

Abstract
Complex I (NADH:ubiquinone oxidoreductase) is the first and largest multimeric complex of the mitochondrial respiratory chain. Human complex I comprises seven subunits encoded by mitochondrial DNA and 38 nuclear-encoded subunits that are assembled together in a process that is only partially understood. To date, mutations causing complex I deficiency have been described in all 14 core subunits, five supernumerary subunits, and four assembly factors. We describe complex I deficiency caused by mutation of the putative complex I assembly factor C20orf7. A candidate region for a lethal neonatal form of complex I deficiency was identified by homozygosity mapping of an Egyptian family with one affected child and two affected pregnancies predicted by enzyme-based prenatal diagnosis. The region was confirmed by microcell-mediated chromosome transfer, and 11 candidate genes encoding potential mitochondrial proteins were sequenced. A homozygous missense mutation in C20orf7 segregated with disease in the family. We show that C20orf7 is peripherally associated with the matrix face of the mitochondrial inner membrane and that silencing its expression with RNAi decreases complex I activity. C20orf7 patient fibroblasts showed an almost complete absence of complex I holoenzyme and were defective at an early stage of complex I assembly, but in a manner distinct from the assembly defects caused by mutations in the assembly factor NDUFAF1. Our results indicate that C20orf7 is crucial in the assembly of complex I and that mutations in C20orf7 cause mitochondrial disease.
Introduction
Mitochondrial energy-generation disorders affect at least 1 in 5000 births1 and can be caused by mutations in nearly 100 different genes.2 Respiratory chain complex I (NADH-ubiquinone oxidoreductase) deficiency (MIM 252010) is the most commonly diagnosed energy-generation disorder.3,4 It has a wide range of clinical presentations and disease onset3,5,6 and has also been implicated in more common neurological disorders such as Parkinson's disease.7
Complex I is the major entry point of electrons into the mitochondrial electron-transport chain and contributes to the establishment of a proton gradient that is required for ATP synthesis. Human complex I is the first and most complicated of the respiratory-chain complexes and is composed of seven subunits encoded by mitochondrial DNA (mtDNA) and 38 nuclear-encoded subunits, which are assembled into a large complex of ∼950 kDa.8 It further assembles into higher-ordered supercomplexes or “respirasomes” with respiratory-chain complexes III and IV.9,10 Although all of its subunits have been identified, we have only a partial understanding of how the massive complex I assembles and which proteins aid this process.11
Mutations causing complex I deficiency have been identified in 23 genes, with most detected in only a small number of families. These genes encode the 14 core subunits of complex I, comprising seven nuclear genes (NDUFS1 [MIM 157655], NDUFS2 [MIM 602985], NDUFS3 [MIM 603846], NDUFS7 [MIM 601825], NDUFS8 [MIM 602141], NDUFV1 [MIM 161015], and NDUFV2 [MIM 600532])12–18 and all seven mtDNA subunit genes.3,19,20 Pathogenic mutations have also been identified in genes encoding five supernumerary subunits, NDUFS4 (MIM 602694),21 NDUFS6 (MIM 603848),22 NDUFA1 (MIM 300078),23 NDUFA1124 and NDUFA2 (MIM 602137);25 three assembly factors, NDUFAF2 (B17.2L) (MIM 609653),26 NDUFAF1 (CIA30) (MIM 606934),27 and C6orf66 (MIM 611776);28 and a putative new assembly factor, C8orf38.29 Mutations in mtDNA appear to account for approximately one-quarter of complex I deficiency,19,20 and mutations in all the reported mtDNA and nuclear genes appear to explain only about half of the cases.30 Therefore, these data suggest that the remaining mutations reside in unknown genes encoding additional factors involved in complex I biogenesis.
In this report, we describe a homozygous mutation in C20orf7 causing complex I deficiency in one affected child and two affected fetuses of an Egyptian family. We show that C20orf7 is a mitochondrial inner-membrane protein with a role in the early stages of complex I assembly. Therefore, we have identified mutations in a fifth assembly factor of complex I as a cause of complex I deficiency.
Material and Methods
Subjects
The clinical presentation of the proband was reported previously.31 He was the first child of first-cousin Egyptian parents and presented with lethal neonatal mitochondrial disease. He was born at 35 weeks gestation and suffered from intrauterine growth retardation. He had minor facial dysmorphism, and other minor dysmorphic features included unusual hair patterning, abnormal toes, and a small sacral pit. Cerebral ultrasound showed agenesis of the corpus callosum and ventricular septation. He also had a congenital left diaphragmatic hernia. Adrenal insufficiency resulted in persistent hypotension. His initial blood lactate level was 3.1 mM on day 5, which rose to 16.5 mM within 24 hr, with pyruvate 0.15 mM (lactate/pyruvate ratio of 110). Cerebrospinal fluid (CSF) lactate at this time was 20.1 mM, with pyruvate 0.34 mM. Urinary alanine was also elevated. He had normal liver and renal function. Histology of skeletal muscle showed no specific abnormalities. He died of cardiorespiratory arrest due to progressive lactic acidosis on day 7 and was diagnosed with an isolated complex I defect. Subsequently, four enzyme-based prenatal diagnoses were undertaken: Two affected pregnancies were terminated and two unaffected pregnancies resulted in healthy children. The proband and affected siblings were reported as patients A, A2, and A3, respectively, in a previous study.22 This study was approved by the Royal Children's Hospital Ethics in Human Research Committee.
Cell Culture and Complex I Enzyme Assays
Cell culture and assays for respiratory-chain enzymes and citrate synthase (CS) were performed as described3,32 with cultured cell mitochondria and post-600 g supernatants prepared from muscle and other tissues. Complex I-linked ATP synthesis was assessed in permeabilized cells by measurement of the ratio of ATP synthesis using glutamate plus malate (complex I substrate) with that using succinate and rotenone (complex II substrate) as described previously.3
Microcell-Mediated Chromosome Transfer
Primary fibroblasts from patients and a healthy control were transduced with a retroviral vector expressing the E6E7 region of type 16 human papilloma virus to extend their life span.33 A cell line from a panel of human-mouse monochromosomal hybrids34 was used as the donor of normal human chromosome 20. Chromosome 20 was transferred into the E6E7-transduced patient cell line by microcell-mediated chromosome transfer (MMCT), as described previously,33 following the modified protocol described elsewhere.22
Genotyping
Genome-wide scans, fine mapping, and genotyping of parental cell lines and clones were performed at the Australian Genome Research Facility (Melbourne, Victoria, Australia) with 377 and 3700 DNA sequencers (Applied Biosystems). Data were analyzed with GeneScan Analysis version 3.1.2 and Genotyper version 2.1 software (Applied Biosystems). Genome-wide scans were performed on purified DNA with the LMS2-HD5 marker set (Applied Biosystems), which has 811 microsatellite markers spread at an average genetic distance of 5 cM. Additional markers were used for fine mapping of the causative locus.
Bioinformatic Analysis
The likelihood of mitochondrial localization of proteins encoded by all RefSeq genes in the candidate region was initially predicted with methods described elsewhere.35 Additionally, candidate genes were compared with a set of 19 genes that we recently identified as showing correlated evolution with a set of complex I subunits.29
Mutation Analysis and DNA Sequencing
Exons from 11 transcripts were sequenced: C20orf7 (AK091060, NM_024120.1, NM_024120.3), C20orf72 (NM_052865), HSPC072 (NM_014162), SNRPB2 (NM_198220, NM_003092), SEC23B (NM_032986, NM_006363, NM_032985), SNX5 (NM_014426, NM_152227), DSTN (NM_006870, NM_0010011546), C20orf133/MACROD2 (NM_080676), ZNF339/OVOL2 (NM_021220), C20orf79 (NM_178483), and RBBP9 (NM_006606, NM_153328). Primary fibroblasts were grown for 24 hr in 100 μg/ml cycloheximide prior to preparation of RNA in order to minimize nonsense-mediated mRNA decay.36 RNA was reverse transcribed into cDNA with Superscript III reverse transcriptase via an RT-PCR kit (Invitrogen). Automated sequencing was performed on shrimp alkaline phosphatase-treated PCR products (ExoSAP-IT; USB Corp.) via a BigDye Terminator Cycle Sequencing kit (Applied Biosystems). The sequencing reactions were purified with DyEx columns (QIAGEN). Electrophoresis was performed on an Applied Biosystems Prism 3100 Genetic Analyzer at Applied Genetic Diagnostics (University of Melbourne). Sequence data were analyzed with Chromas (Technelysium Pty.) software version 1.51 and compared with the RefSeq sequence.
All exons of C20orf7 were amplified from genomic DNA with primers that incorporated at least 30 bp of 5′ and 3′ intronic sequence (Table 1 available online). The entire coding region of C20orf7 was also amplified from cDNA. Wild-type and variant C20orf7 alleles were distinguished by a PCR-RFLP test utilizing a BstNI site introduced by the c.719T→C mutation.
RNA Interference and Measurement of Complex I Activity
Lentiviral vectors (pLKO.1) containing short-hairpin sequences targeted against C20orf7 were obtained from the Broad RNAi Consortium (TRC).37 Virus production and infection of MCH58 human fibroblast cells was performed as recently reported.29 Knockdown efficiency was assessed via real-time PCR (Applied Biosystems Taqman Assays) with beta actin as an endogenous control after growth for ∼2 weeks in puromycin-containing medium. For immunoblot analysis of complex I and complex IV subunits, 10 μg of protein from cleared whole-cell lysate was separated on a 4%–12% SDS-PAGE gel (Invitrogen) and transferred to polyvinylidene fluoride membrane. Membranes were probed with antibodies raised against MTCO2 (Mitosciences), an 8 kDa complex I subunit (Mitosciences), and beta actin (Sigma). Complex I and complex IV activity assays were performed with immunocapture-based dipstick assays (Mitosciences). Fifteen micrograms of protein (complex I) or 30 μg of protein (complex IV) of cleared whole-cell lysate were used for each assay following the manufacturer's protocol. Results were scanned with a BioRad GS-800 scanner and analyzed with Quantity One software.
In Vitro Protein Import into Isolated Mitochondria
Radiolabeled C20orf7 protein was translated from pGEM-4Z plasmid DNA containing the C20orf7 transcript via the TnT Coupled Reticulocyte Lysate System (Promega) in the presence of 35S-methionine and 35S-cysteine (EXPRE35S35S Protein Labeling Mix; Perkin Elmer Life Sciences, USA). Translated protein was incubated with mitochondria isolated from HT1080 cells at 37°C for various times as indicated. Proteinase K treatment and dissipation of mitochondrial membrane potential were performed as described elsewhere.38
C20orf7 Mitochondrial Localization
COS-7 cells were transfected with plasmid DNA containing a FLAG-tagged C20orf7 transcript by use of Lipofectamine 2000 (Invitrogen). Mitochondria were isolated and subjected to either sonication in 100 mM NaCl,10 mM Tris-HCl (pH 7.6) or alkaline extraction in freshly prepared 0.1M Na2CO3 (pH 11.5).39 Membranes were pelleted at 100,000 × g for 30 min at 4°C, and supernatants were precipitated with the addition of 1/5 volume of 72% trichloroacetic acid. After treatments, soluble (S, supernatant) and insoluble (P, pellet) fractions were subjected to SDS-PAGE and western-blot analysis using antibodies against FLAG (to detect C20orf7-FLAG), NDUFB6, and cytochrome c.
Antibodies
Monoclonal antibodies against the complex III subunit core 1, complex IV subunit II, complex V α-subunit, and the complex II 70 kDa subunit were from Molecular Probes, and antibodies to the complex I 8 kDa subunit were from Mitosciences. Antibodies against cytochrome c (BD Biosciences), VDAC1 (Abcam), and FLAG (Sigma) were also obtained from commercial sources. Polyclonal antibodies against NDUFA9 and NDUFB6 were raised in rabbits as previously described.40
Other Methods
Mitochondria were isolated from cultured cells as previously described.41 Radiolabeling of mitochondrial translation products, blue-native (BN) PAGE, and two-dimensional (2D) and Tris-tricine SDS-PAGE were performed as previously described.42 Western blotting was performed with a semidry transfer method.43 Immunoreactive proteins from blots were detected with a ChemiGenius BioImaging system (SynGene) using horse-radish peroxidase coupled secondary antibodies (Sigma) and ECL substrate (GE).
Results
Biochemistry, Homozygosity Mapping, and Candidate-Gene Analysis
Respiratory-chain enzyme analysis showed marked complex I deficiency in skeletal muscle, liver, and skin fibroblasts of the proband, with essentially normal activity of other complexes.31 Prenatal diagnosis was undertaken in four subsequent pregnancies by enzyme and functional analysis of cultured chorionic villus cells. Two affected pregnancies were diagnosed by finding values of complex I activity and complex I-linked ATP synthesis that were < 30% of control values (ATP synthesis not shown). The affected pregnancies were terminated and complex I deficiency was confirmed in fetal skin fibroblasts or tissues. Two unaffected pregnancies resulted in the birth of healthy children (Figure 1).
Figure 1.
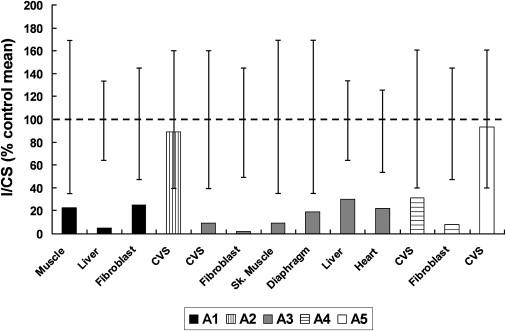
Residual Complex I Activity in Affected and Unaffected Individuals of Family A
Individuals A1, A3, and A4 are affected, and individuals A2 and A5 are unaffected. Complex I activity is expressed relative to citrate synthase activity (I/CS), as a percentage of the control mean for each tissue. The observed ranges for controls are represented by the vertical bars. CVS denotes chorionic villus cells.
Previous analysis with BN-PAGE and western blotting showed the absence of fully assembled complex I in fibroblasts from the proband.22 A 5 cM genome-wide scan of 811 microsatellite markers in the three affected individuals and their parents identified only seven markers that were homozygous in the three affected individuals but not in the parents. In addition, there were 42 autosomal homozygous markers that were not informative, i.e., were homozygous in one or both parents. Three of the seven informative markers were located adjacent to each other on chromosome 20p, making it the largest homozygous region shared exclusively by all three affected individuals and not by healthy family members. Fine mapping of all seven family members with additional microsatellite markers narrowed the candidate region to 9.2 Mb between D20S894 and D20S471 (Figure 2).
Figure 2.
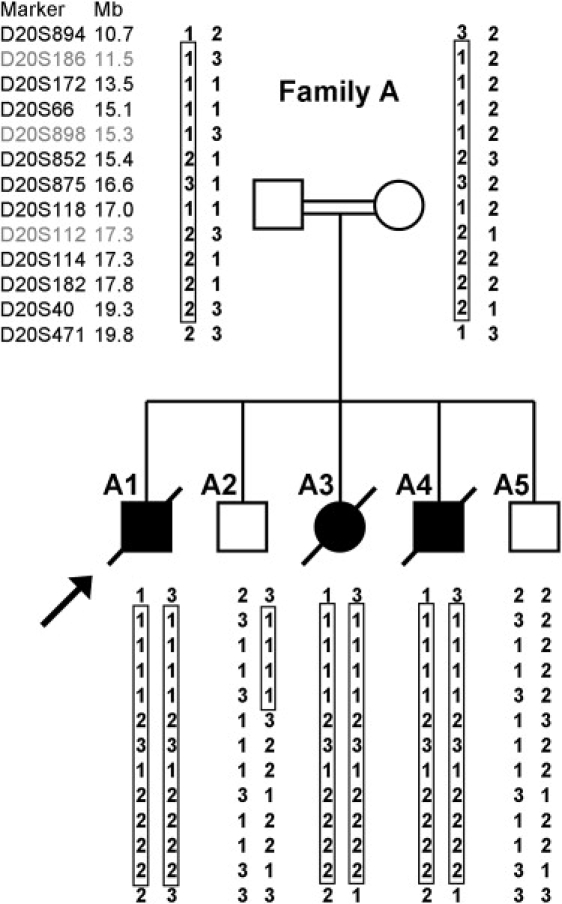
Pedigree and Haplotypes of Family Members
Genotyped markers from the chromosome 20p region between D20S894 and D20S471 are shown to the left, and each individual's allele numbers are listed. The three homozygous markers from the original genome-wide scan are shown in gray. The largest homozygous region shared exclusively by the three affected siblings is boxed, and the proband is indicated by the arrow.
Microcell-mediated chromosome transfer was used to determine whether introducing a normal copy of chromosome 20 into a patient cell line could restore complex I levels. Phenotypic correction was assessed by SDS-PAGE western blotting with antibodies to two complex I subunits that were grossly decreased in patient cell lines. Clones were classified as corrected, uncorrected, or ambiguous on the basis of at least two independent western blots. Figure 3 shows examples for two clones, one of which has corrected complex I subunit levels to near-normal levels whereas the other has not. Of 14 tested clones, four had normalized complex I subunit levels, five did not, and five gave ambiguous results. These data confirm that the causative locus is on chromosome 20. Uncorrected clones were genotyped with microsatellite markers and Affymetrix 50K XbaI SNP chips, which confirmed that at least two clones had deleted all or part of the candidate region (not shown). However, these data did not provide significant narrowing of the candidate region.
Figure 3.
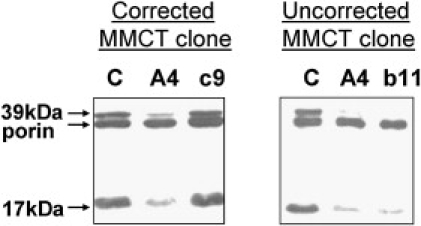
SDS-PAGE Western Blotting of MMCT Clones
Antibodies against complex I 39 kDa (NDUFA9) and 17 kDa (NDUFB6) subunits were used as well as VDAC (porin, as loading control). Detectable protein in one corrected clone (c9) and one uncorrected clone (b11) are shown in comparison to control (C) and patient A4.
To begin searching for a candidate gene in this interval that may underlie the observed complex I deficiency, we performed gene-expression analysis with Compugen 19K Human Oligoarrays and Codelink 50K Human Bioarrays. However, no transcripts within the region showed significant differential expression (data not shown).
The causative mutation for this deficiency is most likely in a gene encoding a mitochondrial protein. We previously described an integrative genomics approach to predict the probability of any gene encoding a mitochondrial-localized protein.35 This approach combines predictions of mitochondrial-targeting sequence, protein-domain enrichment, presence of cis-regulatory motifs, yeast homology, ancestry, tandem-mass spectrometry, coexpression, and transcriptional induction during mitochondrial biogenesis. Eleven of 29 RefSeq genes in the candidate region were predicted to encode possible mitochondrial proteins and were sequenced. Ten of the sequenced genes had either no sequence variants or contained only RefSNPs or variants that were not predicted to change conserved amino acids or affect splicing. Only one gene, C20orf7, contained a putative pathogenic sequence variant. Interestingly, we also identified C20orf7 recently in an independent phylogenetic analysis as being one of 19 genes that had a shared evolutionary history with complex I subunits and were therefore likely to be important for the function of complex I.29
Mutation Analysis of C20orf7 Transcripts
Two NCBI RefSeq transcripts of C20orf7 (NM_024120.3 and NM_001039375.1) are present. Both transcripts encode proteins that are 96.4% identical and are predicted to be imported into mitochondria. NM_024120.3 is slightly longer and has somewhat higher homology to its orthologs (on the basis of BLASTp). Therefore, all nomenclature in this manuscript refers to NM_024120.3 and its protein NP_077025.
Genomic DNA sequencing analysis of the patient revealed a homozygous missense mutation in exon 7 of NM_024120.3: c.719T→C, leading to substitution of leucine 229 in the C20orf7 protein to proline (Figure 4A). Leucine 229 is located within a region of the protein that is highly conserved between different species from human to fungus (Figure 4B), and its mutation to proline is predicted to alter secondary structure by breaking an alpha helix (SOPM,44 data not shown). This putative pathogenic homozygous mutation is also contained within the NM_001039375.1 sequence (Figure 4B: NP_001034464).
Figure 4.
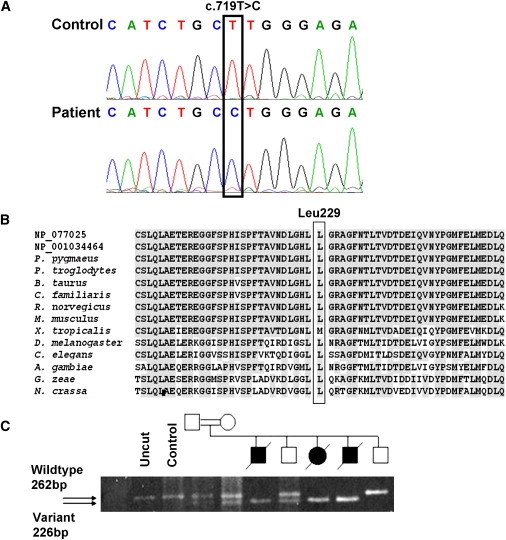
Mutation Analysis of C20orf7
Nomenclature is based on NM_024120.3.
(A) Sequencing chromatograms showing the c.719T→C mutation in the proband's genomic DNA, which predicts a Leucine-to-Proline substitution at codon 229.
(B) Amino acid alignment of C20orf7 orthologs shows that Leu229 and the surrounding region are highly conserved (gray shading). The alignment of the mutated region was generated with ClustalW via sequences obtained from BLASTp with both human transcripts: NP_077025 and NP_001034464. Sequences are from Pongo pygmaeus (orangutan, accession number CAH90789), Pan troglodytes (chimpanzee, XP_514521), Bos taurus (bovine, XP_600505), Canis familiaris (dog, XP_534340), Rattus norvegicus (rat, XP_215857), Mus musculus (mouse, NP_081369), Xenopus tropicalis (frog, NP_001016398), Drosophila melanogaster (fruit fly, AAL29045), A. gambiae str. PEST (mosquito, EAA05526), Gibberella zeae PH-1 (fungus, EAA70416), Neurospora crassa (fungus, CAD11326).
(C) BstNI restriction-enzyme-based analysis of the mutation in genomic DNA from family members shows that only DNA from affected individuals is homozygous for the BstNI site introduced by the mutation. The parents and one unaffected sibling are heterozygous, and one unaffected sib is homozygous for the wild-type allele.
Restriction fragment length polymorphism (RFLP) analysis showed that homozygosity for the c.719T→C substitution segregated with disease in the pedigree (Figure 4C), although the same would be expected for all variants in the candidate region. The c.719T→C substitution was not present in 114 Australian or 210 Egyptian alleles. C20orf7 was sequenced in a further 52 unrelated patients with complex I deficiency in whom no molecular diagnosis had been achieved previously. No further patients with a pathogenic C20orf7 mutation were identified.
C20orf7 Is an Inner-Mitochondrial-Membrane Protein
C20orf7 has a predicted N-terminal mitochondrial leader sequence (Mitoprot II, probability of mitochondrial import = 0.986),45 and its mitochondrial localization is supported by GFP-tagging and microscopy.29 For confirmation of its mitochondrial location, in vitro transcription and translation was performed in the presence of 35S-methionine and 35S-cysteine. Radiolabeled C20orf7 was incubated with isolated mitochondria for various times with import monitored by SDS-PAGE (Figure 5A). The full-length 39 kDa translation product (“p,” precursor) can be seen in the lysate (Figure 5A, lane 1) and associated with the mitochondria (Figure 5A, lanes 2 to 5). C20orf7 was cleaved to its mature form (“m”) after incubation (Figure 5A, lanes 2–4 and 6–8). This mature form is protected from proteinase K treatment (Figure 5A, lanes 6 to 9), indicating that it has been imported into the mitochondria. Furthermore, the import of C20orf7 is dependent on an intact membrane potential (Figure 5A, lanes 5 and 9), suggesting that it resides within the mitochondrial matrix, where it is processed by proteases.39
Figure 5.

Mitochondrial Import and Localization of C20orf7
(A) [35S]-labeled precursor forms of C20orf7 were incubated with mitochondria isolated from HT1080 cells at 37°C for the time points indicated in the presence and absence of a mitochondrial membrane potential (Δψm). Samples were treated with and without proteinase K (PK, 50 mg/ml) before SDS–PAGE and phosphorimager analysis to detect the precursor (p) and mature (m) proteins.
(B) COS-7 cells were transfected with plasmid DNA containing a FLAG-tagged C20orf7 construct. Isolated mitochondria were subjected to either sonication, alkaline (carbonate, Na2CO3) extraction, or no treatment (Con). After treatments, soluble (S) and insoluble (P) fractions were subjected to SDS-PAGE and western-blot analysis using antibodies against FLAG, NDUFB6 17 kDa complex I subunit, and cytochrome c.
The submitochondrial location of C20orf7 was investigated by transfection of COS-7 cells with a FLAG-tagged C20orf7 construct, followed by incubation for 24 hr to allow for protein expression. Mitochondria were isolated and either sonicated or subjected to alkaline extraction with sodium carbonate (Figure 5B). A FLAG antibody was used to detect the C20orf7-FLAG protein. C20orf7 was detected in the pellet (Figure 5B, “p,” lane 1) and not the supernatant (Figure 5B, “s,” lane 2) after sonication, indicating membrane localization. Conversely, the protein was found in the supernatant (Figure 5B, “s,” lane 5) and not the pellet (Figure 5B, “p,” lane 4) after alkaline extraction, suggesting that C20orf7 is only peripherally associated with the membrane. A membrane-arm subunit of complex I (NDUFB6) and the intermembrane space protein cytochrome c were used as controls. The combined import and western-immunoblotting results suggest that C20orf7 resides on the matrix side of the inner membrane, consistent with its putative role in complex I biogenesis.
C20orf7 Knockdown Decreases Complex I Activity
We next tested whether directly knocking down the levels of C20orf7 would cause complex I deficiency. MCH58 cells were stably transduced with lentiviral constructs containing short hairpin sequences. Control experiments utilized hairpins designed against the NDUFAF1 gene encoding the CIA30 complex I assembly factor. Knockdown efficiency was assessed by real-time PCR assays using beta actin as an endogenous control, and more than 80% knockdown was achieved for both genes (data not shown). The activities of complexes I and IV were assessed first by immunocapture-based activity assays, which showed that both C20orf7 and NDUFAF1 hairpins caused a similar gross decrease in complex I activity, with a more modest inhibition of complex IV activity (Figure 6A). SDS-PAGE immunoblotting confirmed that the C20orf7 hairpin caused a gross decrease in the steady-state level of the 8 kDa complex I subunit but no obvious effect on the levels of VDAC or the MTCO2 complex IV subunit (Figure 6B). These data strongly suggest that C20orf7 is crucial for activity and/or assembly of endogenous complex I.
Figure 6.
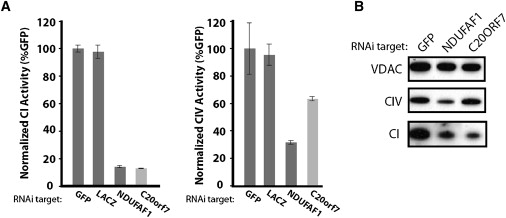
Knockdown of C20orf7 by RNA Interference Disrupts Complex I
(A) MCH58 cells were infected with lentivirus containing constructs encoding hairpins against the indicated targets. After growth for 2 weeks in puromycin-containing selection medium, cells were lysed and immunocapture-based activity assays for complexes I and IV were performed in duplicate. Activity is normalized to GFP control, with error bars representing the observed range of duplicate estimates.
(B) For western analysis, 4 μg of a mitochondria-enriched fraction from the indicated knockdown cell lines were separated by SDS-PAGE, transferred to PVDF membranes, and blotted with antibodies against VDAC1, a complex IV subunit (MTCO2), or a complex I subunit previously shown to correlate with complex I disassembly (8 kDa subunit).
C20orf7 Is Involved Early in Complex I Assembly
Patient A4 fibroblasts had been previously shown to almost completely lack the mature complex I holoenzyme by BN-PAGE immunoblotting.22 To examine whether the loss of complex I is due to an assembly defect or increased turnover, we radiolabeled mtDNA-encoded subunits and monitored their assembly into various complexes with BN-PAGE (Figure 7A). In control fibroblasts, mature complex I is clearly detectable after 24 hr chase (Figure 7A, lane 4), with some mature complex weakly detectable after only 6 hr chase (Figure 7A, lane 3). However, in patient A4 fibroblasts, no complex I was detectable after 24 hr chase (Figure 7A, lane 8), suggesting a defect in the assembly of mtDNA-encoded subunits into complex I. The assembly of complex IV, complex V, and the complex III homodimer (III2) in patient A4 was comparable to the assembly in control fibroblasts (Figure 7A).
Figure 7.
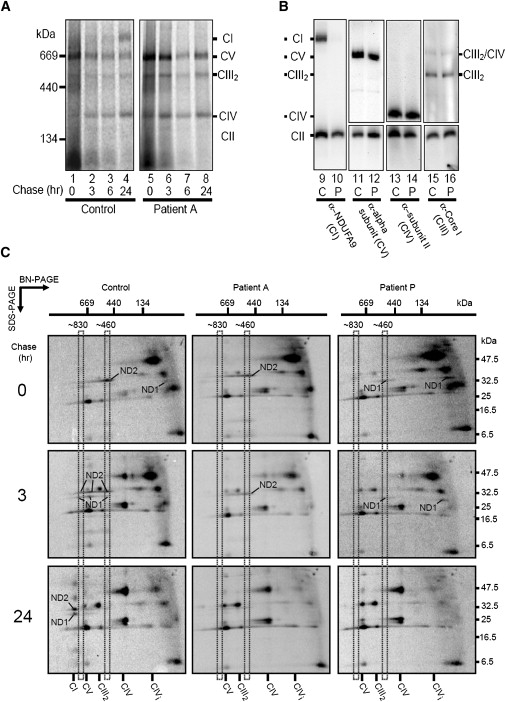
Complex I Assembly Is Impaired in Patient Cells
(A) After radiolabeling of mtDNA-encoded subunits, control (lanes 1–4), or patient A4 (lanes 5–8) fibroblasts were chased for various times as indicated. Mitochondria were isolated, solubilized in 1% Triton X-100, and subjected to BN-PAGE followed by phosphorimage analysis. Complex I (CI), the complex III homodimer (CIII2), complex IV (CIV), and complex V (CV) are indicated.
(B) BN-PAGE and western blot indicating the steady-state levels of the respiratory complexes in mitochondria from control (lanes 9, 11, 13, and 15) and patient A4 (lanes 10, 12, 14, and 16) fibroblasts.
(C) 2D-PAGE analysis of radiolabeled mtDNA-encoded subunits from control (left panels), patient A4 (middle panels), and patient P (NDUFAF1 defect, right panels) fibroblasts at different chase times. Mitochondria were isolated, solubilized in 1% Triton X-100, and subjected to BN-PAGE in the first dimension followed by SDS–PAGE in the second dimension and phosphorimager analysis. The positions of ND1 and ND2 are indicated.
Immunoblot analysis was also performed with antibodies against subunits of the respiratory complexes. Steady-state levels of complex I are reduced in patient A4 (Figure 7B, lane 10) compared to control (Figure 7B, lane 9), whereas levels of complex IV, the complex III dimer (CIII2), complex V, and the complex III2/IV supercomplex were all similar in patient A4 (“P”) compared to control (“C”).
To further examine the potential assembly defect in patient A4 fibroblasts, we analyzed the assembly profiles of radiolabeled mtDNA-encoded subunits by using two-dimensional BN-PAGE analysis.42 It has been previously established that the ND2 subunit of complex I assembles into a ∼460 kDa intermediate complex before assembling into an ∼830 kDa intermediate that includes the ND1 subunit. At later times, these subunits assemble into holocomplex I.11 In control fibroblasts (Figure 7C, left panels), ND1 and ND2 are found in the ∼460 kDa and ∼830 kDa intermediate complexes at early chase times (0 and 3 hr), whereas at 24 hr chase these two subunits assemble into the mature complex (CI). In patient A4, ND2 was present in the ∼460 kDa complex at 0 and 3 hr chase, but it did not assemble into the ∼830 kDa complex (Figure 7C, middle panels). Furthermore, ND1 (or an intermediate complex containing ND1) was not present at these early chase times, suggesting that this assembly step was defective. At 24 hr chase, no complex I had assembled in patient A4 mitochondria, and the ND2 intermediate complex was lost, presumably turned over (Figure 7C, middle panel, bottom). This assembly defect is distinct from that seen in patient P, who harbors a CIA30 (NDUFAF1) defect.27 In this case, the intermediate complex containing ND2 is absent at 0 and 3 hr chase, whereas ND1 is present but remains stalled in an ∼400 kDa complex (Figure 7C, right panels). At 24 hr chase, complex I assembly was not detectable, and both ND1 and ND2 were lost (Figure 7C, right panels). These results suggest that C20orf7 is involved in the assembly or stability of an early complex I assembly intermediate that contains (among others) the ND1 subunit.
Discussion
We studied a consanguineous Egyptian family in which one child and two pregnancies were affected by complex I deficiency. Homozygosity mapping identified a 9.2 Mb region on chromosome 20p as being the largest region that was identical by descent in the three affected siblings. Transfer of a normal copy of chromosome 20 into a patient cell line by microcell-mediated chromosome transfer could correct complex I activity, confirming that the causative gene was located on chromosome 20. Genes within the candidate region were prioritized by bioinformatic analyses, and 11 genes encoding known or predicted mitochondrial proteins were sequenced. Only one possibly pathogenic mutation was identified, a homozygous c.719C→T mutation present in both predicted transcripts of C20orf7. This mutation was not found in 210 Egyptian control alleles and is predicted to cause the substitution of a highly conserved leucine residue to proline, probably affecting C20orf7 protein structure. We showed that C20orf7 was imported into mitochondria, where it is localized on the matrix side of the inner membrane. Knockdown of C20orf7 expression via lentiviral-mediated RNAi caused marked decrease in complex I activity, confirming that lack of C20orf7 can cause complex I deficiency.
Complex IV activity and the amount of the complex IV subunit MTCO2 showed a modest decrease after C20orf7 shRNA treatment, and a more marked decrease after NDUFAF1 shRNA treatment. This raises the possibility that both proteins may play a role in complex IV assembly or that efficient assembly or stability of complex IV may depend on normal complex I or supercomplex assembly. A similar loss of complex IV activity and protein has been noted previously in transgenic Caenorhabditis elegans strains expressing mutations in the NDUFV1 complex I subunit.46 However, this issue remains unresolved because all tissues and cell lines studied from patients with mutations in the C20orf7 or NDUFAF1 genes have shown essentially normal complex IV activity.27 Secondary effects on complex IV assembly may only occur in some tissues and with specific types or severity of complex I defects.
Extensive proteomic studies have led to the conclusion that the complex I holoenzyme comprises 45 known subunits.8 C20orf7 is not one of these subunits, so it presumably plays a role in complex I assembly or maintenance. Steady-state mRNA transcripts for C20orf7 have been detected in 79 human tissues,47 implying that the protein is widely expressed. C20orf7 possesses a predicted S-adenosylmethionine (SAM)-dependent methyltransferase fold, suggesting that it serves to methylate a protein, small molecule or nucleic acid within mitochondria. Interestingly, a complex I subunit, NDUFB3, has been reported to be methylated on at least two highly conserved histidines.48 This raises the interesting possibility that C20orf7 methylates this subunit as a requisite step in the complex I assembly process.
Four complex I assembly factors in which mutations cause complex I deficiency in humans have been identified previously, namely, NDUFAF2, NDUFAF1, C6orf66,11 and our recent identification of C8orf38.29 NDUFAF2 (B17.2L) appears to function in a late stage of complex I assembly. It is not present in the holoenzyme but is associated with a subcomplex of ∼830 kDa that accumulates in patients with mutations in the NDUFS4 subunit.26,49 This subassembly is a productive intermediate in complex I assembly because import of the missing subunit restores complex I assembly.38 NDUFAF1 (CIA30) associates with complex I at earlier stages (500–850 kDa subcomplexes)27 whereas the roles of C6orf6628 and C8orf3829 are not yet clear.
C20orf7 appears to function at an early step in the complex I assembly process but in a different manner to CIA30. C20orf7 is involved in the assembly of an early intermediate of ∼400 kDa, which contains the ND1 subunit, whereas CIA30 appears to associate with assembly intermediates that contain the ND2 subunit. The localization of C20orf7 to the mitochondrial inner membrane, disruption of complex I activity upon silencing of its expression, and the lethal neonatal complex I deficiency arising from a mutation in the coding sequence of C20orf7, strongly confirm its role as a bona fide complex I assembly factor.
Other putative complex I assembly factors have also been identified but have not yet been shown to cause complex I deficiency in humans. Ecsit has been reported to interact with CIA30 in 500 kDa and 850 kDa complexes and its knockdown resulted in severe impairment of complex I assembly and mitochondrial function.50 Deficiency of apoptosis-inducing factor (AIF) in the harlequin mutant mouse causes a complex I assembly defect.51 Prohibitin and PTCD1, the human ortholog of the CIA84 Neurospora crassa complex I assembly factor, have also been identified as possible assembly factors of complex I.11,51–53
Assembly factors could potentially be associated with other pathways or diseases because dual roles have been reported for Ecsit,54 B17.2L,55 and C6orf66.56 These are also involved in regulating cell proliferation. As far as we know, no other roles have as yet been reported for C20orf7, although it was identified as being downregulated in a microarray expression analysis of cells treated with heteroaromatic quinols possessing antitumor activity.57
In conclusion, we have identified C20orf7 as a complex I assembly factor and mutations in C20orf7 as a new cause of complex I deficiency. It is likely that many more assembly factors of complex I may be identified, bearing in mind that complex IV with only 13 subunits requires more than 15 assembly factors for its assembly.58 Further studies are needed to incorporate known and unidentified complex I assembly factors into a more complete complex I assembly model. Identification of additional assembly factors remains a challenge but should increase our understanding not only of complex I biogenesis but also of mitochondrial involvement in other common pathways or diseases.
Supplemental Data
Supplemental Data include one table and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI Reference Sequence (RefSeq), http://www.ncbi.nlm.nih.gov/RefSeq/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We thank Andrew Cuthbert for advice on monochromosomal transfer. This work was supported by grants (D.R.T. and M.T.R.), postdoctoral fellowships (M.McK. and D.M.K.), and a principal research fellowship (D.R.T.) from the Australian National Health and Medical Research Council and a grant from the Australian Research Council (M.T.R.). C.S. was supported by a University of Melbourne postgraduate research scholarship. Grant funding was also received from the Muscular Dystrophy Association (D.R.T.), the Ramaciotti Foundation (M.McK.), and the National Institutes of Health (GM077465) (V.K.M.).
References
- 1.Skladal D., Halliday J., Thorburn D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 2.Kirby D.M., Thorburn D.R. Approaches to finding the molecular basis of mitochondrial oxidative phosphorylation disorders. Twin Res. Hum. Genet. 2008;11:395–411. doi: 10.1375/twin.11.4.395. [DOI] [PubMed] [Google Scholar]
- 3.Kirby D.M., Crawford M., Cleary M.A., Dahl H.H., Dennett X., Thorburn D.R. Respiratory chain complex I deficiency: An underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1264. doi: 10.1212/wnl.52.6.1255. [DOI] [PubMed] [Google Scholar]
- 4.Triepels R.H., Van Den Heuvel L.P., Trijbels J.M., Smeitink J.A. Respiratory chain complex I deficiency. Am. J. Med. Genet. 2001;106:37–45. doi: 10.1002/ajmg.1397. [DOI] [PubMed] [Google Scholar]
- 5.Robinson B.H. Human complex I deficiency: Clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochim. Biophys. Acta. 1998;1364:271–286. doi: 10.1016/s0005-2728(98)00033-4. [DOI] [PubMed] [Google Scholar]
- 6.Loeffen J.L., Smeitink J.A., Trijbels J.M., Janssen A.J., Triepels R.H., Sengers R.C., van den Heuvel L.P. Isolated complex I deficiency in children: Clinical, biochemical and genetic aspects. Hum. Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 7.Schapira A.H. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 8.Carroll J., Fearnley I.M., Skehel J.M., Shannon R.J., Hirst J., Walker J.E. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 9.Schagger H., Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schagger H. Respiratory chain supercomplexes. IUBMB Life. 2001;52:119–128. doi: 10.1080/15216540152845911. [DOI] [PubMed] [Google Scholar]
- 11.Lazarou M., Thorburn D.R., Ryan M.T., McKenzie M. Assembly of mitochondrial complex I and defects in disease. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbamcr.2008.04.015. in press. Published online May 4, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Benit P., Chretien D., Kadhom N., de Lonlay-Debeney P., Cormier-Daire V., Cabral A., Peudenier S., Rustin P., Munnich A., Rotig A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet. 2001;68:1344–1352. doi: 10.1086/320603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeffen J., Elpeleg O., Smeitink J., Smeets R., Stockler-Ipsiroglu S., Mandel H., Sengers R., Trijbels F., van den Heuvel L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. 2001;49:195–201. doi: 10.1002/1531-8249(20010201)49:2<195::aid-ana39>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 14.Benit P., Slama A., Cartault F., Giurgea I., Chretien D., Lebon S., Marsac C., Munnich A., Rotig A., Rustin P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 2004;41:14–17. doi: 10.1136/jmg.2003.014316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Triepels R.H., van den Heuvel L.P., Loeffen J.L., Buskens C.A., Smeets R.J., Rubio Gozalbo M.E., Budde S.M., Mariman E.C., Wijburg F.A., Barth P.G. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann. Neurol. 1999;45:787–790. doi: 10.1002/1531-8249(199906)45:6<787::aid-ana13>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 16.Loeffen J., Smeitink J., Triepels R., Smeets R., Schuelke M., Sengers R., Trijbels F., Hamel B., Mullaart R., van den Heuvel L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998;63:1598–1608. doi: 10.1086/302154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuelke M., Smeitink J., Mariman E., Loeffen J., Plecko B., Trijbels F., Stockler-Ipsiroglu S., van den Heuvel L. Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat. Genet. 1999;21:260–261. doi: 10.1038/6772. [DOI] [PubMed] [Google Scholar]
- 18.Benit P., Steffann J., Lebon S., Chretien D., Kadhom N., de Lonlay P., Goldenberg A., Dumez Y., Dommergues M., Rustin P. Genotyping microsatellite DNA markers at putative disease loci in inbred/multiplex families with respiratory chain complex I deficiency allows rapid identification of a novel nonsense mutation (IVS1nt −1) in the NDUFS4 gene in Leigh syndrome. Hum. Genet. 2003;112:563–566. doi: 10.1007/s00439-002-0884-2. [DOI] [PubMed] [Google Scholar]
- 19.Lebon S., Chol M., Benit P., Mugnier C., Chretien D., Giurgea I., Kern I., Girardin E., Hertz-Pannier L., de Lonlay P. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J. Med. Genet. 2003;40:896–899. doi: 10.1136/jmg.40.12.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McFarland R., Kirby D.M., Fowler K.J., Ohtake A., Ryan M.T., Amor D.J., Fletcher J.M., Dixon J.W., Collins F.A., Turnbull D.M. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann. Neurol. 2004;55:58–64. doi: 10.1002/ana.10787. [DOI] [PubMed] [Google Scholar]
- 21.van den Heuvel L., Ruitenbeek W., Smeets R., Gelman-Kohan Z., Elpeleg O., Loeffen J., Trijbels F., Mariman E., de Bruijn D., Smeitink J. Demonstration of a new pathogenic mutation in human complex I deficiency: A 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am. J. Hum. Genet. 1998;62:262–268. doi: 10.1086/301716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirby D.M., Salemi R., Sugiana C., Ohtake A., Parry L., Bell K.M., Kirk E.P., Boneh A., Taylor R.W., Dahl H.H. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J. Clin. Invest. 2004;114:837–845. doi: 10.1172/JCI20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez-Moreira D., Ugalde C., Smeets R., Rodenburg R.J., Lopez-Laso E., Ruiz-Falco M.L., Briones P., Martin M.A., Smeitink J.A., Arenas J. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann. Neurol. 2007;61:73–83. doi: 10.1002/ana.21036. [DOI] [PubMed] [Google Scholar]
- 24.Berger I., Hershkovitz E., Shaag A., Edvardson S., Saada A., Elpeleg O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008;63:405–408. doi: 10.1002/ana.21332. [DOI] [PubMed] [Google Scholar]
- 25.Hoefs S.J., Dieteren C.E., Distelmaier F., Janssen R.J., Epplen A., Swarts H.G., Forkink M., Rodenburg R.J., Nijtmans L.G., Willems P.H. NDUFA2 complex I mutation leads to Leigh disease. Am. J. Hum. Genet. 2008;82:1306–1315. doi: 10.1016/j.ajhg.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogilvie I., Kennaway N.G., Shoubridge E.A. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest. 2005;115:2784–2792. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunning C.J., McKenzie M., Sugiana C., Lazarou M., Silke J., Connelly A., Fletcher J.M., Kirby D.M., Thorburn D.R., Ryan M.T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;26:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saada A., Edvardson S., Rapoport M., Shaag A., Amry K., Miller C., Lorberboum-Galski H., Elpeleg O. C6ORF66 is an assembly factor of mitochondrial complex I. Am. J. Hum. Genet. 2008;82:32–38. doi: 10.1016/j.ajhg.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.E., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorburn D.R., Sugiana C., Salemi R., Kirby D.M., Worgan L., Ohtake A., Ryan M.T. Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim. Biophys. Acta. 2004;1659:121–128. doi: 10.1016/j.bbabio.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Ellaway C., North K., Arbuckle S., Christodoulou J. Complex I deficiency in association with structural abnormalities of the diaphragm and brain. J. Inherit. Metab. Dis. 1998;21:72–73. doi: 10.1023/a:1005367515701. [DOI] [PubMed] [Google Scholar]
- 32.Rahman S., Blok R.B., Dahl H.H.M., Danks D.M., Kirby D.M., Chow C.W., Christodoulou J., Thorburn D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 34.Cuthbert A.P., Trott D.A., Ekong R.M., Jezzard S., England N.L., Themis M., Todd C.M., Newbold R.F. Construction and characterization of a highly stable human: Rodent monochromosomal hybrid panel for genetic complementation and genome mapping studies. Cytogenet. Cell Genet. 1995;71:68–76. doi: 10.1159/000134066. [DOI] [PubMed] [Google Scholar]
- 35.Calvo S., Jain M., Xie X., Sheth S.A., Chang B., Goldberger O.A., Spinazzola A., Zeviani M., Carr S.A., Mootha V.K. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 36.Lamande S.R., Bateman J.F., Hutchison W., McKinlay Gardner R.J., Bower S.P., Byrne E., Dahl H.H. Reduced collagen VI causes Bethlem myopathy: A heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum. Mol. Genet. 1998;7:981–989. doi: 10.1093/hmg/7.6.981. [DOI] [PubMed] [Google Scholar]
- 37.Root D.E., Hacohen N., Hahn W.C., Lander E.S., Sabatini D.M. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat. Methods. 2006;3:715–719. doi: 10.1038/nmeth924. [DOI] [PubMed] [Google Scholar]
- 38.Lazarou M., McKenzie M., Ohtake A., Thorburn D.R., Ryan M.T. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol. Cell. Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan M.T., Voos W., Pfanner N. Assaying protein import into mitochondria. Methods Cell Biol. 2001;65:189–215. doi: 10.1016/s0091-679x(01)65012-x. [DOI] [PubMed] [Google Scholar]
- 40.Johnston A.J., Hoogenraad J., Dougan D.A., Truscott K.N., Yano M., Mori M., Hoogenraad N.J., Ryan M.T. Insertion and assembly of human tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J. Biol. Chem. 2002;277:42197–42204. doi: 10.1074/jbc.M205613200. [DOI] [PubMed] [Google Scholar]
- 41.Pallotti F., Lenaz G. Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol. 2001;65:1–35. doi: 10.1016/s0091-679x(01)65002-7. [DOI] [PubMed] [Google Scholar]
- 42.McKenzie M., Lazarou M., Thorburn D.R., Ryan M.T. Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal. Biochem. 2007;364:128–137. doi: 10.1016/j.ab.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 43.Harlow E., Lane D. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1999. Using Antibodies: A Laboratory Manual. [Google Scholar]
- 44.Combet C., Blanchet C., Geourjon C., Deleage G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 45.Claros M.G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 46.Grad L.I., Lemire B.D. Mitochondrial complex I mutations in Caenorhabditis elegans produce cytochrome c oxidase deficiency, oxidative stress and vitamin-responsive lactic acidosis. Hum. Mol. Genet. 2004;13:303–314. doi: 10.1093/hmg/ddh027. [DOI] [PubMed] [Google Scholar]
- 47.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carroll J., Fearnley I.M., Skehel J.M., Runswick M.J., Shannon R.J., Hirst J., Walker J.E. The post-translational modifications of the nuclear encoded subunits of complex I from bovine heart mitochondria. Mol. Cell. Proteomics. 2005;4:693–699. doi: 10.1074/mcp.M500014-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Vogel R.O., van den Brand M.A., Rodenburg R.J., van den Heuvel L.P., Tsuneoka M., Smeitink J.A., Nijtmans L.G. Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Mol. Genet. Metab. 2007;91:176–182. doi: 10.1016/j.ymgme.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 50.Vogel R.O., Janssen R.J., van den Brand M.A., Dieteren C.E., Verkaart S., Koopman W.J., Willems P.H., Pluk W., van den Heuvel L.P., Smeitink J.A. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vahsen N., Cande C., Briere J.J., Benit P., Joza N., Larochette N., Mastroberardino P.G., Pequignot M.O., Casares N., Lazar V. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bourges I., Ramus C., Mousson de Camaret B., Beugnot R., Remacle C., Cardol P., Hofhaus G., Issartel J.P. Structural organization of mitochondrial human complex I: Role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem. J. 2004;383:491–499. doi: 10.1042/BJ20040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gabaldon T., Rainey D., Huynen M.A. Tracing the evolution of a large protein complex in the eukaryotes, NADH:ubiquinone oxidoreductase (Complex I) J. Mol. Biol. 2005;348:857–870. doi: 10.1016/j.jmb.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 54.Xiao C., Shim J.H., Kluppel M., Zhang S.S., Dong C., Flavell R.A., Fu X.Y., Wrana J.L., Hogan B.L., Ghosh S. Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis. Genes Dev. 2003;17:2933–2949. doi: 10.1101/gad.1145603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsuneoka M., Teye K., Arima N., Soejima M., Otera H., Ohashi K., Koga Y., Fujita H., Shirouzu K., Kimura H. A novel Myc-target gene, mimitin, that is involved in cell proliferation of esophageal squamous cell carcinoma. J. Biol. Chem. 2005;280:19977–19985. doi: 10.1074/jbc.M501231200. [DOI] [PubMed] [Google Scholar]
- 56.Karp C.M., Shukla M.N., Buckley D.J., Buckley A.R. HRPAP20: A novel calmodulin-binding protein that increases breast cancer cell invasion. Oncogene. 2007;26:1780–1788. doi: 10.1038/sj.onc.1209980. [DOI] [PubMed] [Google Scholar]
- 57.Bradshaw T.D., Matthews C.S., Cookson J., Chew E.H., Shah M., Bailey K., Monks A., Harris E., Westwell A.D., Wells G. Elucidation of thioredoxin as a molecular target for antitumor quinols. Cancer Res. 2005;65:3911–3919. doi: 10.1158/0008-5472.CAN-04-4141. [DOI] [PubMed] [Google Scholar]
- 58.Fontanesi F., Soto I.C., Horn D., Barrientos A. Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am. J. Physiol. Cell Physiol. 2006;291:C1129–C1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.