

Abstract
Lysyl hydroxylase 3 (LH3, encoded by PLOD3) is a multifunctional enzyme capable of catalyzing hydroxylation of lysyl residues and O-glycosylation of hydroxylysyl residues producing either monosaccharide (Gal) or disaccharide (Glc-Gal) derivatives, reactions that form part of the many posttranslational modifications required during collagen biosynthesis. Animal studies have confirmed the importance of LH3, particularly in biosynthesis of the highly glycosylated type IV and VI collagens, but to date, the functional significance in vivo of this enzyme in man is predominantly unknown. We report here a human disorder of LH3 presenting as a compound heterozygote with recessive inheritance. One mutation dramatically reduced the sugar-transfer activity of LH3, whereas another abrogated lysyl hydroxylase activity; these changes were accompanied by reduced LH3 protein levels in cells. The disorder has a unique phenotype causing severe morbidity as a result of features that overlap with a number of known collagen disorders.
Main Text
Lysyl hydroxylase (LH, E.C. 1.14.11.4), a member of the 2-oxoglutarate-dependent dioxygenase family, is an enzyme modifying lysyl residues in collagens,1,2 highly specialized proteins universally distributed in human body. LH has three isoforms (LH1, LH2, and LH3) with LH activity characterized in various tissues of higher species.3 In addition to lysyl hydroxylase activity,4,5 LH3 (encoded by PLOD3) has also collagen galactosyltransferase6 (GT, E.C. 2.4.1.50) and glucosyltransferase7,8 (GGT, E.C. 2.4.1.66) activities in vitro and in vivo.9,10 The multifunctionality of LH3 distinguishes it from the other LH isoforms. LH3 is evolutionarily the oldest of the LH isoforms,11 and this ancestral gene in lower-level species such as C. elegans is also multifunctional.3,6,8 LH3 is found in serum and in tissues, where this enzyme is also detected in extracellular space.12 Our recent data reveal the importance of extracellular LH3 in cell growth10 in vitro, but the in vivo function of LH3 in the extracellular space remains to be elucidated. LH isoforms have no strict sequence specificity, but there is a clear preference for some sequences to be bound and hydroxylated by certain isoforms in vitro.13 LH1 and LH3 have been shown to be responsible for hydroxylation of lysines destined for the triple helical region of collagens, and LH2 has been shown to function as a telopeptide lysyl hydroxylase.14–16 Our data suggest that LH3 is particularly responsible for hydroxylation of lysine residues in which hydroxylysines are further glycosylated.17
Analyses of the pyridinium crosslinks of collagen in urine provide a useful method of detecting certain connective tissue disorders. During such monitoring, we detected one patient lacking the disaccharide derivative of pyridinoline (Glc-Gal-PYD), a component that in normal urine comprises ∼15% of free pyridinoline crosslinks:18 There was no compensatory increase in the monosaccharide derivative, Gal-PYD (Figure 1). Further analysis of the urine sample also revealed abnormalities in hydroxylysine glycosides with a decreased ratio of disaccharide to monosaccharide (0.8) compared with that for two age-matched controls (1.3). The measured total hydroxyproline concentration in the urine was within the normal range at 287 μmol/mmol creatinine. The lack of glycosylated collagen crosslinks in the patient's urine suggested a defect of the enzyme glycosylating hydroxylysine residues, for which LH3 was the prime candidate. The suggestion was supported by our recent data from LH3-manipulated mouse lines9 revealing that LH3 is most likely to be solely responsible for GGT activity in mouse embryos. Our results also indicated that GGT activity correlated with the amount of LH3 in mouse tissues.12 In addition, the recent data17 from LH3-knockout embryonic fibroblasts indicated a smaller molecular size of types I, IV, and VI collagens in immunoblot analysis; such a finding suggests a loss of hydroxylysine glycosylations in polypeptides and thus confirms the role of LH3 in hydroxylysine glycosylation.
Figure 1.
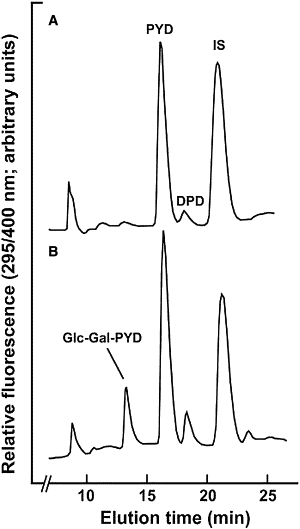
Collagen Crosslink Analysis
Patterns of collagen-derived pyridinium crosslinks in unhydrolyzed urine from (A) proband and (B) age-matched healthy control, analyzed as described previously.26 Hydroxylysine glycosides were analyzed by LC/MS/MS with multiple reaction monitoring27 with a ABI QTRAP 3200 instrument. After prefractionation of urines by partition chromatography on cellulose columns, the crosslink components were separated by RP-HPLC and quantified by their natural fluorescence (ex. 295: emm 400 nm) in relation to an internal standard (IS), O-acetyl-pyridinoline (DPD, deoxypyridinoline). Note the absence of Glc-Gal-PYD in (A): When present, Gal-PYD elutes as a partially resolved peak 0.5 min later than Glc-Gal-PYD.
The proband is female and the fourth liveborn infant of a healthy, nonconsanguineous couple of European descent. Pregnancy was complicated by intrauterine growth retardation (IUGR). Delivery was at 40 weeks, gestation and the infant was small for dates at 1.95 Kg (<0.3rd centile). Length and head circumference were also <0.3rd centile. Postnatally, growth continued below −2 standard deviations. Craniofacial characteristics included shallow orbits, a small nose, downturned corners of the mouth, low-set ears, and a generally flat facial profile (Figures 2A–2C). A right-sided diaphragmatic eventration was repaired when the patient was 5 months of age. The diaphragm was noted to be friable; the initial repair failed and a second surgical repair was needed. Skeletal problems included bilateral talipes equinovarus requiring surgical correction and flexion contractures at the proximal interphalangeal joints of the first, second, and third fingers. Knee joints appeared prominent. Scoliosis progressed from age 7 of the patient. No underlying vertebral malformations were found on X-rays. Platyspondyly with inferior beaking of the second lumbar vertebra, small capital femoral epiphyses, a J-shaped sella turcica, and small odontoid were noted, but no unifying diagnosis that could explain these features was suggested. Osteopenia and healed fractures of the left clavicle, right femur, and right humerus were seen. Skeletal muscle bulk was poor, but no investigations of muscle function were clinically indicated. Profound bilateral sensorineural deafness (no responses at >100 dB on brain-stem evoked responses) was treated with cochlear implantation. Visual acuities were reduced with myopia (right −2/−0.75 × 90, left −2.25/−0.50 × 180). Eye abnormalities were shallow anterior chambers, flat retinae, and cataracts requiring surgery at age 7 of the patient. Skin, hair, and nail abnormalities all presented in the first year of her life. Skin creases on both palms were reduced (Figure 2D). Blistering affecting all toes and fingers and blistering of the pinnae was present (Figure 2D). This healed without scarring. A skin biopsy taken outside the blistered areas was normal on light microscopy. The skin appeared to be thin and poorly perfused but with no significant abnormalities of healing or scarring. Hair was described as coarse in texture and rapidly growing with an unusual distribution (thick over the vertex and sparse at the hairline). All nails were hypoplastic, although the small, dysplastic fingernails were described as “rapidly-growing with brittle tips” (Figure 2E). Teeth were normal.
Figure 2.
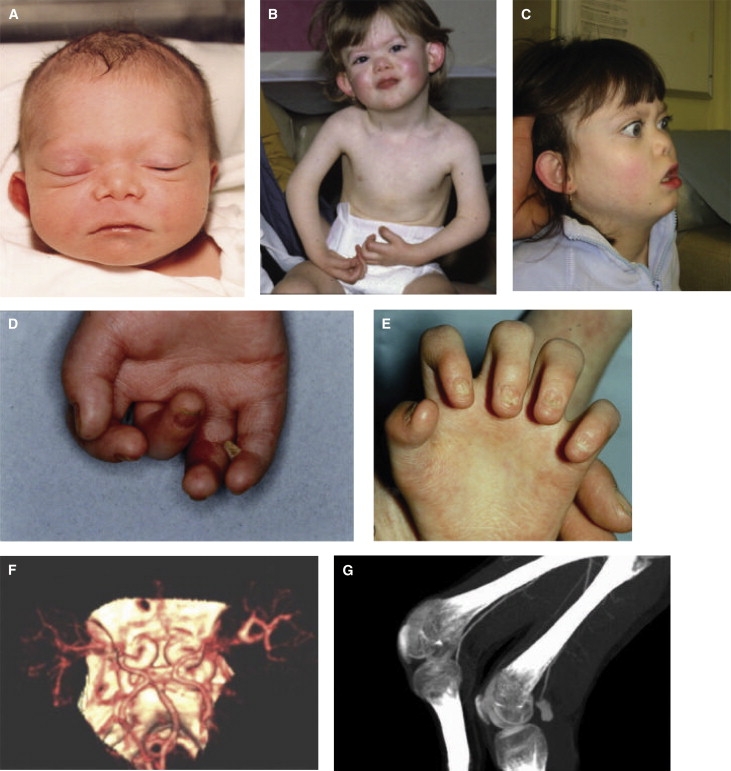
Clinical Findings of the Proband
(A) Patient at 1 month of age shows a short, upturned nose, long philtrum, and downturned angles of the mouth.
(B) Patient at 3 years of age shows protruding, simple pinnae, and flexion contractures of both elbows and all interphalangeal joints.
(C) Lateral photograph of a patient at age 14 years showing shallow orbits, flat facial profile, short nose with anteverted nares and low-set, simple ear.
(D) Palmar aspect of the right hand shows superficial peeling of the skin overlying the fourth and fifth middle phalanges (1 year).
(E) By age 14 of the patient, palmar skin shows reduced creases and all fingernails show evidence of progressive atrophy, unrelated to blistering.
(F) CT angiogram showing aneurysmal dilatation of arteries.
(G) Popliteal angiogram demonstrates aneurysmal dilatation of left popliteal vessels. Informed consent was obtained from the family involved in this study.
Bruising occurred readily but prothrombin and partial thromboplastin times and platelet counts were all found to be normal. In the second decade, spontaneous vascular ruptures occurred. At the time when the patient was 11 years old, a spontaneous cerebral arterial haemorrhage presented with hemiplegia. A large hematoma that was present in the white matter of the left cerebral hemisphere and involved both limbs of the internal capsule and basal ganglia was demonstrated by computed tomography (CT). A spontaneous rupture of a right popliteal aneurysm (demonstrated by CT angiography, Figure 2F) presented with pain and swelling. A subsequent CT scan when the patient was 14 years of age demonstrated gross dilatation of both internal carotids (Figure 2G). Development was globally delayed. She walked independently at 4 years of age. Communication was by sign language (Makaton and British Sign Language) that, by the time that the patient was 16 years old, was used to communicate basic needs and to ask simple questions. Social skills were also delayed and joint contractures impeded development of fine motor skills. Chromosomes showed a normal female pattern, 46,XX, with no evidence of mosaicism. Asymmetry of the palate raised the possibility of mosaicism; however, skin chromosomes were normal in 98/100 cells with a marker chromosome of unidentified origin in 2/100 cells representing possible culture artifact.
One male sibling was stillborn at 28 weeks' gestation. Intrauterine growth retardation complicated pregnancy. He had a pointed chin and small, round, low-set ears (Figure 3A). His middle fingers were long and flexed with the index finger overlapping (Figure 3B). Toe nails were hypoplastic. Autopsy revealed a porencephalic cyst with dilatation of the cerebral ventricles. Extensive petechial haemorrhages were seen on the skin. Chromosomes showed a normal male pattern, 46,XY, in cultured fibroblasts from a postmortem biopsy.
Figure 3.
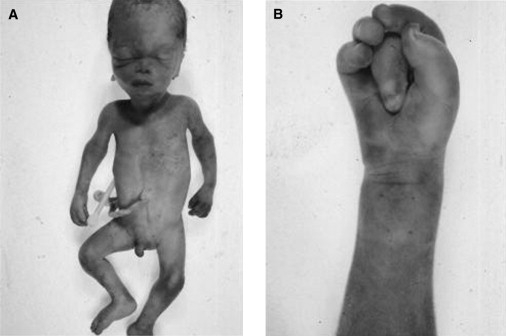
Fetal Photos
Postmortem photographs of stillborn fetus (A), showing a large cranial vault caused by cerebral ventricular dilatation. The male fetus is small for gestational age, with a short, upturned nose and extensive petechial skin haemorrhages, although delivery was not traumatic (that may reflect vascular fragility). (B) shows the right hand with flexion contractures of proximal interphalangeal joint, worst on the middle finger.
The first direct evidence that LH3 may be responsible for the disorder came from measurements indicating markedly reduced values of GGT activity in the proband's serum (Figure 4A) and in lymphoblastoid cells (Figure 4B) compared to controls. Western analysis revealed a low concentration of LH3 protein in lymphoblastoid cells compared to control cells (Figure 4C). In order to see possible mutations in LH3, total RNA was isolated from Epstein-Barr virus-immortalized lymphoblastoid cells of the proband. cDNA was synthesized with Dynabeads (Dynal) purified mRNA as a template in oligo(dT)-primed synthesis. The PCR amplified fragments of the cDNA were used for direct sequencing. LH3 cDNA (NM_001084.4) analysis (Figures 5A and 5B) revealed two heterozygous changes in the coding nucleotide sequence: one nucleotide transition c.668A→G (mutation 1) resulting in the amino acid substitution, p.Asn223Ser, and one nucleotide deletion c.2071 delT (mutation 2) causing a translational frameshift and generating a premature translational stop codon in the sequence (p.Cys691AlafsX9). The protein product resulting from mutation 2 consists of 698 amino acids, with Cys691 being the first changed amino acid. The premature termination does not make nonsense-mediated decay operative because the allele product could be seen in lymphoblastoid cells of the patient after digestion of cDNA fragments with the NarI restriction enzyme used to indicate deletion of the T in the cDNA sequence (data not shown, see also Figure 5D). This mutated allele produced a protein that was truncated by 40 amino acids and that deviated from the LH3 sequence by the eight terminal amino acids at the C-terminal end of the molecule. Both of the mutations are located in conserved regions of the LH3 amino acid sequence, and as indicated earlier,3,6–8 they are located in the regions responsible for glycosyltransferase and lysyl hydroxylase activities, respectively (Figure 6). Furthermore, Asn223 is highly conserved among species. Both nucleotide changes generated a new restriction site to the nucleotide sequence of PLOD3, so that restriction analysis could be utilized as the basis for a carrier test for the mother and father. Our restriction analysis indicated that mutation 1 was inherited from the father and mutation 2 was inherited from the mother (Figures 5C and 5D).
Figure 4.
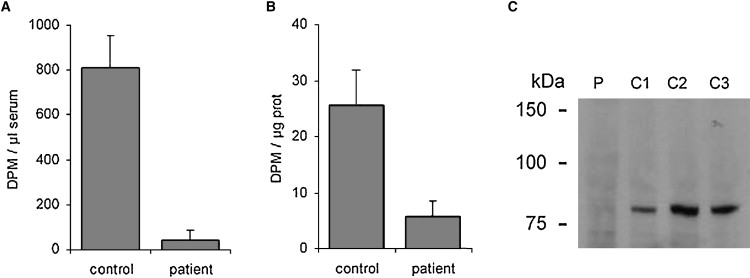
GGT Activity and LH3 Protein Level of the Proband
GGT activity in proband's serum (A) and in lymphoblastoid cells (B). The activity in the cells is expressed per μg soluble protein and compared with a mean value of two control cell lines. The activity in serum is expressed per μl serum and compared with a mean value of five control sera. The SD is indicated by the bars. LH3 protein level was analyzed by western-blot analysis (C) from lymphoblastoid cells of the patient (P) and from three control lymphoblastoid cells (C1-C3). A total of 2 mg soluble protein from lymphoblastoid cells was pulled down with Concanavalin A-sepharose (GE Healthcare) and applied to the lanes. Molecular-weight markers are indicated. A GGT activity assay was performed as described elsewhere.28 The proteins were fractionated under reducing conditions by 7.5% SDS-PAGE, transferred onto an Immobilon-P membrane (Millipore), and incubated with a polyclonal PLOD3 antibody (Proteintech Group). Anti-rabbit-IgG-HRP (P.A.R.I.S biotech) was used for secondary detection with ECL+ system (GE Healthcare) and Biomax XAR X-ray film (Kodak).
Figure 5.
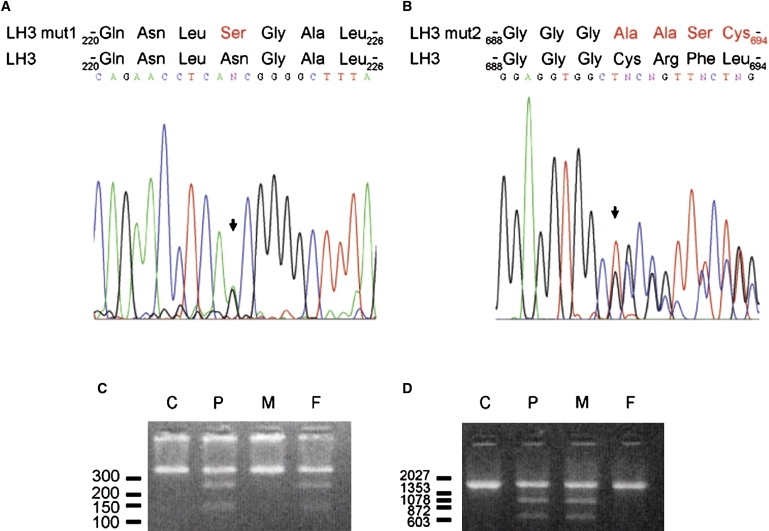
Mutation Analysis of LH3
Sequence analysis of LH3 cDNA of the patient revealed a heterozygous nucleotide transition c.668A→G (arrow in [A]) generating amino acid change p.Asn223Ser in the sequence (mutation 1), and a heterozygous one nucleotide deletion c.2071 delT (arrow in [B]) causing p.Cys691AlafsX9 frameshift in the amino acid sequence (mutation 2). We used digestion of PCR amplified DNA fragment by restriction enzymes to screen the mutations in DNA of parents. We used oligonucleotides (A20 5′-aagagagagaaaagtgagaaat-3′ and A15 5′-tgtgcaacagatgccagac-3′) from introns 5 and 6 to screen mutation 1. Oligonucleotides (A7 5′-gagcagcgagagcctcctac-3′ and A1 5′-actgtctccagctcaggcaat-3′) from intron 17 and 3′ downstream region of PLOD3 were employed for identification of mutation 2. Digestion of genomic DNA fragment by BbvCI (NEB) (C) or NarI (NEB) (D) and analysis of the fragments by agarose gel after ethidium bromide staining indicated that the mutation 1 was inherited from the father and mutation 2 was inherited from the mother. DNA markers are indicated (bp). C, control; P, patient; M, mother; F, father.
Figure 6.
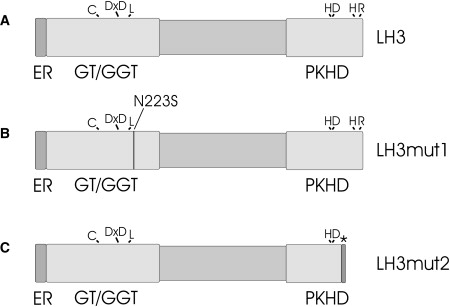
Domain Representation of Human LH3
(A) Putative N-terminal ER signal sequence (ER, amino acids 1–24) is followed by glycosyltransferase (GT/GGT) domain (25–280), which is responsible for GT/GGT activities and has structural similarities with glycosyltransferases.3 Lysyl hydroxylase/prolyl 4-hydroxylase (PKHD) domain (565–738) shares homology with 2-oxoglutarate-dependent dioxygenases and includes a 2-oxoglutarate and ironII-dependent oxygenase domain. Domain borders are approximated by sequence comparisons. Amino acids that are important for activity are indicated in the corresponding domain. DxD motif is a typical domain of glycosyltransferases and is also crucial for LH3 activity. LH3mut1 (B) is a protein produced by mutation 1 (c.668A→G) resulting p.Asn223Ser amino acid substitution. LH3mut2 (C) is a result of mutation 2 (c.2071 delT), in which one nucleotide deletion causes a frameshift and premature stop-codon formation (p.Cys691AlafsX9). The size of a LH3mut2 protein is 698 amino acids. The asterisk represents the last eight amino acids of LH3mut2 differing from LH3 as a result of frameshift and premature stop codon formation.
To ascertain the consequences of these mutations on LH3 activities, we used in vitro mutagenesis with the QuickChange II site-directed mutagenesis kit (Stratagene) to generate mutation 1 and mutation 2 in LH3 cDNA and to produce corresponding recombinant proteins in Sf9 insect cells. The production of recombinant proteins was followed by western-blot analysis, and the recombinant LH3 was analyzed in LH, GT, and GGT activity assays. The recombinant protein with mutation 1 resulted in a higher apparent molecular mass for most molecules compared to that for the wild-type LH3 (Figure 7A), a difference that was not observed when the recombinant proteins were treated with EndoH for removal of asparagine-linked oligosaccharides before western-blot analysis (Figure 7B). These data suggested that mutation 1 is generating a new glycosylation site for LH3 at Asn221; this generation would have been predicted because the amino acid substitution p.Asn223Ser changes Asn-x-Asn to Asn-x-Ser, the latter being a well-known Asn-linked glycosylation motif of proteins.19 This abnormality in glycosylation does not, however, affect the LH3 glucosyltransferase activity (Figure 7C) and therefore does not explain reduction of GGT activity of the patient. There was also a mobility shift in the apparent molecular mass of the recombinant protein with mutation 2 (Figure 7A), with a 40 amino acid deletion being clearly observed in the western blot. Our enzyme activity assays (Figure 8) indicated that mutation 1 reduced substantially the GT and GGT activities of LH3, and it also reduced the LH activity to approximately half of the normal level. Mutation 2 resulted in complete loss of the LH activity for LH3, as well as reduced its GT and GGT activities. As seen in the lymphoblastoid cells of the patient (Figure 4C), the amount of LH3 was decreased in cells, thereby leading to a phenotype that exhibited low LH3 content and in which the LH3 has markedly reduced lysyl hydroxylase and glycosyltransferase activities compared to normal. On the basis of results obtained from mouse studies,9,17 it seems probable that phenotypic consequences are mainly resulting from decreased glycosyltransferase function and to a lesser extent from decreased LH function. Complete loss of GT/GGT function is lethal in mouse,9 suggesting that complete loss of GT/GGT activities would be lethal also in humans.
Figure 7.
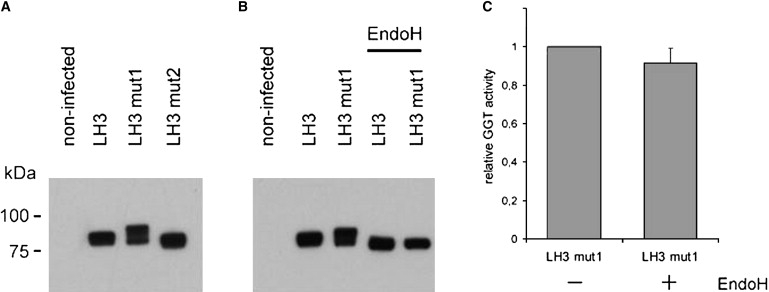
LH3 with Mutation 1 or Mutation 2 Produced in Insect Cells
(A) Immunoblot analysis of recombinant LH3, LH3mut1, and LH3mut2, stained with c-myc antibody.
(B) Immunoblot of EndoH-treated (1 hr at 37°C with 50 U EndoH from NEB) LH3 and LH3mut1 shows removal of asparagine-linked oligosaccrarides, seen as a mobility shift of the bands.
(C) Removal of N-linked glycans by Endo H has no effect on GGT activity of LH3mut1. We used 10 μg soluble protein of insect cell supernatant in the analysis. GGT activity result is a mean value of four independent experiments; SD is indicated by the bars. Expression of human LH3 cDNA was carried out in baculovirus transfer vector pFastBacI in the BAC-TO-BAC (Invitrogen) expression system.29 cDNA (coding amino acids 25–738) of LH3 was inserted to the vector in-frame downstream from ER signal sequence from LH1 and c-myc epitope tag. Insect cells were harvested 72 hr after infection and homogenized into a solution containing 0.1 M glycine, 1% Igepal CA-630 (Sigma), and 20 mM Tris-HCl (pH 7.4). The homogenate was sonicated and then centrifuged at 15,000 × g for 20 min and the soluble fraction was used for western-blot analysis. Proteins were transferred from SDS-PAGE gel to Hybond-LFP (GE Healthcare) PVDF membrane. Anti c-myc antibody 9E10 (Santa Cruz) and anti-mouse-IgG-AlexaFluor488 (Molecular Probes) were used with Typhoon 9400 (GE Healthcare) for fluorescence detection.
Figure 8.
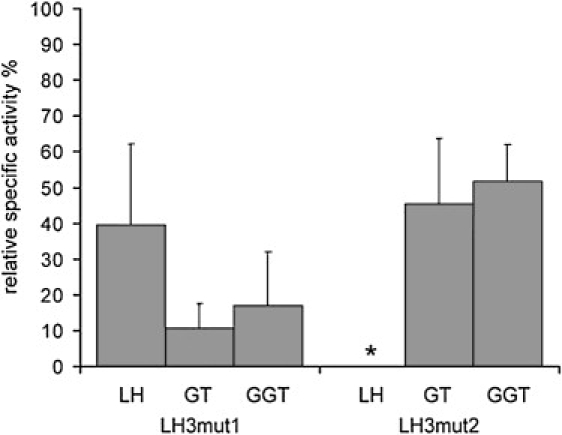
Enzyme Activity Analysis of Recombinant LH3 with and without Mutation 1 or Mutation 2
The 15,000 × g supernatant of insect cell homogenate was used for activity assays. LH, GT, and GGT activities were assayed,28 and an amount of LH3 protein was measured with monoclonal anti c-myc antibody 9E10 (Santa Cruz), anti-mouse-IgG-AlexaFluor488 (Molecular Probes) and Typhoon 9400 (GE Healthcare) for fluorescence detection of immunoblot analysis. Quantification of immunostained LH3 protein levels was performed with ImageQuant TL 7.0 software, GE Healthcare. Relative specific activities of mutation 1 (LH3mut1) and mutation 2 (LH3mut2) were calculated and compared to activities of nonmutated LH3. The data show dramatic reduction of glycosyltransferase activities of LH3mut1 and LH activity of LH3mut2. Activities are expressed as the percentage from activity of nonmutated LH3. n = 4 independent measurements, except n = 3 in GT of LH3mut1. The SD indicated by the bars. The asterisk represents activity below detection range.
As shown in our LH3-knockout studies,9 it was the GGT activity of LH3 and not its LH activity that is important for embryonal development. Lack of the GGT activity causes embryonic lethality, and with higher GGT activity the embryos survived longer and had less severe defects than knockout embryos. Skin blisters and dilatation of blood vessels were present, and the lethality was probably a consequence of the endothelial cell-layer ruptures and haemorrhage often seen in the embryos. This paper describes a severe phenotype of PLOD3 mutations in man. Intrauterine death of an affected male could have been the result of cerebral vascular rupture, as suggested by the large porencephalic cyst found at autopsy. His surviving sister (proband) is severely disabled, with clinical signs of several discrete conditions that are normally caused by mutations in genes encoding different types of collagen. No tissue samples for further collagen studies were available, but an interesting comparison can be made on clinical grounds. Similarities between her phenotype and Stickler syndrome20 (MIM 108300 and MIM 604841) were noted (Table 1) at an early age. The flat facial profile and myopia together with the prominent knee joints would be consistent with this diagnosis. Profound sensorial deafness is a feature of Stickler syndrome type II (MIM 604841). By contrast, growth retardation, nail and skin abnormalities, osteopenia, joint contractures, and spontaneous vascular rupture are not seen in Stickler syndrome. Spontaneous vascular rupture is a feature of Ehlers-Danlos syndrome type IV (EDS IV [MIM 130050]),21 a disorder of type III collagen characterized by thin skin with easy bruising and talipes (all clinical features seen in the patient). Arterial rupture and osteopenia are both features of EDS VI (MIM 225400),21 whereas other major features of this disease, namely joint laxity, congenital scoliosis, scleral fragility, and the characteristic facial appearance, were absent in this patient. Although osteopenia is a feature of EDS VI, caused by PLOD1 mutations, associated fractures are more in keeping with Bruck syndrome (MIM 609220), caused by PLOD2 mutations (see also Table 1). Rather than joint laxity, contractures that fall within the spectrum of disease seen in patients with EDS VI and Bruck syndrome are observed. We used imaging to investigate our patient's clinical symptoms and found arterial dilation in the regions of previous vascular rupture (Figures 2F and 2G). The anatomy of other large vessels has not been studied. Abnormalities in the ratio of urinary pyridinoline and deoxypyridinoline crosslinks characteristic of EDS VI were also absent. Urinary Glc-Gal-pyridinoline has been suggested as a specific marker of synovial tissue breakdown in arthritic diseases,22 but other studies indicate that this component is derived primarily from bone degradation.23 The association of joint contractures and fractures is seen in Bruck syndrome.14 None of the potential diagnoses discussed, however, can explain all features of the present case, and other phenotypic characteristics, some unique such as skin blistering, are not associated with any of the conditions discussed so far. The skin blistering that occurred from infancy to approximately 5 years of age occurred over areas exposed to friction, flexor aspects of the fingers and the pinnae. There was some resemblance to the dystrophic type of epidermolysis bullosa (MIM 131750),24 some types of which are caused by mutations in collagen genes. Nail abnormalities are also similar to those that can be seen in epidermolysis bullosa (MIM 131750). Developmental delay is not a feature of the collagen disorders summarized in Table 1. Although multiple sensory impairments and limitation of physical mobility caused by joint contractures make accurate development assessment challenging, the patient has some degree of global developmental delay.
Table 1.
Clinical Symptoms of PLOD3 Mutations
| Clinical Symptom | Other Collagen Disorder with Common Feature | Mutated Gene |
|---|---|---|
| Flat facial profile | Stickler syndrome | COL2A1, COL11A1, and COL11A2 |
| Deafness | Stickler syndrome | COL2A1, COL11A1, and COL11A2 |
| Myopia | Stickler syndrome | COL2A1, COL11A1, and COL11A2 |
| Prominent knee joint | Stickler syndrome | COL2A1, COL11A1, and COL11A2 |
| Arterial rupture | EDSIV | COL3A1 |
| Arterial rupture | EDSVI | PLOD1 |
| Osteopenia | EDSVI | PLOD1 |
| Osteopenia | Bruck syndrome | PLOD2 |
| Joint contractures and fractures | Bruck syndrome | PLOD2 |
| Skin blistering | Epidermolysis bullosa | COL7A1 and COL17A1 |
| Cataract | Stickler syndrome | COL2A1, COL11A1, and COL11A2 |
| Nail abnormalities | Epidermolysis bullosa | COL7A1 |
There is some overlap of clinical feature with the recently described Spondylocheiro dysplastic form of EDS.25 Children with this recessive condition showed postnatal growth retardation, whereas PLOD3 mutations caused prenatal and postnatal growth retardation. Hand appearances are similar with small joint contractures and marked thenar and hypothenar muscle wasting in both conditions. Broad large joints and thin skin with poor healing and easy bruising are also similar. Osteopenia and talipes equinovarus are described in both conditions. Important differences are the absence of severe myopia, cataracts, deafness, skin blistering, progressive scoliosis, arterial rupture, and fractures in the Spondylocheiro dysplastic form of EDS, in which survival into the third decade is described. Unlike other types of EDS, which are caused by mutations in collagen or PLOD genes, mutations in the zinc-transporter gene SLC39A13 are causative. These are postulated to interfere with hydroxylation of lysyl and prolyl residues.
In summary, our data establish a syndrome of congenital malformations severely affecting many tissues and organs and revealing features of several collagen disorders, most of them involving type II collagen. The findings suggest that the failure of lysyl hydroxylation and hydroxylysyl carbohydrate addition, which affects many collagens, is the molecular basis of this syndrome. No similar cases have been identified in literature searches. To our knowledge, this is the first inherited syndrome in which mutations are localized in the PLOD3 gene. The small nucleotide changes in PLOD3 result in amino acid changes in the LH3 polypeptide chain localized in regions responsible for LH3 glycosyltransferase and lysyl hydroxylase activities, respectively. The consequences of these mutations to LH3 enzyme activities and protein levels are dramatic, leading to a phenotype in which LH3 concentration is low and all LH3 activities, LH, GT, and GGT, are remarkably reduced. Because of its apparent pleiotropic effects, LH3 aberrations may in fact contribute more commonly to connective tissue disorders than what is currently recognized. Measurements of serum GGT activity or urinary collagen crosslink patterns now provide tools for detecting such disorders.
Acknowledgments
We thank the family participating in this study, clinicians for sample collection, Piia Mäkelä for technical help, and Robert Winqvist for providing control lymphoblastoid cells. The work was supported by research grants to R.M., research grants by the Academy of Finland (grant 109784), by Biocenter Oulu, and by Sigrid Juselius Foundation, a graduate school position to A.S. by Biocenter Oulu, and a graduate school position to M.R. by Glycoscience Graduate School. Orion-Farmos Research Foundation is acknowledged for a grant to A.S. S.R. is indebted to the Scottish Government Rural and Environment Research and Analysis Directorate for support. R.M. designed this study, supervised A.S., and obtained the funding; S.R. made initial observations of urinary abnormalities; A.S. designed and performed all other laboratory work with the help of the technician funded by R.M.; M.R. designed the oligos for DNA sequencing, H.C., P.F., C.M., and H.G. performed phenotype assessment, H.C., P.F., and C.M. provided clinical photographs, and H.C., A.S., S.R., and R.M. contributed to the writing of this paper.
Web Resources
The URLs for data presented herein are as follows:
Entrez Gene, http://www.ncbi.nlm.nih.gov/sites/entrez/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
References
- 1.Kielty C.M., Grant M.E. The collagen family: Structure, assembly, and organization in the extracellular matrix. In: Royce P.M., Steinmann B., editors. Connective Tissue and Its Heritable Disorders. Molecular Genetics and Medical Aspects. Wiley-Liss Inc; New York: 2002. pp. 103–147. [Google Scholar]
- 2.Kivirikko K.I., Myllylä R., Pihlajaniemi T. Hydroxylation of proline and lysine residues in collagen and other animal and plant proteins. In: Harding J.J., Crabbe J.C., editors. Post-Translational Modifications of Proteins. CRC Press; Boca Raton: 1992. pp. 1–51. [Google Scholar]
- 3.Myllylä R., Wang C., Heikkinen J., Juffer A., Lampela O., Risteli M., Ruotsalainen H., Salo A., Sipilä L. Expanding the lysyl hydroxylase toolbox: new insights into the localization and activities of lysyl hydroxylase 3 (LH3) J. Cell. Physiol. 2007;212:323–329. doi: 10.1002/jcp.21036. [DOI] [PubMed] [Google Scholar]
- 4.Valtavaara M., Szpirer C., Szpirer J., Myllylä R. Primary structure, tissue distribution and chromosomal localization of a novel isoform of lysyl hydroxylase (lysyl hydroxylase 3) J. Biol. Chem. 1998;273:12881–12886. doi: 10.1074/jbc.273.21.12881. [DOI] [PubMed] [Google Scholar]
- 5.Passoja K., Rautavuoma K., Ala-Kokko L., Kosonen T., Kivirikko K.I. Cloning and characterization of a third human lysyl hydroxylase isoform. Proc. Natl. Acad. Sci. USA. 1998;95:10482–10486. doi: 10.1073/pnas.95.18.10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C., Luosujärvi H., Heikkinen J., Risteli M., Uitto L., Myllylä R. The third activity for lysyl hydroxylase 3 (LH3): Galactosylation of hydroxylysyl residues in collagens in vitro. Matrix Biol. 2002;21:559–566. doi: 10.1016/s0945-053x(02)00071-9. [DOI] [PubMed] [Google Scholar]
- 7.Heikkinen J., Risteli M., Wang C., Latvala J., Rossi M., Valtavaara M., Myllylä R. Lysyl hydroxylase 3 is a multifunctional protein possessing collagen glucosyltransferase activity. J. Biol. Chem. 2000;275:36158–36163. doi: 10.1074/jbc.M006203200. [DOI] [PubMed] [Google Scholar]
- 8.Wang C., Risteli M., Heikkinen J., Hussa A.-K., Uitto L., Myllylä R. Identification of amino acids important for the catalytic activity of the collagen glucosyltransferase associated with the multifunctional lysyl hydroxylase 3 (LH3) J. Biol. Chem. 2002;277:18568–18573. doi: 10.1074/jbc.M201389200. [DOI] [PubMed] [Google Scholar]
- 9.Ruotsalainen H., Sipilä L., Vapola M., Sormunen R., Salo A.M., Uitto L., Mercer D.K., Robins S.P., Risteli M., Aszodi A. Glycosylation catalyzed by lysyl hydroxylase 3 (LH3) is essential for basement membranes. J. Cell Sci. 2006;119:625–634. doi: 10.1242/jcs.02780. [DOI] [PubMed] [Google Scholar]
- 10.Wang C., Kovanen V., Raudasoja P., Eskelinen S., Pospiech H., Myllylä R. The glycosyltransferase activities of lysyl hydroxylase 3 (LH3) in the extracellular space are important for cell growth and viability. J. Cell. Mol. Med. 2008;12:1–18. doi: 10.1111/j.1582-4934.2008.00286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruotsalainen H., Sipilä L., Kerkelä E., Pospiech H., Myllylä R. Characterization of cDNAs for mouse lysyl hydroxylase 1, 2 and 3, their phylogenetic analysis and tissue-specific expression in the mouse. Matrix Biol. 1999;18:325–329. doi: 10.1016/s0945-053x(99)00016-5. [DOI] [PubMed] [Google Scholar]
- 12.Salo A.M., Wang C., Sipilä L., Sormunen R., Vapola M., Kervinen P., Ruotsalainen H., Heikkinen J., Myllylä R. Lysyl hydroxylase 3 (LH3) modifies proteins in the extracellular space, a novel mechanism for matrix remodelling. J. Cell. Physiol. 2006;207:644–653. doi: 10.1002/jcp.20596. [DOI] [PubMed] [Google Scholar]
- 13.Risteli M., Niemitalo O., Lankinen H., Juffer A.H., Myllylä R. Characterization of collagenous peptides bound to lysyl hydroxylase isoforms. J. Biol. Chem. 2004;279:37535–37543. doi: 10.1074/jbc.M405638200. [DOI] [PubMed] [Google Scholar]
- 14.Bank R.A., Robins S.P., Wijmenga C., Brealau-Siderius L.J., Bardoel A.F., van der Sluijs H.A., Pruijs H.E., TeKoppele J.M. Defective collagen crosslinking in bone, but not in ligament or cartilage, in Bruck syndrome: indications for a bone-specific telopeptide lysyl hydroxylase on chromosome 17. Proc. Natl. Acad. Sci. USA. 1999;96:1054–1058. doi: 10.1073/pnas.96.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercer D.K., Nicol P.F., Kimbembe C., Robins S. Identification, expression and tissue distribution of the three rat lysyl hydroxylase isoforms. Biochem. Biophys. Res. Commun. 2003;307:803–809. doi: 10.1016/s0006-291x(03)01262-2. [DOI] [PubMed] [Google Scholar]
- 16.Zuurmond A.M., van der Slot-Verhoeven A.J., van Dura E.A., De Groot J., Bank R.A. Minoxidil exerts different inhibitory effects on gene expression of lysyl hydroxylase 1, 2 and 3: implications for collagen cross-linking and treatment of fibrosis. Matrix Biol. 2005;24:261–270. doi: 10.1016/j.matbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Sipilä L., Ruotsalainen H., Sormunen R., Baker N.L., Lamande S.R., Vapola M., Wang C., Sado C., Aszodi A., Myllylä R. Secretion and assembly of type IV and VI collagens depend on glycosylation of hydroxylysines. J. Biol. Chem. 2007;282:33381–33388. doi: 10.1074/jbc.M704198200. [DOI] [PubMed] [Google Scholar]
- 18.Robins S.P., Duncan A., Riggs B.L. Direct measurement of free hydroxypyridinium cross-links of collagen in urine as new markers of bone resorption in osteoporosis. In: Christiansen C., Overgaard K., editors. Osteoporosis 1990. Osteopress ApS; Copenhagen: 1990. pp. 465–468. [Google Scholar]
- 19.Helenius A., Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 20.Snead M.P., Yates J.R. Clinical and molecular genetics of Stickler syndrome. J. Med. Genet. 1999;36:353–359. [PMC free article] [PubMed] [Google Scholar]
- 21.Steinmann B., Royce P.M., Superti-Furga A. The Ehlers-Danlos syndrome. In: Royce P.M., Steinmann B., editors. Connective Tissue and Its Heritable Disorders. Molecular Genetics and Medical Aspects. Wiley-Liss Inc; New York: 2002. pp. 431–523. [Google Scholar]
- 22.Garnero P., Gineyts E., Christgau S., Finck B., Delmas P.D. Association of baseline levels of urinary glucosyl-galactosyl- pyridinoline and type II collagen C-telopeptide with progression of joint destruction in patients with early rheumatoid arthritis. Arthritis Rheum. 2002;46:21–30. doi: 10.1002/1529-0131(200201)46:1<21::AID-ART10061>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 23.Young-Min S., Cawston T., Marshall N., Coady D., Christgau S., Saxne T., Robins S., Griffiths I. Biomarkers predict radiographic progression in early rheumatoid arthritis and perform well compared with traditional markers. Arthritis Rheum. 2007;56:3236–3247. doi: 10.1002/art.22923. [DOI] [PubMed] [Google Scholar]
- 24.Uitto J., Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin. Dermatol. 2005;23:33–40. doi: 10.1016/j.clindermatol.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Giunta C., Elçioglu N.H., Albrecht B., Eich G., Chambaz C., Janecke A.R., Yeowell H., Weiss M., Eyre D.R., Kraenzlin M., Steinmann B. Spondylocheiro dysplastic form of Ehlers-Danlos syndrome—an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008;82:1290–1305. doi: 10.1016/j.ajhg.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pratt D.A., Daniloff Y., Duncan A., Robins S.P. Automated analysis of the pyridinium crosslinks of collagen in tissue and urine using solid-phase extraction and reversed-phase high-performance liquid chromatography. Anal. Biochem. 1992;207:168–175. doi: 10.1016/0003-2697(92)90519-d. [DOI] [PubMed] [Google Scholar]
- 27.Casetta B., Romanello M., Moro L. A rapid and simple method for quantitation of urinary hydroxylysyl glycosides, indicators of collagen turnover, using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2000;14:2238–2241. doi: 10.1002/1097-0231(20001215)14:23<2238::AID-RCM157>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 28.Kivirikko K.I., Myllylä R. Post-translational enzymes in the biosynthesis of collagen: intracellular enzymes. Methods Enzymol. 1982;82:245–304. doi: 10.1016/0076-6879(82)82067-3. [DOI] [PubMed] [Google Scholar]
- 29.Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Smith J.A., Seidman J.G., Struhl K. John Wiley & Sons; New York: 1989. Current Protocols in Molecular Biology. [Google Scholar]