

Abstract
Recurrent episodes of life-threatening myoglobinuria in childhood are caused by inborn errors of glycogenolysis, mitochondrial fatty acid beta-oxidation, and oxidative phosphorylation. Nonetheless, approximately half of the patients do not suffer from a defect in any of these pathways. Using homozygosity mapping, we identified six deleterious mutations in the LPIN1 gene in patients who presented at 2–7 years of age with recurrent, massive rhabdomyolysis. The LPIN1 gene encodes the muscle-specific phosphatidic acid phosphatase, a key enzyme in triglyceride and membrane phospholipid biosynthesis. Of six individuals who developed statin-induced myopathy, one was a carrier for Glu769Gly, a pathogenic mutation in the LPIN1 gene. Analysis of phospholipid content disclosed accumulation of phosphatidic acid and lysophospholipids in muscle tissue of the more severe genotype. Mutations in the LPIN1 gene cause recurrent rhabdomyolysis in childhood, and a carrier state may predispose for statin-induced myopathy.
Main Text
Recurrent episodes of myoglobinuria are frequent features of congenital metabolic myopathies. The differential diagnosis includes defects in glycogen breakdown, mitochondrial fatty acid beta oxidation, and oxidative phosphorylation.1 Diagnostic investigations including ischemic stress tests, fatty acid oxidation studies in lymphocytes, and histological, electron microscopic, and enzymatic determination of the mitochondrial respiratory chain in muscle fail to detect the primary defect in approximately half of the patients.2,3 In order to identify new etiologies, we used homozygosity mapping in patients who suffer from life-threatening episodes of rhabdomyolysis and who originate from small consanguineous families.
Three patients of Arab-Muslim origin (partly depicted in Figure 1) were the subjects of this study. They were all born at term, their birth and early motor and cognitive development were uneventful. All suffered from recurrent episodes of myoglobinuria, and these episodes were invariably precipitated by febrile illnesses and lasted 7–10 days. The first episode occurred around the second birthday in patients 2257 and 2120 and at 7 years in patient 2572. The presenting symptoms were generalized weakness, inability to walk, myalgia, and dark urine. On examination, marked sensitivity over the thighs and calf muscles was present, but there was no swelling, localized warmth, or redness. At presentation, the patients refused to lift the legs against gravity but retracted the legs in response to painful stimuli, muscle strength was markedly reduced, and the patellar and Achilles reflexes could not be elicited bilaterally. The strength, tone, and reflexes of the upper limbs were usually preserved. The central nervous system was typically spared during the episodes; however, when first admitted at age 18 months, patient 2120 was stuporous and had generalized muscle hypotonia; the EEG recording was abnormal and showed slow background activity along with theta and delta activity mainly above the occipital regions. During the episodes, plasma creatine kinase (CK) levels were markedly elevated (peak levels 180,000-450,000 U, N < 150) with overt myoglobinuria. Lactate dehydrogenase and aspartate aminotransferase were concomitantly elevated (peak levels 24,540 u/liter and 12,620 u/liter, respectively). The following tests gave normal results: CBC, electrolytes, alkaline phosphatase, coagulation studies, albumin and total proteins, cholesterol, triglycerides, urea, creatinine, uric acid, blood gasses, plasma lactate, total and free carnitine, blood acylcarnitine profile, plasma amino acids, beta-oxidation of labeled palmitate, CPT2 activity in lymphocytes, and urinary organic acids. Normal results were also obtained by inter-ictal electromyography, brain CT scan, abdominal ultrasound, and echocardiogram. In a biopsy of patient 2120's quadriceps two months after an episode of rhabdomyolysis, the enzymatic activities of the five mitochondrial respiratory-chain complexes and the pyruvate dehydrogenase complex were normal. The muscle contained no excess fat or glycogen, and the cytoarchitecture was preserved.
Figure 1.
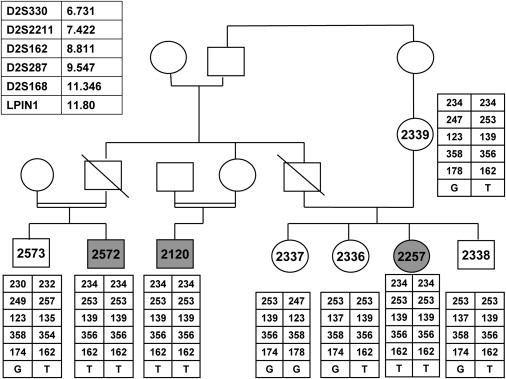
The Family Pedigree and the Haplotypes along the Critical Region on Chromosome 2
Patients' symbols are filled. Numbered symbols represent individuals whose DNA samples were available for analysis. The polymorphic microsatellite markers and their chromosomal locations (in Mb) are given in the upper left panel. Only siblings whose DNA samples were available were included in this figure.
During the episodes, treatment with intravenous fluids and alkalinization was sufficient to maintain normal plasma creatinine and urinary output. Between episodes, the patients enjoyed normal health. Their weight was at the 25th–40th percentile, and their height was at the 10th–25th percentile. At the time of writing, a physical examination of the patients, aged 8–10 years, gave normal results, including normal fat distribution (Figure 2), muscle strength, and deep-tendon reflexes. The patients participated in their age-appropriate physical activities, and plasma CK levels were normal. Their intellectual abilities were reportedly average, but patient 2120 at 9 years suffered from mild learning disabilities.
Figure 2.
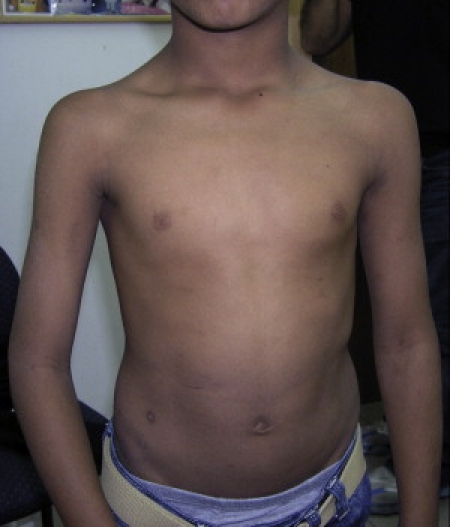
Patient 2572, Age 10 Years
Note Normal Fat Distribution
All experiments involving DNA of the patients, their relatives, healthy controls, and patients' cells were approved by the Hadassah Ethical Review Committee. The DNA of the three patients was analyzed with GeneChip Human Mapping 250K Nsp Array from Affymetrix, as previously described.4 Patient 2572 was homozygous for nine regions of more than 5 Mb, and patient 2257 was homozygous for seven regions. Both patients were homozygous and shared identical haplotype over 788 markers (rs2609168–rs779379) on chromosome 2 at 6.14–13.00 Mb. Using polymorphic microsatellite markers that spanned this region, we confirmed homozygosity for the same allele in the three patients and in none of their unaffected relatives (Figure 1). Within this 6.85 Mb genomic region there were 46 ORFs. and one of them, LPIN1 [MIM 605518], was further analyzed because of its predominant expression in muscle. Sequence analysis of its 19 coding exons and the flanking intronic regions revealed a homozygous G-to-T mutation at nucleotide 643; the result was that Glu215 was replaced by a termination codon (E215X). The LPIN1 mRNA:beta-actin mRNA ratio in the patients' fibroblasts determined by real-time PCR was only 5.8% ± 4.7% of the control. This mutation was not detected in 81 anonymous individuals of Arab-Muslim origin or in 85 anonymous individuals of Ashkenazi-Jewish origin.
The LPIN1 sequence analysis in an additional 22 patients who suffered from recurrent rhabdomyolysis resulted in the identification of five additional mutations, all deleterious, in four unrelated patients. The clinical course of these patients was essentially similar: They had normal early development and episodes of myoglobinuria precipitated by catabolic stress. All developed normally between episodes. Their family history, clinical course, and genotypes are given in Table 1. The following mutations were not detected in 75 anonymous individuals of French origin or in 75 anonymous individuals of Maghreb origin: Arg800X, c.297+2T > C, c.1259+2T > C, and a ∼2Kb genomic deletion that includes exons 18 and 19.
Table 1.
The Clinical, Biochemical, and Molecular Findings in the Index Patients and in an Additional Four Patients Who Were Identified by Screening of a Cohort of 22 Unrelated Patients for Mutations in the LPIN1 Gene
| Patient (Age at Time of Writing) | Age at First Episode (Number of Episodes) | Peak CK Level (u/liter) | Family History | Muscle Findings | Genotype (Mutation at the DNA Level) | Fibroblasts' mRNA |
|---|---|---|---|---|---|---|
| 2120, 2257, 2572 (8–10 years) | 2–7 years (1–5) | 180,000–450,000 | consanguineous, Arab-Muslim, seven healthy siblings | normal lipid content | E215X/ E215X (c.643G > T) | 6% |
| 2714 (5 years) | 3 years (2) | 20,000 | consanguineous, Palestinian-Muslim, three healthy siblings | normal lipid content | R388X/ R388X (c.1162C > T) | not available |
| L.R. (4 years) | 15 months (6) | 100,000 | French, nonconsanguineous | moderate lipid accumulation | R800X (c.2398C > T) / genomic deletion of exon 18 and 19a | 44% |
| D.A. (5 years) | 27 months (3) | 200,000 | Mauritanien, consanguineous | moderate lipid accumulation | c.297+2T > C/ c.297+2T > Cb | 74% |
| P.N. (8 years) | 18 months (4) | 80,000 | French, nonconsanguineous. A brother died of rhabdomyolysis at 16 months | not available | c.1259+2T > Cc/ genomic deletion of exon 18 and 19a | Not available |
Patients L.R. and P.N. were heterozygous for an ∼2Kb genomic deletion that included exon 18 and 19. The borders of this deletion were not determined.
Patient D.A. was homozygous for a c.297+2T > C mutation that was associated with the activation of a cryptic splice site in exon 1 and consequent deletion of the last 106 bp of this exon.
Patient P.N. was heterozygous for c.1259T > C mutation that resulted in skipping of exon 8.
The LPIN1 gene encodes Lipin-1, a phosphatidic acid phosphatase (EC 3.1.3.4) that consists of 890 amino acids with two conserved domains, an N-terminal region that spans the first 114 residues, and the catalytic domain LNS2 (Lipin/Ned1/Smp2) at amino acids 674–830. The mutations identified in our patients, which create a stop codon at residues 215, 388, and 800 or produce frame shifts as a result of exons' being skipped, are thus predicted to result in truncated proteins lacking catalytic activity.
Nearly 20 years ago, mouse strains with spontaneous mutations, Gly84Arg and a complex genomic rearrangement in the murine ortholog of the LPIN1 gene, were described. These mutants had a distinctive phenotype (fld, fatty liver dystrophy) characterized by dystrophic epididymal fat pads with reduced plasma leptin levels and insulin resistance, transient fatty liver, and severe hypertriglyceridemia resulting in a considerable death rate and peripheral neuropathy.5,6 In contrast, transgenic mice overexpressing the LPIN1 gene had a tendency to accelerate dietary-induced obesity. In view of these complementary phenotypes, it was suggested that subtle genetic variations in LPIN1 expression levels might contribute to the range of adiposity in human populations.7 The normal fat distribution and average weight percentiles in our patients exclude such a role for the human LPIN1 gene. After the identification of the mutations in the patients, we reviewed their files and noted normal plasma glucose, triglyceride, cholesterol, LDL, and HDL levels even during times of severe rhabdomyolysis. Cholesterol, LDL, HDL, and fasting glucose levels in plasma samples of their three mothers, aged 35-45, were also within the control range for age, and there were no cases of diabetes in the extended family. Thus, the clinical manifestations of LPIN1 mutations in humans, unlike in mice, are confined to skeletal muscle. This exclusive phenotype might be explained by the nearly ubiquitous expression of two other LPIN genes, LPIN2 and LPIN3, encoding closely related proteins.8 LPIN1 expression is most prominent in skeletal muscle and adipose tissue in both humans and mice. However, the human LPIN2 is similarly expressed in adipose tissue, which probably accounts for the lack of lipodystrophy in our patients.
Phosphatidic acid phosphatase catalyzes the conversion of phosphatidic acid (phosphatidate) to diacylglycerol (DAG) in the triacylglycerol (TAG) synthesis pathway9 (Figure 3). Because of this prominent role in triglyceride metabolism and the exclusive muscle involvement in patients with deleterious LPIN1 mutations, we hypothesized that LPIN1 mutations when combined with other factors attenuating lipid metabolism would result in overt myopathy. To this end, we determined LPIN1 sequence in six adults who developed significant clinical myopathy with elevated plasma CK levels while they were being treated with statins for hypercholesterolemia. In two of the patients, we found nucleotide substitution in exons encoding the catalytic site: One patient, of Arab origin, carried a synonymous change, GGG to GGC at codon 750, which is predicted by the ESEfinder program to abolish a binding site of the SC35 splicing factor, and the other patient, of Ashkenazi origin, carried an A-to-G mutation at nucleotide 2306, resulting in replacement of the highly conserved glutamic acid at codon 769 by glycine (E769G). An additional patient of Ashkenazi origin carried a C-to-T mutation at nucleotide 1828, resulting in Pro610Ser, which was not predicted to be pathogenic. Because the E769G mutation was not present in the 14 individuals who did not develop muscle symptoms while being treated with statins, we further studied the pathogenic nature of this mutation by using a yeast strain harboring a deletion of the gene encoding PAH1,10 the yeast homolog of human LPIN2. This deletion mutant grew very slowly in glycerol medium, a phenotype that was partially complemented by expression of the wild-type human LPIN1 gene or the LPIN1-P610S gene11 (Figure 4). In contrast, yeast PAH1 deletion mutants expressing human LPIN1-E769G hardly supported growth on glycerol medium, indicating that this mutation is of functional significance. This mutation was not detected in 85 anonymous individuals of Ashkenazi-Jewish origin or in 81 anonymous individuals of Arab-Muslim origin.
Figure 3.
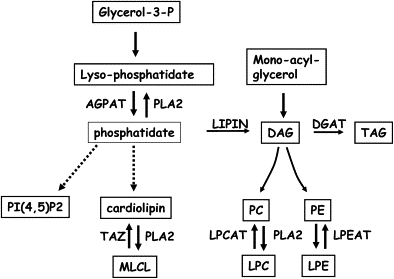
Triacylglycerol and Phospholipid Biosynthetic Pathway
Abbreviations are as follows: AGPAT, 1-acyl-sn-glycero-3-phosphate acyltransferase; DAG, diacylglycerol; DGAT, diacylglycerol acyltransferase; Glycerol-3-P, glycerol-3-phosphate; LPC, lyso-phosphatidylcholine; LPE, lyso-phosphatidylethanolamine; PLA2, phospholipase A2; LPEAT, lysophosphatidylethanolamine acyltransferase; LPCAT, lysophosphatidylcholine acyltransferase; LIPIN, phosphatidic acid phosphatase; MLCL, Monolyso-cardiolipin; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI(4,5)P2, phosphatidylinositol 4,5-diphosphate; TAG, triacylglycerol; TAZ, 1-palmitoyl-2-linoleoyl-phosphatidylcholine:monolysocardiolipin linoleoyltransferase.
Figure 4.
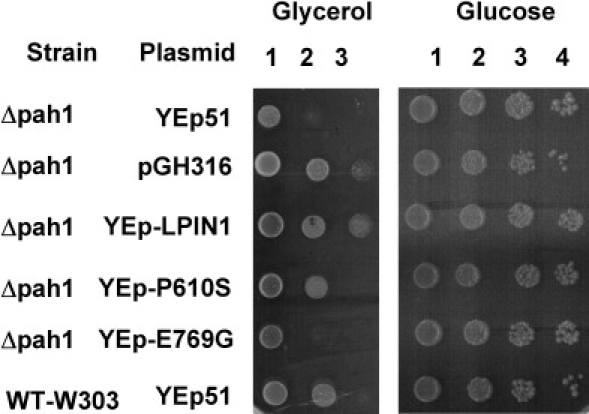
Human LPIN1 but Not LPIN1- E769G Can Partially Complement a Δpah1 Yeast Strain
Wild-type (WT-W303) and Δ pah1 strains harboring an empty plasmid (Yep51) or plasmids encoding derivatives of the normal human LPIN1 (YEp-LPIN1), the mutant human LPIN1 (YEp-E769G or Yep-P610S) or the normal yeast PAH1 (pGH316) were grown overnight at 30°C in glucose medium. Cultures at a density of 4 × 107 cells/ml were serially diluted at 10-fold intervals (numbered 1, 2, 3 and 4 for 101, 102, 103 and 104 dilutions, respectively) and 10 μl of each was spotted onto plates containing 2% glucose or 2% glycerol. The cells on glucose and glycerol medium were incubated at 30°C for 3 and 7 days, respectively.
Statins or 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors are widely used and are generally well tolerated; however, their administration is associated with myalgia in 1%–7% of the patients and with severe myopathy in 0.5% of the patients.12,13 Although the mechanism of this drug-induced myotoxicity is unknown, a strong association was found between mild, reversible statin-induced myopathy and a SNP within the SLCO1B1 gene.14 Moreover, a substantial proportion of the patients who developed myopathy were found to carry a mutation in genes responsible for one of the three common metabolic myopathies (Carnitine palmitoyl transferase 2 deficiency, McArdle disease, and myoadenylate deaminase deficiency). The importance of screening for such risk factors is underscored by the persistence of the muscle symptoms in the majority of the patients despite cessation of the lipid-lowering therapy.15 Given that an LPIN1 mutation has been detected in an individual who developed severe statin-induced myopathy, screening of a larger number of statin-treated patients and controls is warranted.
Phosphatidic acid phosphatase is located at an intersection of phospholipid metabolism; DAG is the precursor of the plasma membrane phospholipids phosphatidyl-choline (PC) and phosphatidyl-ethanolamine (PE), and phosphatidate is the precursor of cardiolipin, the major phospholipid in mitochondrial membranes (Figure 3). Abnormal phospholipid content was found in the peripheral nerves of the fld mutant mice and was proposed to account for the ataxia and tremor in these animals.16 To clarify the effect of the deleterious mutations in the LPIN1 gene in muscle, we determined the phospholipid content, as previously described17, in muscle tissues of patient 2120 and patient D.A. while they were asymptomatic. The results of this analysis, presented in Table 2, revealed normal PE, PC, and cardiolipin content and slightly increased phosphatidate level. In patient 2120 but not in D.A., markedly elevated amounts of lyso-phospholipids, including lyso-phosphatidate, lyso-PE, lyso-PC, and the lyso-form of cardiolipin, monolyso-cardiolipin (MLCL), were found. The source of these lyso-phospholipids is a remodeling process (Lands' cycle) that is catalyzed by phospholipase A2s and lysophospholipid acyltransferases. This rapid turnover of the sn-2 acyl moiety of the phospholipids is essential for membrane asymmetry and diversity.18–20 Alterations of the lyso-phospholipid levels are expected to be harmful to the plasma membrane and possibly to the mitochondrial membranes. The importance of normal levels of DAG, phosphatidate, and its metabolite, phosphatidylinositol 4,5-diphosphate (Figure 3), is further underscored by their role as potent lipid second messengers. Based on our findings, we propose that a defect in phosphatidic acid phosphatase results in lyso-phosphatidate accumulation that competes with lyso-PE, lyso-PC, and MLCL for their respective acyltransferases (LPEAT, LPCAT, and TAZ in Figure 3) and that this leads to the accumulation of these lysophospholipids. Further accumulation of lyso-phospholipids during stress periods may result in acute rhabdomyolysis because these metabolites not only act as detergents but may also perturb signal transduction. The normal phospholipid content in the muscle of patient D.A is probably the result of a splice-site mutation that allows the production of a small but significant amount of normal transcript in muscle; this, in turn, could attenuate the lyso-phosphatide accumulation in unstressful periods. The lack of muscle tissue from additional patients currently precludes definitive conclusions.
Table 2.
Phospholipid Content in Muscle
| Phospholipid Level (nmol/mg Protein) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Muscle | LPE | PE | LPC | PC | MLCL | CL | LPA | PA |
| 2120 | 0.45 | 2.7 | 5.9 | 27 | 3.0 | 3 | 1.2 | 0.5 |
| D.A. | 0.12 | 5.7 | 0.1 | 21 | 0.3 | 9 | u.d | 2.2 |
| Shipment controls (n = 3) | 0.03 | 2.4 | 0.1 | 18 | 0.3 | 6 | u.d | 0.2 |
| Disease controls (n = 85)a | n.d. | n.d. | 0.2 ± 0.2 | 24 ± 12 | 0.2 ± 0.1 | 4 ± 3 | n.d. | n.d. |
Abbreviations: n.d., not determined; u.d., undetectable.
Data from patients with mitochondrial myopathies collected for an unrelated research project. Hence, not all phospholipids were quantified.
In summary, deleterious mutations in LPIN1 are associated with recurrent episodes of rhabdomyolysis in early childhood, possibly because of secondary lyso- phospholipid accumulation. In view of the finding of a milder LPIN1 mutation in a patient who developed statin-induced myopathy, screening of larger cohort of patients and controls is warranted.
Acknowledgments
We gratefully acknowledge the collaboration of the patients' families and the dedicated assistance of Yehudit Karp. We thank Arnold Munnich and Ron Wanders for creating the international collaborations, George M. Carman for kindly providing S. cerevisiae strains and plasmids, Jan Smeitink and Richard Rodenburg for kindly providing the disease control muscle samples, and Saul Yedgar for fruitful discussions. This work was supported by grants of the Prinses Beatrix Fonds (grant number WAR05-0126) and Barth syndrome foundation to F.M.V. and by Fondation pour la Recherche Médicale to P.d.L.
Web Resources
URLs for data presented herein are as follows:
ESEfinder, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
References
- 1.Dubowitz V. Metabolic myopathies. In: Dubowitz V., editor. Muscle disorders in childhood. Second Edition. Sounders; London: 1995. pp. 177–314. [Google Scholar]
- 2.Tein I., DiMauro S., DeVivo D.C. Recurrent childhood myoglobinuria. Adv. Pediatr. 1990;37:77–117. [PubMed] [Google Scholar]
- 3.Tonin P., Lewis P., Servidei S., DiMauro S. Metabolic causes of myoglobinuria. Ann. Neurol. 1990;27:181–185. doi: 10.1002/ana.410270214. [DOI] [PubMed] [Google Scholar]
- 4.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-tRNA synthetase gene is associated with ponto-cerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langner C.A., Birkenmeier E.H., Ben-Zeev O., Schotz M.C., Sweet H.O., Davisson M.T., Gordon J.I. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J. Biol. Chem. 1989;264:7994–8003. [PubMed] [Google Scholar]
- 6.Péterfy M., Phan J., Xu P., Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- 7.Phan J., Reue K. Lipin, a lipodystrophy and obesity gene. Cell Metab. 2005;1:73–83. doi: 10.1016/j.cmet.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Donkor J., Sariahmetoglu M., Dewald J., Brindley D.N., Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 2007;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- 9.Han G.S., Wu W.I., Carman G.M. The Saccharomyces cerevisiae Lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 2006;281:9210–9218. doi: 10.1074/jbc.M600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han G.S., Siniossoglou S., Carman G.M. The cellular functions of the yeast lipin homolog PAH1p are dependent on its phosphatidate phosphatase activity. J. Biol. Chem. 2007;282:37026–37035. doi: 10.1074/jbc.M705777200. [DOI] [PubMed] [Google Scholar]
- 11.Stein I., Peleg Y., Even-Ram S., Pines O. The single translation product of the FUM1 gene (fumarase) is processed in mitochondria before being distributed between the cytosol and mitochondria in Saccharomyces cerevisiae. Mol. Cell. Biol. 1994;14:4770–4778. doi: 10.1128/mcb.14.7.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ucar M., Mjörndal T., Dahlqvist R. HMG-CoA reductase inhibitors and myotoxicity. Drug Saf. 2000;22:441–457. doi: 10.2165/00002018-200022060-00003. [DOI] [PubMed] [Google Scholar]
- 13.Shek A., Ferrill M.J. Statin-fibrate combination therapy. Ann. Pharmacother. 2001;35:908–917. doi: 10.1345/aph.10315. [DOI] [PubMed] [Google Scholar]
- 14.SEARCH Collaborative Group. Link E., Parish S., Armitage J., Bowman L., Heath S., Matsuda F., Gut I., Lathrop M., Collins R. SLCO1B1 variants and Statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 15.Vladutiu G.D., Simmons Z., Isackson P.J., Tarnopolsky M., Peltier W.L., Barboi A.C., Sripathi N., Wortmann R.L., Phillips P.S. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve. 2006;34:153–162. doi: 10.1002/mus.20567. [DOI] [PubMed] [Google Scholar]
- 16.Langner C.A., Birkenmeier E.H., Roth K.A., Bronson R.T., Gordon J.I. Characterization of the peripheral neuropathy in neonatal and adult mice that are homozygous for the fatty liver dystrophy (fld) mutation. J. Biol. Chem. 1991;266:11955–11964. [PubMed] [Google Scholar]
- 17.Valianpour F., Mitsakos V., Schlemmer D., Towbin J.A., Taylor J.M., Ekert P.G., Thorburn D.R., Munnich A., Wanders R.J., Barth P.G., Vaz F.M. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 2005;46:1182–1195. doi: 10.1194/jlr.M500056-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Lands W.E. Metabolism of glycerolipides: A comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958;231:883–888. [PubMed] [Google Scholar]
- 19.Nakagawa Y., Waku K. The metabolism of glycerophospholipid and its regulation in monocytes and macrophages. Prog. Lipid Res. 1989;28:205–243. doi: 10.1016/0163-7827(89)90013-1. [DOI] [PubMed] [Google Scholar]
- 20.Farooqui A.A., Horrocks L.A., Farooqui T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids. 2000;106:1–29. doi: 10.1016/s0009-3084(00)00128-6. [DOI] [PubMed] [Google Scholar]