

Abstract
The H syndrome is a recently reported autosomal-recessive disorder characterized by cutaneous hyperpigmentation, hypertrichosis, hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism, short stature, hallux valgus, and fixed flexion contractures of the toe joints and the proximal interphalangeal joints. Homozygosity mapping in five consanguineous families resulted in the identification of mutations in the SLC29A3 gene, which encodes the equilibrative nucleoside transporter hENT3. Three mutations were found in 11 families of Arab and Bulgarian origin. The finding of several different mutations in a small geographic region implies that the H syndrome might be rather common. The identification of mutations in the SLC29A3 gene in patients with a mild clinical phenotype suggests that this is a largely underdiagnosed condition and strongly suggests that even oligosymptomatic individuals might have the disorder.
Main Text
The H syndrome is a recently described autosomal-recessive genodermatosis with systemic manifestations. The term H syndrome refers to the major clinical and laboratory findings of hyperpigmentation and hypertrichosis, hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism, low height, and occasionally, hyperglycemia.1 We recently reported on ten Arab patients (nine males and one female) originating from six consanguineous families from a small region near Jerusalem. These patients suffered from hyperpigmented, hypertrichotic, and indurated cutaneous patches involving the middle and lower parts of the body. The onset of the cutaneous lesions was at the first or second decade of life. Additional systemic features were sensorineural hearing loss, cardiac anomalies, hepatosplenomegaly, scrotal masses, endocrinopathy (short stature, gynecomastia), exophthalmos with normal thyroid function, angiopathy (varicosities, dilated lateral scleral vessels, and facial telangiectases), and camptodactyly. Laboratory evaluation revealed growth-hormone deficiency and hypergonadotrophic hypogonadism with azoospermia. Histopathological examination of involved skin showed hyperpigmentation of the basal layer with seborrheic keratosis-like acanthosis, infiltration of CD68-positive histiocytes, and a perivascular mononuclear infiltrate with plasma cells and mast cells throughout the dermis and subcutaneous fat.1 The syndrome may not be limited to our region because seven other patients, six males and one female with a similar phenotype, have already been reported from other parts of the world.2–4 The subjects of the present study were the ten previously reported patients and an additional nine patients: three males and four females from three families (G–I), originating from the same geographic area, and one male (Family J) from 50 Km away and a previously reported Bulgarian patient (Family K)3 (Figure 1).
Figure 1.
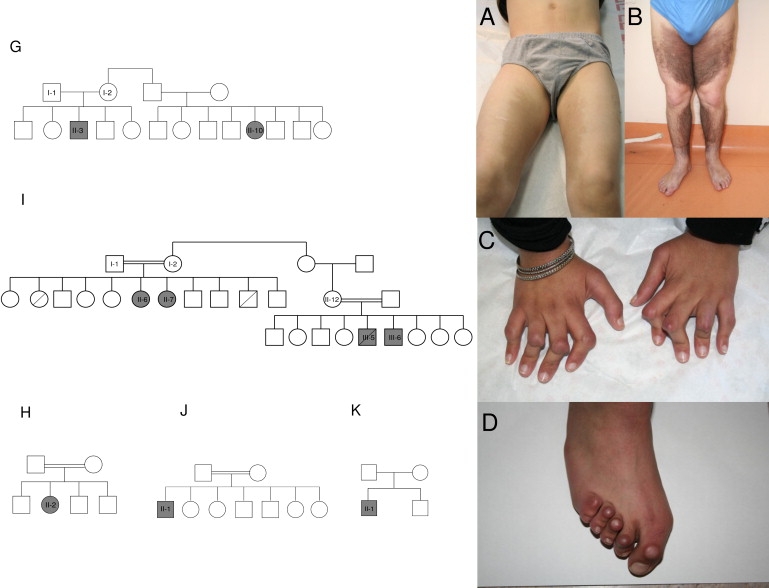
Pedigrees and Clinical Findings of Four Novel Families with H Syndrome and of a Previously Reported Bulgarian Family
Affected individuals are marked by gray symbols.
Pedigrees for families G–K are shown at left.
(A) Hyperpigmentation of inner thighs in patient GII-3.
(B) More diffuse and extensive hyperpigmentation in patient KII-1.
(C) Fixed flexion contractures of proximal interphalangeal joints in patient HII-2.
(D) Hallux valgus and fixed flexion contractures of toe joints in patient III-6.
The clinical findings of patients from families G-J (Figure 1 and Table 1) were similar to those of the previously reported patients.1 Additional clinical features were prominent: Hallux valgus with fixed flexion contractures of the toe joints (8/10 patients) and fixed flexion contractures of the hands' proximal interphalangeal joints (7/9) appeared during the first decade of life. More prominent changes occurred in female patients, and some patients displayed lateral tibial torsion (4/9) and myelofibrosis (1/9). Reviewing the clinical findings1 we now find it likely that the previously reported camptodactyly is in fact a milder form of the contractures of the proximal interphalangeal joints. Another exceptional finding was noted in the female patient III-7, whose indurated and hyperpigmented patches not only were present at the middle and lower half of the body but also extended to the trunk and neck with a reticular and annular morphology. This patient had hard subcutaneous masses with overlying hyperpigmentation in the mons pubis, a finding that has previously been reported only in males. Interestingly, two other female patients; GII-10 and HII-2, lacked the classical cutaneous findings of hyperpigmentation, hypertrichosis, and induration of the lower limbs. At the time of writing, all the patients were alive, except for patient IIII-5, who died at nine years of age, probably because of patent ductus arteriosus with pulmonary hypertension and recurrent pneumonia. This patient had also suffered from hearing loss, short stature, and hepatomegaly. The oldest surviving patient (HII-2) was a 50-year-old female with short stature, marked skeletal changes of the hands and feet, and hypogonadism manifesting as delayed puberty and primary amenorrhea. Another unique feature in this patient was red cell aplasia that developed at the age of 29 and necessitated multiple blood transfusions and splenectomy and was attributed to myelofibrosis. Re-examination of members of the previously reported families B and D1 identified two more affected patients (BII-6 and DII-7). At the age of 18 and 22 years, the only abnormalities in these patients were dilated lateral scleral vasculature and prominent gynecomastia.
Table 1.
Clinical Features of Patients in Families G–J
| Patient | GII-3 | GII-10 | HII-2 | III-6 | III-7 | IIII-5 | IIII-6 | JII-1 |
|---|---|---|---|---|---|---|---|---|
| Age at examination (years) | 13 | 20 | 50 | 20 | 17 | Died at age 9 | 13 | 12 |
| Sex | male | female | female | female | female | male | male | male |
| Height(m)/Weight (kg) | 1.32/36 | 1.63/53 | 1.46/46 | 1.65/62 | 1.60/55 | known short stature | 1.37/34 | 1.10/23 |
| Cutaneous hyperpigmentation, hypertrichosis and induration | genitals, inner thighs, shins | − | − | inner and posterior thighs, shins | inner thighs, shins, hyperpigmented reticulate patches over trunk | NA | lower back and abdomen, genitals, thighs, shins, dorsum of foot | mid-abdomen and back, genitals, thighs, shins, dorsum of foot, forearms |
| Genital examination | subcutaneous masses, micropenis | − | − | − | subcutaneous masses | NA | subcutaneous masses, micropenis | subcutaneous masses, micropenis |
| lack of secondary sexual signs | ||||||||
| Hepatosplenomegaly | hepatomegaly | − | hepatomegaly | − | − | hepatomegaly | + | + |
| Heart examination | mild pulmonic stenosis | 2/6 systolic murmura | − | mild pericardial effusion | − | patent ductus arteriosus with right to left shunt, left superior vena cava to coronary sinus | pulmonary hypertension | 2/6 systolic murmura |
| Hearing loss | + | + | − | + | + | + | − | + |
| Hallux valgus | + | + | + | + | + | + | + | − |
| severe rocker bottom deformity of foot | ||||||||
| Fixed flexion contracture of proximal interphalangeal joints | + mild | + | + | + | + | NA | + | + |
| Lateral tibial torsion | − | − | + | + | + | − | − | + |
| Varicose veins | + | + | + | + | + | NA | − | − |
| stasis ulcers | ||||||||
| Exophthalmos | − | − | + | + | − | NA | − | − |
| Dilated lateral scleral vasculature | + | + | − | + | + | NA | + | + |
| Facial telangiectases | − | + | + | + | + | + | + | − |
| Mental retardation | − | − | − | − | − | − | mild | − |
| Other findings | arcus senilis hematologic illness | arcus senilis | arcus senilis, gynecomastia |
NA, not available for examination.
Echocardiography was unavailable.
Blood samples were collected from 17 patients and 22 healthy family members of the ten families after consent was obtained from the Institutional Review Board and the patients. GeneChip Human Mapping 50K and 250K SNP Array of Affymetrix was used for performing a genome-wide analysis of single-nucleotide polymorphisms with the DNA samples of patients AII-8, CII-2, CII-4, DII-2, EII-1, and FII-6. A single region of homozygosity shared by five patients from four families (C, D, E, and F) was identified in chromosome 10q21.3–q22.1 (haplotype cluster rs7894724–rs7081747). This region was not shared by patient AII-8. We next genotyped all members of the six families for polymorphic microsatellites (D10S1665, D10S529, D10S537, D10S1650, and D10S606) spanning the homozygous region on chromosome 10. The analysis revealed two distinct allele patterns segregating with the disorder. One of these was in family B, and the other was in families C–F, for whom a key recombination event occurred between markers D10S529 and D10S537 in family C but not in families D, E, and F (Figure 2). Patient AII-8 was a compound heterozygote of both haplotypes and had a recombination event between markers D10S1665 and D10S529. The recombination event in family C enabled us to narrow the interval of linkage to a 1.53 Mb region, flanked by markers D10S529 and D10S606 (71,509,225–73,039,899). This region contained 21 known genes, and we were able to exclude 14 of them by direct sequencing: AIFM2 [gene ID 84883], ADAMTS14 [MIM 607506], EIF4EBP2 [MIM 602224], C10ORF54 [gene ID 64115], PPA1 [MIM 179030], H2AFY2 [gene ID 55506], LRRC20 [gene ID 55222], NODAL [MIM 601265], PRF1 [MIM 170280], TYSND1 [MIM 611017], SAR1A [MIM 607691], PSAP [MIM 176801], NPFFR1 [gene ID 64106], and C10ORF27 [gene ID 219793]. Sequencing of the six exons of the SLC29A3 gene [gene ID 55315] with intronic primers (Table S1 in the Supplemental Data available online) revealed homozygosity for two missense mutations in exon 6: a c.1279G > A mutation, changing glycine 427 to serine (G427S) in all patients of families B and I, and a c.1309G > A mutation, changing glycine 437 to arginine (G437R) in all patients of families C–H. The only patient in family A, AII-8, was a compound heterozygote for both mutations (Figure 3 and Table 2). This patient resides in a village located between families B and C, which have the c.1279G > A and c.1309G > A mutations, respectively. Sequence analysis of exon 6 in the DNA sample of patient JII-1 revealed homozygosity for a single-nucleotide deletion, c.1045delC, which is predicted to result in a frameshift and the generation of a stop codon at residue 404 of the protein (Figure 3). Restriction digestion analysis with PvuII was used for detecting the G427S mutation after the introduction of a mismatch four bases upstream the mutation in the reverse primer. The Eco1471 (StuI) restriction enzyme was used for detecting the G437R mutation. All mutations segregated with the disease within the families, and no other mutations were identified in the other five exons of the SLC29A3 gene.
Figure 2.
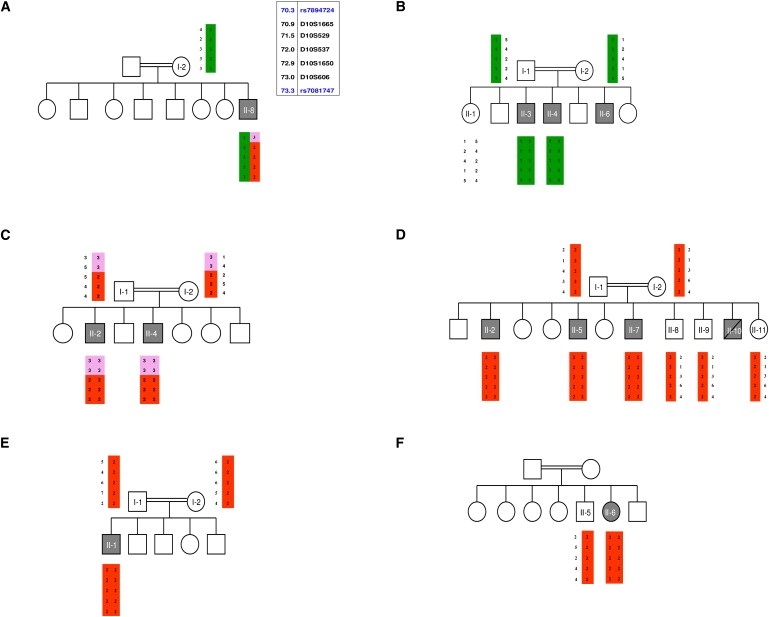
Haplotype Analysis Performed in Six Families with H Syndrome
Affected individuals are marked by gray symbols. The disease-associated haplotypes in the linked interval on chromosome 10q21.3–q22.1 are shown by vertical bars (red and green) in families A–F. Alleles excluded by recombination events are marked in pink. The physical locations of the polymorphic microsatellites are given near the haplotype for individual AI-2.
Figure 3.
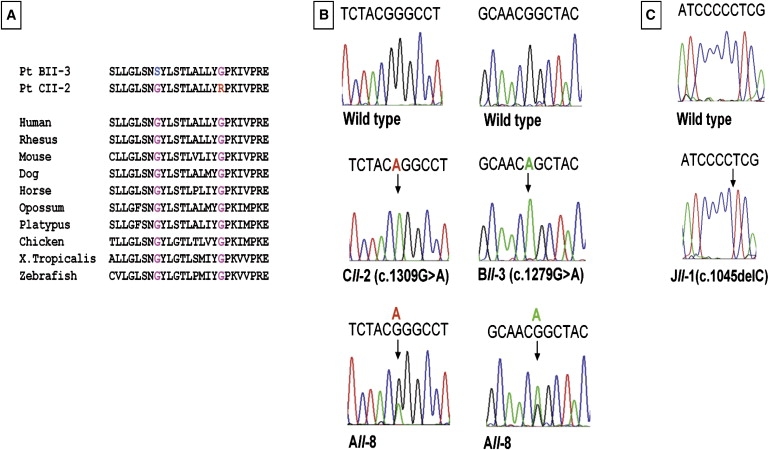
Mutations c.1279G > A, c.1309G > A, and c.1045delC in the SLC29A3 Gene
(A) Conservation among species of amino acid positions affected by G427S and G437R missense mutations. The mutated amino acids are marked by blue and red, respectively.
(B) The single base-pair substitution mutations are shown (vertical arrow) in DNA samples of patients CII-2 (c.1309G > A), BII-3 (c.1279G > A), and AII-8 (c.1309G > A; c.1279G > A). The wild-type is shown in the upper panel.
(C) The c.1045delC mutation in patient JII-1 is shown in the lower panel, and the wild-type is shown in the upper panel.
Table 2.
Mutations in SLC29A3 in 11 Families with Autosomal-Recessive H Syndrome
| Family | Mutation |
|---|---|
| A | G427S; G437R |
| B, I, K | G427S |
| C–H | G437R |
| J | c.1045delC |
Subsequently, we analyzed the six exons of the SLC29A3 gene in a previously reported Bulgarian patient with a similar phenotype. The patient originated from a nonconsanguineous family, and his parents came from the same small town. He suffered from growth retardation, hepatomegaly, gynecomastia, hypogonadism, diabetes mellitus, malabsorption, and skin changes consisting of induration, hyperpigmentation, and hypertrichosis on the anterior aspect of both thighs, lower abdomen, and scrotum.3 Re-examination of the patient at 23 years revealed additional H-syndrome-characteristic features, including facial telangiectases, mild proptosis, and bilateral camptodactyly. Sequence analysis revealed homozygosity for the G427S mutation due to the same nucleotide change as in the index families A, B, and I. However, the microsatellite haplotype was completely different (Table S2), indicating that the Bulgarian mutation arose independently.
362 and 212 control chromosomes of individuals of the same geographical and ethnic background as families A–I were examined for the presence of the c.1279G > A and c.1309G > A mutations, respectively. Four and two heterozygous individuals, respectively, were identified, indicating a 1% frequency of both diseased alleles in this population. None of the mutations was found in 60 chromosomes derived from individuals of Jewish origin or in 76 control chromosomes of individuals of European or Bulgarian origin.
The SLC29A3 gene encodes the human equilibrative nucleoside transporter 3 (hENT3), a member of the equilibrative nucleoside transporter (ENT) family that mediates passive sodium-independent transport of nucleosides. Members of this family possess 11 transmembrane helices, with a cytoplasmic N terminus and an extracellular C terminus.5 hENT3 is a 475 amino acid protein, and both Gly427 and Gly437 residues are part of the tenth transmembrane domain (TMD), which is highly conserved from C. elegans to humans. Mutations in this TMD are therefore likely to perturb the transporter function. Similarly, the single-nucleotide deletion located at the eighth TMD is expected to result in a truncated protein lacking the tenth and 11th TMDs and to disrupt the eighth and ninth TMDs. Nucleoside transporters are essential for nucleotide synthesis by salvage pathways in cells that lack de novo pathways and are the route of cellular uptake for cytotoxic nucleosides used in cancer and viral chemotherapy.6 Two families of nucleoside transporters are known in mammals: the concentrative nucleoside transporter (SLC28) family, which consists of active, sodium-dependent transporters found mainly in specialized epithelial tissues7,8, and the ENT family. Of the four members of the ENT family, ENT1, 2, and 4 are primarily located at the cell surface, although ENT1 has also been found in liver mitochondria.9 hENT3 is widely expressed in adult and fetal human tissues; the highest levels are detected in placenta, uterus, ovary, spleen, lymph node, and bone marrow. Transport experiments with hENT3 expressed in Xenopus oocytes revealed that this transporter has a broad selectivity for nucleosides. hENT3 cellular sublocalization is still in doubt: the protein was found to colocalize with lysosomal markers in cultured human cells and was suggested to be a late endosomal/lysosomal transporter involved in the export of nucleosides from the lysosomal interior.10 A recent abstract suggested the localization of this transporter to the mitochondria in multiple cell types.11 Gene silencing of hENT3 in a placental cell line significantly decreased both cellular and mitochondrial uptake of nucleosides, nucleobases and dideoxynucleosides suggesting it may also play a role in the cell membrane.11
The present lack of certainty regarding the exact localization of hENT3 is not clarified by the clinical features characteristic of the H syndrome. Both mitochondrial and lysosomal disorders are known for their extreme clinical heterogeneity, for the involvement of multiple organs, and for their progressive course.12,13 Many features of the H syndrome are prevalent among patients with mitochondrial respiratory-chain defects, including the sensorineural hearing loss, cardiomyopathy, hepatomegaly, endocrinopathy (especially the short stature, delayed puberty, and early menopause), anemia, and hyperglycemia. Cutaneous involvement is rare but has been described infrequently in patients with mitochondrial disorders resulting from both mitochondrial and nuclear-DNA mutations. These include disorders of pigmentation (both hyper- and hypopigmentation), hypertrichosis, erythematous eruptions, alopecia, hair-shaft abnormalities, acrocyanosis, and multiple lipomatosis.14,15 Lysosomal disorders13 are characterized by mesenchymal and reticuloendothelial involvement, which are also prominent in the H syndrome. However, and most importantly, the lack of a storage material argues against a lysosomal pathogenesis in this disorder. Further characterization of the localization and function of normal and mutated hENT3 is required.
The identification of three different mutations, all in the sixth exon of the gene, in ten affected families residing in a small geographic region, and the resultant carrier rate of 1%, is striking. This relatively high carrier rate combined with the marked phenotypic heterogeneity within our families, some of which include mildly affected patients, implies that H syndrome may be a largely underdiagnosed condition. The occurrence of the same mutation (G427S) in different populations indicates that the mutation might reside in a hot spot in the SLC29A3 gene. We conclude that sequence analysis of the SLC29A3 gene, or at least of its sixth exon, is warranted in all patients presenting with any unexplained combination of the above symptoms.
Acknowledgments
We thank the family members for their participation in this study and Sofia Babay and Ruth Bargal for expert technical assistance. This study was supported in part by the Authority for Research and Development, Hebrew University of Jerusalem (A.Z.), the Hadassah Medical Center physician scientist program, the Hadassah-Hebrew University joint research fund (V.M.P.), and the North American Hair Research Society (Z.A.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GDB database, http://www.gdb.org
Ensembl genome browser, http://www.ensembl.org
UCSC genome browser, http://www.genome.ucsc.edu
References
- 1.Molho-Pessach V., Agha Z., Aamar S., Glaser B., Doviner V., Hiller N., Zangen D.H., Raas-Rothschild A., Ben-Neriah Z., Shweiki S. The H syndrome: A new genodermatosis characterized by indurated, hyperpigmented and hypertrichotic skin with systemic manifestations. J. Am. Acad. Dermatol. 2008;59:79–85. doi: 10.1016/j.jaad.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Hamadah I.R., Banka N. Autosomal recessive plasma cell panniculitis with morphea-like clinical manifestation. J. Am. Acad. Dermatol. 2006;54:S189–S191. doi: 10.1016/j.jaad.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 3.Marina S., Broshtilova V. POEMS in childhood. Pediatr. Dermatol. 2006;23:145–148. doi: 10.1111/j.1525-1470.2006.00201.x. [DOI] [PubMed] [Google Scholar]
- 4.Prendiville J., Rogers M., Kan A., de Castro F., Wong M., Junker A., Becknell C., Schultz K. Pigmented hypertrichotic dermatosis and insulin dependent diabetes: Manifestations of a unique genetic disorder? Pediatr. Dermatol. 2006;24:101–107. doi: 10.1111/j.1525-1470.2007.00352.x. [DOI] [PubMed] [Google Scholar]
- 5.Hyde R.J., Cass C.E., Young J.D., Baldwin S.A. The ENT family of eukaryote nucleoside and nucleobase transporters: Recent advances in the investigation of structure/function relationships and the identification of novel isoforms. Mol. Membr. Biol. 2001;18:53–63. [PubMed] [Google Scholar]
- 6.Leung G.P., Tse C.M. The role of mitochondrial and plasma membrane nucleoside transporters in drug toxicity. Expert Opin. Drug Metab. Toxicol. 2007;3:705–718. doi: 10.1517/17425255.3.5.705. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin S.A., Mackey J.R., Cass C.E., Young J.D. Nucleoside transporters: Molecular biology and implications for therapeutic development. Mol. Med. Today. 1999;5:216–224. doi: 10.1016/S1357-4310(99)01459-8. [DOI] [PubMed] [Google Scholar]
- 8.Gray J.H., Owen R.P., Giacomini K.M. The concentrative nucleoside transporter family, SLC28. Pflugers Arch. 2004;447:728–734. doi: 10.1007/s00424-003-1107-y. [DOI] [PubMed] [Google Scholar]
- 9.Lai Y., Tse C.M., Unadkat J.D. Mitochondrial expression of the human equilibrative nucleoside transporter 1 (hENT1) results in enhanced mitochondrial toxicity of antiviral drugs. J. Biol. Chem. 2004;279:4490–4497. doi: 10.1074/jbc.M307938200. [DOI] [PubMed] [Google Scholar]
- 10.Baldwin S.A., Yao S.Y., Hyde R.J., Ng A.M.L., Foppolo S., Barnes K., Ritzel M.W.L., Cass C.E., Young J.D. Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. J. Biol. Chem. 2005;280:15880–15887. doi: 10.1074/jbc.M414337200. [DOI] [PubMed] [Google Scholar]
- 11.Govindarajan R., Blonski M., Ming Tse C.M., Wang J., Unadkat J.D. The human equilibrative nucleoside transporter-3 is a mitochondrial transporter that transports anti-HIV dideoxynucleoside drugs. FASEB J. 2008;22:1132.2. [Google Scholar]
- 12.Munnich A., Rustin P., Rotig A., Chretien D., Bonnefont J.P., Nuttin C., Cormier V., Vassault A., Parvy P., Bardel J. Clinical aspects of mitochondrial disorders. J. Inherit. Metab. Dis. 1992;15:448–455. doi: 10.1007/BF01799603. [DOI] [PubMed] [Google Scholar]
- 13.Vellodi A. Lysosomal storage disorders. Br. J. Haematol. 2005;128:413–431. doi: 10.1111/j.1365-2141.2004.05293.x. [DOI] [PubMed] [Google Scholar]
- 14.Birch-Machin M.A. Mitochondria and the skin. Clin. Exp. Dermatol. 2000;25:141–146. doi: 10.1046/j.1365-2230.2000.00605.x. [DOI] [PubMed] [Google Scholar]
- 15.Bodemer C., Rotig A., Rustin P., Cormier V., Niaudet P., Saudubray J.M., Rabier D., Munnich A., de Prost Y. Hair and skin disorders as signs of mitochondrial disease. Pediatrics. 1999;103:428–433. doi: 10.1542/peds.103.2.428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.