing CcO, which contains four filled CL binding sites, slightly decreased the yields of subunits VIIa, VIIb/ VIIc, and VIII (Figure 5A), but the errors associated with these decreases were nearly as large as the values themselves. The poor labeling of these subunits is almost certainly due to incomplete exchange of bis-(AzC12)-CL for two endogenous CL that are tightly bound near these subunits ([14C]-acetyl-CL binds at the two high-affinity sites only if the endogenous CL has been removed [13]). The inability of bis-(AzC12)-CL to exchange for endogenous CL would explain why the photolabeling of these subunits is not significantly higher than the nonspecific photolabeling of other transmembrane subunits, that is, subunits IV and VIc. However, subunit VIa within monomeric, CL-containing CcO was efficiently photolabeled by bis-(AzC12)-CL. The yield of subunit VIa decreased by 88% ± 8% after photoactivation (Figures 4B and 5A) indicating almost complete exchange of the photolabel for endogenous CL bound near this subunit. High-efficiency photolabeling of subunit VIa by bis-(AzC12)-CL occurred only with monomeric CcO; with dimeric CcO, only 13% of VIa was modified suggesting that dimerization blocks access of bis-(AzC12)-CL to this binding site. Photolabeling of subunit VIa also does not occur with CL-free CcO since the 11-subunit CL-free form of CcO does not contain subunit VIa.
Figure 4.

Detection of bis-(AzC12)-CL photolabeling of cytochrome c oxidase subunits by RP-HPLC subunit analysis. Percent photolabeling of each subunit was indirectly determined by quantifying the percent of each subunit that was unmodified after normalizing the two chromatograms to make the percent yield of subunit Va equal. Panel A shows RP-HPLC analysis of CL-free CcO subunits before (thin line) and after (thick line) photoactivation of bound bis-(AzC12)-CL. In this experiment, the yields of subunits IV, VIIa, VIIb/VIIc, and VIII decreased by 19%, 47%, 39%, and 12%, respectively. Panel B shows RP-HPLC analysis of CL-containing CcO subunits before (thin line) and after (thick line) photoactivation of bound bis-(AzC12)-CL. In this experiment, the yields of subunits IV, VIIa, VIIb/VIIc, and VIII decreased by 9%, 21%, 12%, and 17%, respectively. In both panels, the chromatogram for photolabeled CcO (thick line) has been offset by 0.6 min to improve clarity and to facilitate its comparison with the chromatogram for the unlabeled CcO control (thin line). In each analysis, 100 μg (0.5 nmol) of CcO was loaded on the RP-column, and the subunit content was quantitatively determined according to Liu et al. (30).
Figure 5.

Photolabeling of cytochrome c oxidase by bis-(AzC12)-CL or SAND-gly-CL. Panel A shows photolabeling of CL-free (filled bars) and CL-containing CcO (open bars) by bis-(AzC12)-CL. Panel B shows photolabeling of CL-free (filled bars) and CL-containing CcO (open bars) by SAND-gly-CL. Percent photolabeling was calculated from the decreased area of the RP-HPLC elution peak for enzyme that had been reconstituted with either bis-(AzC12)-CL or SAND-gly-CL, purified by HiTrap Q ion exchange chromatography to remove free ligand, and exposed or not exposed to intense visible light. Average values and standard deviations were calculated from three separate paired experiments. The percent labeling of subunit VIa is based upon the total decreased HPLC yield of subunit VIa plus Ac-VIa. In each analysis, 100 μg (0.5 nmol) of CcO was loaded on the RP-column, and the subunit content was quantitatively determined according to Liu et al. (30).
Photolabeling of CL-free and CL-containing CcO by SAND-gly-CL was qualitatively similar to that obtained with bis-(AzC12)-CL, but only subunit VIIa (50% ± 17%) had a decreased subunit yield that was significantly greater than other transmembrane subunits (Figure 5B). The yields of subunits VIIb/VIIc decreased by 13% ± 7%, which would correspond to 26% decrease in VIIc, if VIIb is not labeled (mass spectrometry analysis of SAND-gly-CL labeled CcO proved this to be the case; refer to next section). Total incorporation of SAND-gly-CL into CL-free CcO was significantly less than bis-(AzC12)-CL, only 0.7 mol per mol of enzyme. The lower labeling efficiency is probably due to the attachment of the arylazido group to the polar headgroup of CL via a long hydrocarbon spacer. This position would permit more mobility of the arylazido group and facilitate reaction of the photolabel in a hydrophilic environment with exposure to solvent and detergent, thereby decreasing photolabeling of the protein (31). Photolabeling of CL-containing CcO by SAND-gly-CL was fairly ineffective, suggesting very slow or poor exchange of SAND-gly-CL with bound endogenous CL (the yield of subunit VIa decreased by 9% ± 1%, but this not significantly greater than the nonspecific labeling of other subunits).
Reduction of the internal disulfide bond within SAND-gly-CL after photolabeling greatly facilitates identification of photolabeled products by RP-HPLC and RP-HPLC–ESI mass spectrometry. The resulting ANBET-modified subunits (Δmass = 239 Da, λmax = 374 nm) are much less hydrophobic than the initial SAND-gly-CL labeled subunits and, therefore, can be analyzed by RP-HPLC. Two new elution peaks with λmax = 374 nm were detected by RP-HPLC subunit analysis of SAND-gly-CL labeled CcO after exposure to dithiothreitol, suggesting that at least two CcO subunits contained ANBET (two peaks were detected at 374 nm; one eluted slightly before and the other at the same time as unmodified subunit VIIa). The most likely candidates to have been photolabeled are VIIa, VIIb, VIIc, or VIII since an ANBET-labeled subunit would be somewhat more hydrophobic and, therefore, have a slightly longer HPLC retention time than an unmodified subunit. RP-HPLC subunit analysis of CL-containing CcO, after photolabeling by SAND-gly-CL and reduction by dithiothreitol, gave very similar results. Both of the elution peaks with λmax = 374 nm and retention times between 43 and 46 min were detected suggesting chemical modification of the same two subunits.
The two SAND-gly-CL photolabeled CcO subunits were positively identified as ANBET-modified subunits by RP-HPLC–ESI/MS after reduction of the internal disulfide by dithiothreitol. With unlabeled 13-subunit CL-containing CcO, all 10 nuclear-encoded subunits were detected by on-line mass spectrometric analysis of the C18-RP column effluent. The ESI/MS derived molecular mass of each subunit agrees very well with the value calculated from its known amino acid sequence (Table 1). Selected ion retrieval was then used to detect ANBET-modified subunits within SAND-gly-CL photolabeled CcO that had been exposed to dithiothreitol. An ANBET-modified subunit should have a molecular mass 239 Da greater than the parent subunit. By this approach, significant elution peaks were detected corresponding to ANBET-modified subunits VIIa and VIIc (Figure 6). Ions used for the detection of the ANBET-modified subunits were as follows: unmodified subunit VIIa, m/z 1335.6 (5+) and 1669.5 (4+); ANBET-VIIa, m/z 1383.6 (5+) and 1729.2 (4+); unmodified subunit VIIc, m/z 1361.2 (4+) and 1814.5 (3+); and ANBET-VIIc, m/z 1420.9 (4+) and 1894.3 (3+). The molecular mass calculated from the multiply charged ions of each ANBET-labeled subunit (ANBET-VIIa, 6912.0 Da; ANBET-VIIc, 5679.7 Da) is in excellent agreement with that predicted for addition of an ANBET moiety of 239 Da (Table 1). As would be expected after covalent attachment of a relatively small apolar group, ANBET-VIIa and ANBET-VIIc each eluted slightly later than the corresponding unmodified subunit. No evidence was obtained for chemical modification of any other CcO subunit.
Table 1.
Molecular Masses of CcO Subunits before and after SAND-gly-CL Photolabeling and Dithiothreitol Cleavage of the Linker Disulfide
| subunit | calculated massa | unlabeled CcOb | ANBET-modified CcOc |
|---|---|---|---|
| IV | 17152.7 | 17152 | 17152 |
| Va | 12436.1 | 12436 | 12436 |
| Vb | 10670.1 | 10670 | 10670 |
| VIa | 9532.8 | 9534 | 9534 |
| Ac—VIb | 10067.3 | 10065 | 10065 |
| VIc | 8479.0 | 8479 | 8479 |
| Ac—VIc | 8522.0 | 8521 | 8521 |
| VIIa | 6673.8 | 6674 | 6674 |
| VIIa* | 6912.8 | d | 6912 |
| VIIb | 6357.2 | 6360 | 6360 |
| VIIc | 5441.4 | 5442 | 5441 |
| VIIc* | 5680.4 | d | 5680 |
| VIII | 4961.8 | 4963 | 4961 |
Calculated from the known amino acid sequences.
Unlabeled CcO or SAND-gly-CL before disulfide cleavage by dithiothreitol.
Analysis of CL-free CcO after SAND-gly-CL photolabeling and cleavage of the SAND-disulfide by dithiothreitol. Covalent attachment of the ANBET moiety should increase the molecular mass by 239 Da.
Not present.
Figure 6.

RP-HPLC–ESI/MS analysis of ANBET-modified CcO subunits. Eleven-subunit CL-free CcO was photolabeled with SAND-gly-CL, treated with dithiothreitol, and analyzed by RP-HPLC–ESI/MS, as described in Methods. The top panel shows the base peak chromatogram (plot of the intensity of the most abundant ion in each spectrum) for components eluting in the time range of interest (32−45 min). The lower four panels contain selected ion retrieval traces generated by the sum of the intensities of the indicated multiply charged ions. The resulting molecular mass of each species is indicated on the right side of each panel. Elution peaks labeled VIIa* and VIIc* are the ANBET-modified subunits, that is, +239 Da compared to unlabeled subunit VIIa and VIIc.
DISCUSSION
Two photoreactive arylazido derivatives of CL, bis-(AzC12)-CL and SAND-gly-CL, were synthesized and used to identify subunits located adjacent to the three to four CL binding sites of bovine heart cytochrome c oxidase. The two analogues are structurally different. Arylazido photolabels are attached at the ends of two of the four fatty acyl chains of CL in bis-(AzC12)-CL, while a single arylazido group is attached via a fairly long tether (length > 15 Å) to the 2′-hydroxyl of the CL polar headgroup in SAND-gly-CL. The two derivatives are, therefore, expected to label quite different regions of CcO near the CL binding sites. SAND-gly-CL also contains a cleavable disulfide within the linker region, while bis-(AzC12)-CL does not. Reduction of this disulfide after photolabeling permits RP-HPLC elution of labeled subunits, thereby facilitating subsequent identification of photolabeled subunits by mass spectrometry. The position of the aryl azide attachment to CL is not critical for its binding to CcO. Long chain fatty acids or large fluorescent groups linked to the 2′-hydroxyl of the bridge glycerol or arylazido groups placed at the end of one of the acyl groups do not alter CL binding to CcO (5).
Subunits near the CL-binding sites within two types of detergent-solubilized CcO were investigated: (1) intact CL-containing 13-subunit CcO, which has two high-affinity CL binding sites and one to two low-affinity sites (all are occupied by endogenous CL) and (2) CL-free 11-subunit CcO, which has only the two high-affinity CL bindings sites (CL was completely removed from both sites). CL-free CcO is also missing subunits VIa and VIb because they dissociate upon CL removal (12). Photolabeling differences between the two types of CcO enabled us to identify which CcO subunits are adjacent to the high- and low-affinity CL binding sites.
Photolabeled subunits were identified by two approaches: (1) RP-HPLC subunit analysis to quantify the amount of a subunit that remains unmodified; (2) RP-HPLC–ESI/MS to directly identify the resulting chemically modified subunit (the latter approach could only be used with SAND-gly-CL). Similar results were obtained by both approaches.
Subunits VIIa, VIIc, and possibly small amounts of subunit VIII were photolabeled in CL-free CcO by each arylazido-CL derivative indicating that the high-affinity CL binding sites are located near these subunits (refer to Figure 5). The fact that labeling patterns were qualitatively similar when the arylazido group was attached at the end of the hydrocarbon tails or to the bridge glycerol via a rather long tether suggests that CL is bound parallel to and between the transmembrane helices of these subunits. Photolabeling of these subunits was very low with the 13-subunit CL-containing form of CcO, indicating that the two arylazido-CL derivatives did not exchange well with endogenous CL tightly bound at the high-affinity sites located near these subunits. The other major difference in the photolabeling of the two forms of CcO was the very strong photolabeling of subunit VIa by bis-(AzC12)-CL (refer to Figure 5A), indicating that the arylazido-CL derivatives freely exchanged for the endogenous CL occupying this site, at least in monomeric CcO. Since intact 13-subunit CcO contains one to two additional lower-affinity CL binding sites not present in the 11-subunit CL-free preparation, we conclude that the lower-affinity site(s) is located adjacent to subunit VIa. Similar strong labeling of subunit VIa by SAND-gly-CL did not occur (refer to Figure 5B). The rather poor photolabeling of subunit VIa by SAND-gly-CL may be because the arylazido group is at the end of a long, flexible, somewhat hydrophilic linker, allowing it to swing away from subunit VIa and possibly react with solvent or detergent. It is, of course, possible that the bulkiness of the linker and arylazido group prevent association of the derivative at the CL binding site. Two facts argue against the latter explanation: (1) SAND-gly-CL did photolabel subunits VIIa, VIIc, and possibly VIII; therefore, the bulkiness of the linker and arylazido group does not prevent association of the derivative CL binding sites near these subunits. (2) Attachment of long chain fatty acids or large hydrophobic fluorescent groups to the hydroxyl group on the bridge glycerol of CL does not perturb the binding of CL to CcO. In an interesting supplementary finding, we found that efficient bis-(AzC12)-CL photolabeling of subunit VIa does not occur with dimeric CcO (only 13% is modified). The CL bound near subunit VIa must be buried between the two CcO molecules within dimeric CcO, thereby preventing free exchange of the photolabel for endogenous CL.
Upon the basis of these data, together with our previous results (5, 12, 13), we conclude that one CL (CL1) binds near subunit VIa to stabilize the association of VIa with remainder of CcO and, therefore, is required for CcO dimerization. Two other CLs (CL2 and CL3) bind near subunits VIIa, VIIc, and possibly VIII and influence the electron transport activity of CcO. The chemical photolabeling results identifying the binding sites for CL1 and CL2 are consistent with the known location of two CLs within the crystal structure of each CcO monomer (PDB ID code 1V54 referenced in 14). However, our data indicate that in addition to CL2, another CL (CL3) binds near subunit VIIa, although it was not detected in the crystal structure. We, therefore, propose that two CL sites are located near subunit VIIa, which we have labeled site VIIa-A and site VIIa-B (Figure 7). Furthermore, we believe that it is the binding of CL3 to site VIIa-B (the site not detected in the crystal structure) that alters the electron transport activity of CcO.
Figure 7.
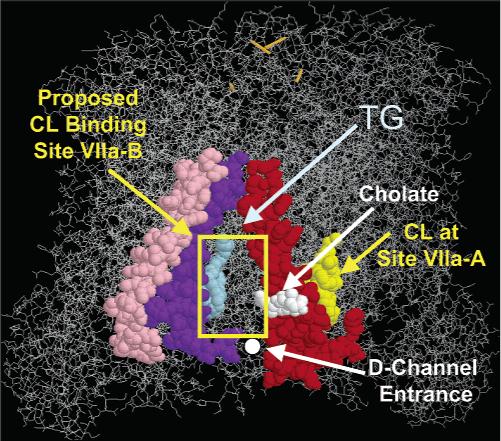
Cardiolipin binding sites near subunit VIIa. Two CL binding sites are proposed to be located adjacent to subunit VIIa. CL bound at the first site (site VIIa-A) is visible in the three-dimensional crystal structure of CcO. A second CL binding site (site VIIa-B) is proposed to be located between subunits VIIa (red) and VIIc (purple), in contact with subunit I (wireframe) and close to subunit VIII (pink). The CL headgroup would be bound adjacent to the entrance to the D-channel. A single CL (yellow), cholate (white), and triglyceride (TG, light blue) are located close to these subunits within the crystal structure. The figure was prepared using the atomic coordinates of CcO in the Protein Data Bank, PDB ID code 1v54 (14).
Cardiolipin Bound at Site VIIa-A
Eleven-subunit CL-free CcO contains two high-affinity CL binding sites. Removal of CL from one or both of these sites results in a 50% decrease in the kcat of CcO (5, 12). The present data indicate that both of these CL are bound near subunits VIIa, VIIc, and possibly VIII. Subunits VIIa and VIIc are definitely photolabeled by both bis-(AzC12)-CL and SAND-gly-CL. However, the evidence that subunit VIII is photolabeled is weaker. Quantitative RP-HPLC subunit analysis suggests that both arylazido-CL derivatives sometimes react with subunit VIII (the HPLC yield of VIII decreased in two of three experiments), but ANBET-modified subunit VIII was not detected by HPLC–ESI/MS. These photolabeling results are consistent with the location of one of the two CLs resolved within the crystal structure of dimeric CcO. This is the CL nestled between subunits VIIa and III. Arylazido-CL derivatives bound at this site would certainly photolabel subunit VIIa. However, it is unclear how removal of CL from this site could decrease the rate of electron transport by 2-fold.
Cardiolipin Bound at Site VIIa-B
We propose that a another CL (CL3) binds between subunits VIIa and VIIc (site VIIa-B, Figure 7) and it is CL bound at this site that alters CcO electron transport activity. The evidence for this second site is as follows. (1) Previous CL binding data indicate that two, not one, CLs bind to 11-subunit CL-free CcO (13). (2) Only subunits VIIa, VIIc, and possibly VIII are photolabeled by either arylazido-CL derivative. (3) It is nearly impossible to reconcile photolabeling of subunit VIIc and perhaps subunit VIII if bis-(AzC12)-CL binds only at site VIIa-A. The hydrocarbon tails are not long enough to reach either subunit VIIc or VIII if bis-(AzC12)-CL is bound behind subunit VIIa at this site. We, therefore, propose that the second high-affinity CL (CL3) binds to a hydrophobic patch on subunit I between subunits VIIa and VIIc (the yellow rectangle labeled site VIIa-B in Figure 7), a position occupied by a triglyceride and a cholate molecule in the crystal structure. A number of basic amino acid side chains are located near the N-terminus of subunits VIIa and VIIc, (e.g., Lys10, His21 and Arg33 of VIIa; His2, Lys9, and Arg20 of VIIc), which would facilitate binding of an anionic PL near this location. Binding of CL at this site would explain the photolabeling of both subunit VIIa and subunit VIIc and would also help to explain the 50% decrease in electron transport activity that occurs upon removal of the two tightly bound CLs (12). Binding of CL3 at site VIIa-B would place the polar headgroup of CL near the entrance to the D-channel, possibly serving as a proton antenna to facilitate proton input into the binuclear center [one pKa of CL is ∼7 (32)]. This would provide a plausible model for the functional role of CL in regulating CcO activity. Proton import into the binuclear center is rate limiting; therefore, if CL does facilitate proton access to the D-channel, its removal would result in decreased CcO activity.
Cardiolipin Bound near Subunit VIa
We previously predicted that one to two CLs must be loosely bound near either subunit VIa or VIb since both of these subunits dissociate during removal of the one to two lower-affinity CLs (12). This prediction is supported by the present data. Subunit VIa is strongly photolabeled by bis-(AzC12)-CL when it is exchanged into 13-subunit CcO, which contains subunit VIa. The fact that bis-(AzC12)-CL freely exchanged for endogenous CL bound at this site is fully consistent with the prediction that the lower-affinity binding sites of CcO are located near subunit VIa (CL1). It is presently not known whether one or two CLs bind near VIa. Previous CL binding studies indicated that the 13-subunit form of CcO contains two additional lower-affinity CL binding sites (13); however, many preparations of CcO contain three, not four, CLs per monomer suggesting that there is only one lower-affinity site (all preparations contain two high-affinity CL binding sites). The present data only prove that at least one of these low-affinity sites must be located near subunit VIa. Photolabeling of subunit VIa, however, is consistent with the location of the second CL resolved within the crystal structure of dimeric CcO. This CL is located at the dimer interface in contact with subunits III and VIa of one monomer and subunits I and II of the other monomer. This location is consistent with both the photolabeling data and the fact that CL can be selectively extracted from monomeric CcO by relatively low concentrations of dodecylmaltoside (12).
In summary, at least three cardiolipin binding sites are located on each CcO monomer. One CL (CL1) binds near subunit VIa, and CL binding at this site stabilizes the association of subunit VIa and, therefore, the CcO dimer. Another CL (CL2) binds behind subunit VIIa and may stabilize the association of subunit VIIa with subunit I. CL1 and CL2 bound at these sites have been resolved in the CcO crystal structure. We propose that the third CL (CL3) binds on subunit I, between subunits VIIa and VIIc. It is CL3 bound at this third site (site VIIa-B) that increases CcO electron transport activity, possibly by acting as a proton antenna to facilitate proton import into the binuclear center.
ACKNOWLEDGMENT
The authors thank Dr. LeAnn K. Robinson for her editorial help in preparing the manuscript. The authors also thank Drs. Jon Hosler and Andrej Musatov for invaluable discussions about these data.
Footnotes
This work was supported by grants from National Institute of Health (Grant NIH GMS 24795) and The Robert A. Welch Foundation (Grant AQ1481).
Abbreviations: CcO, bovine heart cytochrome c oxidase; CL, cardiolipin (diphosphatidylglycerol); PG, phosphatidylglycerol; bis-(AzC12)-CL, CL with the 2′ and 2″ fatty acids replaced by 12-[N-(4-azido-2-nitrophenyl)]-aminododecanoic acid; SAND-gly-CL, 2-[m-azido-o-nitrobenzamido]-ethyl-1,3′-dithiopropionyl-glycyl-CL; SAND, sulfosuccinimidyl-2-[m-azido-o-nitrobenzamido]-ethyl-1,3′-dithiopropionate; ANBET, 2-[m-amino-o-nitrobenzamido]-ethanethiol; ANPA, 12-[N-(4-azido-2-nitrophenyl)]-aminododecanoic acid; THP, tetrahydropyranyl; dilyso-THP-CL, 2-tetrahydropyranyl-dilyso-CL; pTSA, p-tolunesulfonic acid; DCC, N,N-dicyclohexylcarbodiimide; DMAP, 4-(N,N-dimethylamino)pyridine; DMF, N,N-dimethylformamide; TFA, trifluoroacetic acid; Et3N, triethylamine; PLA2, phospholipase A2 isolated from Crotalus atrox; TFA, trifluoroacetic acid; RP-HPLC, reversed-phase high-performance liquid chromatography; TLC, thin-layer chromatography; ESI/MS, electrospray ionization mass spectrometry.
CcO nomenclature is according to Kadenbach et al. (1).
REFERENCES
- 1.Kadenbach B, Jarausch J, Hartmann R, Merle P. Separtation of Mammalian Cytochrome c Oxidase into 13 Polypeptides by a Sodium Dodecyl Sulfate-Gel Electrophoresis Procedure. Anal. Biochem. 1983;129:517–521. doi: 10.1016/0003-2697(83)90586-9. [DOI] [PubMed] [Google Scholar]
- 2.Ioannou PV, Golding BT. Cardiolipins: Their Chemistry and Biochemistry. J. Lipid Res. 1979;17:279–318. doi: 10.1016/0079-6832(79)90010-7. [DOI] [PubMed] [Google Scholar]
- 3.Hoch FL. Cardiolipins and Biomembrane Function. Biochim. Biophys. Acta. 1992;1113:71–133. doi: 10.1016/0304-4157(92)90035-9. [DOI] [PubMed] [Google Scholar]
- 4.Schlame M, Rua D, Greenberg ML. The Biosynthesis and Functional Role of Cardiolipin. Prog. Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 5.Robinson NC. Functional Binding of Cardiolipin to Cytochromec Oxidase. J. Bioenerg. Biomembr. 1993;25:153–163. doi: 10.1007/BF00762857. [DOI] [PubMed] [Google Scholar]
- 6.Gomez B,, Jr., Robinson NC. Phospholipase Digestion of Bound Cardiolipin Reversibly Inactivates Cytochrome bc1. Biochemistry. 1999;38:9031–9038. doi: 10.1021/bi990603r. [DOI] [PubMed] [Google Scholar]
- 7.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Architecture of Succinate Dehydrogenase and Reactive Oxygen Species Generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 8.Hostetler KY. Effect of Thyroxine on the Activity of Mitochondrial Cardiolipin Synthase in Rat Liver. Biochim. Biophys. Acta. 1991;1086:139–140. doi: 10.1016/0005-2760(91)90165-e. [DOI] [PubMed] [Google Scholar]
- 9.Paradies G, Ruggiero FM, Dinoi P, Petrosillo G, Guagliariello E. Decreased Cytochrome Oxidase Activity and Changes in Phospholipids in Heart Mitochondria from Hypothyroid Rats. Arch. Biochem. Biophys. 1993;307:91–95. doi: 10.1006/abbi.1993.1565. [DOI] [PubMed] [Google Scholar]
- 10.Dowhan W, Mileykovskaya E, Bogdanov M. Diversity and Versatility of Lipid–Protein Interactions Revealed by Molecular Genetic Approaches. Biochim. Biophys. Acta. 2004;1666:19–39. doi: 10.1016/j.bbamem.2004.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson NC, Strey F, Talbert L. Investigation of the Essential Boundary Layer Phospholipids of Cytochrome c Oxidase Using Triton X-100 Delipidation. Biochemistry. 1980;19:3656–3661. doi: 10.1021/bi00557a003. [DOI] [PubMed] [Google Scholar]
- 12.Sedlák E, Robinson NC. Phospholipase A2 Digestion of Cardiolipin Bound to Bovine Cytochrome c Oxidase Alters both Activity and Quaternary Structure. Biochemistry. 1999;38:14966–14972. doi: 10.1021/bi9914053. [DOI] [PubMed] [Google Scholar]
- 13.Robinson NC, Zborowski J, Talbert LH. Cardiolipin-Depleted Bovine Heart Cytochrome c Oxidase: Binding Stoichiometry and Affinity for Cardiolipin Derivatives. Biochemistry. 1990;29:8962–8969. doi: 10.1021/bi00490a012. [DOI] [PubMed] [Google Scholar]
- 14.Tomitake T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. The low-Spin Heme of Cytochrome c Oxidase As the Driving Element of the Proton-Pumping Process. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musatov A, Robinson NC. Cholate-Induced Dimerization of Detergent- or Phospholipid-Solubilized Bovine Cytochrome c Oxidase. Biochemistry. 2002;41:4371–4376. doi: 10.1021/bi016080g. [DOI] [PubMed] [Google Scholar]
- 16.Fry M, Green DE. Cardiolipin Requirement by Cytochrome Oxidase and the Catalytic Role of Phospholipid. Biochem. Biophys. Res. Commun. 1980;93:1238–1245. doi: 10.1016/0006-291x(80)90622-1. [DOI] [PubMed] [Google Scholar]
- 17.Kuppe A, Mrsny RJ, Shimizu M, Firsan SJ, Keana JFW, Griffith OH. Labeling of Bovine Heart Cytochrome c Oxidase with Analogues of Phospholipids: Synthesis and Reactivity of a New Cardiolipin Benzaldehyde Probe. Biochemistry. 1987;26:7693–7701. doi: 10.1021/bi00398a024. [DOI] [PubMed] [Google Scholar]
- 18.Fowler WT, Lambeth JD, Powell GL. Photoreactive Cardiolipin Analogues. Chem. Phys. Lipids. 1988;47:261–271. doi: 10.1016/0009-3084(88)90049-7. [DOI] [PubMed] [Google Scholar]
- 19.Dale M, Robinson NC. Photoaffinity Labeling of the Cardiolipin Binding Site of Cytochrome c Oxidase. FASEB J. 1988;2:A774. [Google Scholar]
- 20.Fowler LR, Richardson SH, Hatefi Y. A Rapid Method for the Preparation of Highly Purified Cytochrome Oxidase. Biochim. Biophys. Acta. 1962;64:170–173. doi: 10.1016/0006-3002(62)90770-9. [DOI] [PubMed] [Google Scholar]
- 21.Mahapatro SN, Robinson NC. Effect of Changing the Detergent Bound to Bovine Cytochrome c Oxidase upon Its Individual Electron-Transfer Steps. Biochemistry. 1990;29:764–770. doi: 10.1021/bi00455a025. [DOI] [PubMed] [Google Scholar]
- 22.Dale MP, Robinson NC. Synthesis of Cardiolipin Derivatives with Protection of the Free Hydroxyl: Its Application to the Study of Cardiolipin Stimulation of Cytochrome c Oxidase. Biochemistry. 1988;27:8270–8275. doi: 10.1021/bi00421a042. [DOI] [PubMed] [Google Scholar]
- 23.van Gelder BF. Optical Properties of Cytochromes from Beef Heart Mitochondria, Submitochondrial Vesicles, and Derived Preparations. Methods Enzymol. 1978;53:125–128. doi: 10.1016/s0076-6879(78)53020-6. [DOI] [PubMed] [Google Scholar]
- 24.Wells MA, Hanahan DJ. Phospholipase A. I. Isolation and Characterization of Two Enzymes from Crotalus amadanteus Venom, (1969) Biochemistry. 1969;8:414–424. doi: 10.1021/bi00829a057. [DOI] [PubMed] [Google Scholar]
- 25.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 26.Marinetti GV. Chromatographic separation, identification, and analysis of phosphatides. J. Lipid Res. 1962;3:1–20. [Google Scholar]
- 27.Gomez B, Jr., Robinson NC. Quantitative Determination of Cardiolipin in Mitochondrial Electron Transferring Complexes by Silicic Acid High-Performance Liquid Chromatography. Anal. Biochem. 1999;267:212–216. doi: 10.1006/abio.1998.2998. [DOI] [PubMed] [Google Scholar]
- 28.Rammler DH, Khorana HG. Studies on Polynucleotides. XX. Amino Acid Acceptor Ribonucleic Acids (1). The Synthesis and Properties of 2′(or 3″)-0-(DL-Phenylalanyl)-adeno-sine, 2′(or 3′)-0-(DL-Phenylalany1)-uridine and Related Compounds. J. Am. Chem. Soc. 1963;85:1997–2002. [Google Scholar]
- 29.Robinson NC. Silicic Acid HPLC of Cardiolipin, Mono-, and dilysocardiolipin, and Several of their Chemical Derivatives. J. Lipid Res. 1990;31:1513–1516. [PubMed] [Google Scholar]
- 30.Liu Y-C, Sowdal LH, Robinson NC. Separation and quantitation of cytochrome c oxidase subunits by Mono-Q fast protein liquid chromatography and C18 reverse-phase high-performance liquid chromatography. Arch. Biochem. Biophys. 1995;324:135–142. doi: 10.1006/abbi.1995.9917. [DOI] [PubMed] [Google Scholar]
- 31.Brunner J. New Photolabeling and Cross-linking Methods. Annu. Rev. Biochem. 1993;62:483–514. doi: 10.1146/annurev.bi.62.070193.002411. [DOI] [PubMed] [Google Scholar]
- 32.Kates M, Syz J-Y, Gosser D, Haines TH. pH-Dissociation Characteristics of Cardiolipin and Its 2′-Deoxy Analogue. Lipids. 1993;28:877–882. doi: 10.1007/BF02537494. [DOI] [PubMed] [Google Scholar]