

Abstract
Human movement disorders represent a significant and unresolved societal burden. Among these, the most prevalent is Parkinson’s disease (PD), a disorder afflicting millions worldwide. Despite major advances, stemming primarily from human genetics, there remains a significant gap in our understanding of what factors underlie disease susceptibility, onset, and progression. Innovative strategies to discern specific intracellular targets for subsequent drug development are needed to more rapidly translate basic findings to the clinic. Here we briefly review the recent contributions of research using the nematode roundworm Caenorhabditis elegans as a model system for identifying and characterizing gene products associated with PD. As a microscopic but multicellular and genetically tractable animal with a well-defined nervous system and an experimentally tenable lifespan, C. elegans affords significant advantages to researchers attempting to determine causative and therapeutic factors that influence neuronal dysfunction and age-associated neurodegeneration. The rapidity with which traditional genetic, large-scale genomic, and pharmacological screening can be applied to C. elegans epitomizes the utility of this animal for disease research. Moreover, with mature bioinformatic and functional genomic data readily available, the nematode is well positioned to play an increasingly important role in PD-associated discoveries.
Physicists and astronomers have long posited the concept of a ‘wormhole’ as a means of rapid interstellar travel by analogy with how a worm could eat a hole from one end of an apple, through the center to the other end, and create a shortcut through the intervening space. Unfortunately, bending the fabric of space and time is not typically considered a viable option to more rapidly explore cures for neurodegenerative diseases (and would likely result in a poorly received grant application). The biological equivalent of the space exploration program, the human genome project, has unleashed an age of genomic, proteomic, metabolomic, and bioinformatic analyses that has generated a wealth of datasets primed for subsequent discovery. This exponential growth demands the development of functional means for exploiting this treasure trove of biological information. In this regard, biomedical researchers are literally turning to a worm to accelerate the path toward therapeutic advances and get to the core of mechanisms underlying Parkinson’s disease (PD).
While drugs to treat the symptoms of PD have been prescribed for decades (e.g. L-DOPA), an unmet need for innovative strategies to discern disease etiology and treatments that either halt or reverse progression remains. Application of a microscopic nematode roundworm toward gaining insights into a human neurodegenerative disorder may seem impractical; yet, C. elegans affords many advantages for such research, as it has a defined cell lineage, completed genome sequence, and lifespan of only 2 weeks. As opposed to the human brain, which is estimated to have over 100 billion nerve cells, this nematode contains precisely 302 neurons for which a defined neuronal connectivity map has been determined. In this regard, C. elegans is ideal for investigation of diseases associated with neuronal dysfunction and ageing, and represents a model system that is poised to bridge the gap between basic and translational research.
Interpretation of disease-associated data obtained in invertebrate systems requires downstream validation in mammals prior to establishing the therapeutic significance of any findings. The likelihood of a positive outcome is significant, however, due to the evolutionary conservation of metazoan genomes. For example, human homologs have been identified for at least 50% of C. elegans genes. Remarkably, these include all orthologs of reported genes linked to familial forms of PD (Fig. 1), with the one exception being the gene encoding α-synuclein. Despite this difference, ectopic overexpression of both wildtype and mutant (A53T) human α-synuclein led to motor deficits or DA neuron loss in C. elegans (Lasko et al., 2003). Subsequent reports also demonstrated phenotypic changes associated with driving α-synuclein expression from a variety of pan-neuronal and specific neuronal promoters (Cao et al., 2005; Ved et al., 2005; Kuwahara et al., 2006). The translational utility of C. elegans is perhaps best demonstrated by the characterization of conserved neuroprotective genes that block α-synuclein-associated degeneration in nematodes (Cooper et al., 2006; Gitler et al., 2008; Hamamichi et al., 2008).
Fig. 1. PD-associated gene products and their prospective sites of cellular function.

PD is hypothesized to be a consequence of the dysfunction of intersecting and compensatory protein degradation components, including those associated with the lysosome and autophagy, as well as those associated with the ubiquitin proteasome system (UPS). Inefficient clearance and enhanced misfolding, or expression, of α-synuclein has been shown to block intracellular trafficking and increase cytotoxicity. Accordingly, mutations in Parkin (an E3 ubiquitin ligase), ubiquitin C-terminal hydrolase-1 (UCHL-1), as well as in a lysosomal ATPase termed ATP13A2 and the Gaucher’s disease-related protein glucocerebrosidase (GBA), have further implicated protein degradation pathways in PD. PD-associated mutations in genes linked with mitochondrial function, such as those encoding DJ-1 and the PTEN-induced kinase (PINK1), presumably affect the production of reactive oxygen species (ROS), which accelerate protein damage and neurodegeneration. While mutations in the leucine-rich repeat kinase 2 (LRRK2) protein now represent the most common heritable cause of PD, the function of this large, 2527 amino acid multidomain-containing protein remains undefined.
The rich, 40-year history of C. elegans genetics provides a legacy of behavioral and neuroanatomical information that researchers draw upon to elucidate relationships between gene function and PD-associated mechanisms (Bargmann, 1998). Using traditional genetic suppressor and enhancer screening, investigators have begun to dissect genetic contributors to DA neuron development and function (Chase et al., 2004). A limbless simple invertebrate will certainly never recapitulate the phenotypic behaviors associated with the tremor and dyskinesia of PD, yet this nematode does afford opportunities for accurate quantification of factors that influence DA neuron survival. Loss of DA neurons in C. elegans is not lethal and leads only to a subtle behavioral deficit where affected animals display an inability to discriminate local mechanosensory cues (Sawin et al., 2000). Unfortunately, this behavior, while quantifiable, is not robust enough to be easily used in extensive screening paradigms.
Mammalian and primate modeling approaches to PD have traditionally involved use of neurotoxins to induce DA neuron loss and evaluate the consequences of neurodegeneration. Among these toxins are 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxydopamine (6-OHDA), as well as the pesticides rotenone and paraquat. Worms are susceptible to these toxins (Nass et al., 2002; Braungart et al., 2004; Cao et al., 2005); moreover, the defined and transparent neuroanatomy of C. elegans includes precisely eight neurons that produce DA, thereby enabling an unparalleled level of quantitative analysis of neurodegeneration and protection by use of fluorescent protein labeling (Fig. 2). Furthermore, these animals can be evaluated in specific genetic or transgenic backgrounds to screen for factors that confer either neuroprotection or enhancement of degeneration. One neuroprotective gene product termed TOR-2, a human torsinA-related protein with molecular chaperone-like activity (Caldwell et al., 2008), was shown to deter multiple forms of toxic insults to DA neurons, including 6-OHDA, excess intracellular DA production, and α-synuclein overexpression (Cao et al., 2005). The neuroprotective capacity of torsin-like proteins renders them intriguing candidates for subsequent drug targeting and validation in mammalian systems, where human torsinA is endogenously expressed in DA neurons (Augood et al., 1998).
Fig. 2. Anterior dopamine neurons of C. elegans.
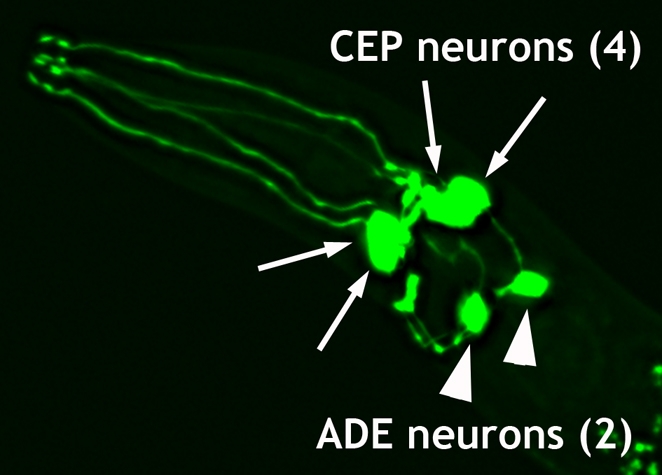
Cell bodies and processes of the six anterior DA neurons including two pairs of the CEP (cephalic) neurons and one pair of ADE (anterior deirid) neurons, as illuminated using GFP driven from the dopamine transporter promoter (Pdat-1::GFP). Two additional DA neurons (PDEs) are found in the posterior (not shown).
Directed and comprehensive screens have been undertaken to define chemical and genetic effectors of worm DA neuron sensitivity to 6-OHDA (Nass et al., 2005; Maranova and Nichols, 2007). These studies identified a collection of intragenic mutations in the gene sequence encoding the key protein required for DA reuptake into presynaptic neurons, the DA transporter (DAT-1). Structural and functional relationships revealed through this work have implications not only for PD, but also for other disorders associated with DAT function (e.g. depression, ADHD). It has also been shown that worm dat-1 knockout mutants exhibit diminished α-synuclein-dependent degeneration, which indicates a possible role for DAT in maintaining an important balance of intraneuronal levels of DA, the dysregulation of which may contribute to cytotoxicity (Cao et al., 2005).
Elegant studies in Drosophila have demonstrated the capacity for model systems to reveal unsuspected relationships between PD gene products, such as PINK1 and Parkin, and their impact on mitochondrial integrity and morphology (Clark et al., 2006; Park et al., 2006; Poole et al., 2008). Likewise, the ability of worm research to link genotype to phenotype in this manner is accelerating our understanding of the underlying nature of PD. Ved et al. investigated the functional consequences of rotenone-induced stress and reported that pan-neuronal overexpression of human αsynuclein (wild-type or A53T mutant), RNAi knockdown of a C. elegans DJ-1 ortholog (B0432.2), or a deletion mutant of the C. elegans parkin ortholog (pdr-1), all produced similar patterns of mitochondrial vulnerability in response to pharmacological challenges associated with complex I inhibition (Ved et al., 2005). Furthermore, the effect of rotenone, which led to mortality in these animals, was more severe in α-synuclein-expressing strains and the pdr-1 deletion mutant than in control animals of wild-type background. Initial work on worm PDR-1 revealed that this Parkin ortholog, an E3 ubiquitin ligase, cooperates with conserved degradation machinery to mediate ubiquitin conjugation (Springer et al., 2005). Co-expression of C. elegans pdr-1 and α-synuclein variants in human cell cultures showed that a truncated protein derived from an in-frame pdr-1 (lg103) deletion allele causes aggregation of α-synuclein, similar to Parkin variants isolated from PD patients. This altered gene product also resulted in proteotoxicity and hypersensitivity to ER stress (Springer et al., 2005). These examples demonstrate how mutant analysis in C. elegans offers a powerful strategy for uncovering functional relationships between gene products with key roles in PD pathogenesis in mammalian cells.
C. elegans has been recently used to show a protective role for human LRRK2 (leucine-rich repeat kinase 2) against mitochondrial toxicity induced by rotenone, an effect potentiated by knockdown of an endogenous worm ortholog, lrk-1 (Wolozin et al., 2008). These data are surprising when considering a PD-associated G2019S mutant version of LRRK2 that exhibits increased kinase activity was also found to be protective; kinase activity has been suggested to contribute to the pathogenesis associated with LRRK2 neurotoxicity (West et al., 2007). An initial report on lrk-1 function in C. elegans indicated a role for the product of this gene in establishing neuronal polarity (Sakaguchi-Nakashima et al., 2007). As mutations in LRRK2 now represent the most common genetic cause of PD (Haugarvoll et al., 2008), further insights gleaned from C. elegans on this important, yet relatively uncharacterized, protein family will undoubtedly be informative.
The capacity for genomic-scale screening has brought C. elegans to the forefront of modern functional analysis (Kamath and Ahringer, 2003). In contrast to mammals, where individual knockouts of mice are expensive and time consuming, worms are an efficient and economical alternative. Hamamichi et al. took a hypothesis-driven approach toward identification of putative genetic modifiers of PD via a multi-tiered screen for C. elegans genes that impact the misfolding of transgenic human α-synuclein, as well as neuroprotection, in vivo (Hamamichi et al., 2008). This study encompassed RNAi knockdown of nearly 900 bioinformatically prioritized gene targets, comprising components of cellular pathways implicated in protein folding or degradation, as well as gene products that are either co-expressed or interact with worm orthologs of familial PD genes. From this varied, but biased, ‘guilt by association’ list of targets, 20 candidate genes emerged as having the greatest propensity to influence α-synuclein misfolding. Internal validation was evident within this short list, as included were the worm homologs of two established recessive PD genes (DJ-1, PINK1), as well as a gene (ULK2) shown to be one of only six identified in a genome-wide association study of polymorphisms in PD patients (Fung et al., 2006). Importantly, overexpression of select cDNAs in C. elegans DA neurons revealed that five of seven gene products tested, chosen from the primary effectors of α-synuclein misfolding identified in the RNAi screen, exhibited significant protection from age- and dose-dependent neurodegeneration induced by α-synuclein (Hamamichi et al., 2008). Thus, application of C. elegans in this context uncovered functionally evaluated targets identified by the two primary clinical criteria associated with PD: α-synuclein accumulation and DA neurodegeneration.
These genes, which included uncharacterized proteins, as well as regulators of autophagy, lysosomal function, cellular trafficking, and G-protein signaling, now represent outstanding candidates for strategically targeted drug development and validation in mammalian systems. A more recently conducted unbiased genome-wide RNAi screen for genetic factors that influence αsynuclein inclusion formation in C. elegans also yielded an over-representation of genes encoding components of vesicular trafficking, as well as specific age-associated genes (e.g. sir-2.1) (van Ham et al., 2008). Similar validation of the effect that these additional targets may have on DA neuron survival will likely reveal functionally significant relationships as well. Associations between PD and lysosomal degradation are of growing interest following the identification of a lysosomal ATPase, ATP13A2 (PARK9), as a gene product linked to hereditary early-onset PD with dementia (Ramirez et al., 2006), in addition to the discovery of a higher incidence of PD in patients with mutations in the Gaucher’s disease-associated gene, GBA, encoding the lysosomal storage-enzyme glucocerebrosidase. (Aharon-Peretz et al., 2004; Clark et al., 2007). It is easy to envision an expansion of such gene-specific data and the application of C. elegans toward investigating the functional consequences of single-nucleotide polymorphisms (SNPs) found in human populations, as these may potentially lead to genetic biomarkers of disease susceptibility.
The burgeoning promise that a microscopic worm may contribute toward the goal of drug discovery for PD has become a tangible reality. Containing a rudimentary nervous system, unlike single-celled organisms, but more amenable to transgenic analysis and drug screening than mammals, C. elegans serves as an excellent intermediary bottleneck in translational research pipelines to characterize therapeutic gene and drug targets across animal models. Concerted efforts toward therapeutic development have been initiated that exploit the high-throughput screening capabilities of yeast cells to define numerous gene targets of interest, followed by subsequent evaluation of their neuroprotective capacity in worms, fruit flies and rat neuronal cultures (Cooper et al., 2006; Gitler et al., 2008). This approach has already revealed that overproduction of αsynuclein leads to a cytotoxic blockage in intracellular vesicular trafficking that can be alleviated by specific members of the Rab protein family (Cooper et al., 2006; Gitler et al., 2008). Thus, through the employment of a combination of powerful model systems, not only has a prospective cellular cause of PD been illuminated, but targeted screens for small molecules that protect against underlying functional aspects of neurodegeneration are now possible.
When envisioning future directions whereby C. elegans will benefit PD research, several inescapable advantages of this model system should be considered. The most obvious of these is the well-established use of the worm for investigating mechanisms of aging (Kenyon, 2005). Indeed, the only definitive risk factor for PD is aging, where PD symptomatically affects over 2% of people above age 65. Extensive studies on worm longevity mutants (e.g. daf-2 or age-1) have demonstrated that evolutionarily conserved mechanisms are shared between invertebrates and mammals. Microarray and genomic-scale RNAi analyses conducted in age-extending backgrounds have yielded significant insights into gene sets that implicate dietary restriction and insulin-like signaling pathways as crucial mediators of lifespan (Murphy et al., 2003; Panowski et al., 2007). The vital, but poorly understood, interface between aging and age-associated neurodegenerative disease represents an exciting frontier that is readily explored using C. elegans.
Furthermore, in the advent of the microRNA (miRNA) revolution, our nascent understanding of putative regulatory roles for miRNAs in neuron function will likely soon interface with our understanding of neurological disease and PD (Kosik, 2006; Kim et al., 2007). Considering the pioneering role worm research has played in defining miRNA function, a universe of possibilities exist, as worm researchers are well-poised to explore relationships between miRNAs, ageing and neuroprotective gene activity (Ibanez-Ventoso, 2007).
Advantages of C. elegans as a model for Parkinson’s disease.
All orthologs of genes linked to familial PD have been identified in C. elegans, except α-synuclein
Ectopic overexpression of α-synuclein has neurotoxic effects in C. elegans, which are blocked by neuroprotective genes
C. elegans has a simple nervous system (302 neurons compared with over 100 billion in humans) that is amenable to quantitative analysis of neurodegeneration
C. elegans is susceptible to toxins commonly used to model neurodegeneration
C. elegans studies predict relationships between cellular signaling, trafficking and protein degradation pathways, which are being tested for their susceptibility to targeted drug development for PD
Finally, although the primary advances in PD etiology have come through human genetics, the largely idiopathic nature of PD remains linked to inexplicable environmental causes. C. elegans research into innate immunity and cellular stress response has already provided mechanistic insights into organismal defenses and environmental influences on homeostasis (Kim et al., 2002; Mohri-Shiomi and Garsin, 2008). An expansion of neurotoxicity studies conducted in C. elegans is warranted and may yield a greater understanding of the interplay between genetic composition and environmental factors such as heavy metals, pesticides and other untested exposures.
The worm is unquestionably a powerful system, yet it has its limitations: the anatomical and functional connectivity of the neuronal circuitry within this simple nematode cannot recapitulate the complex features of mammalian dopamine neurons or mimic the precise behavioral deficits associated with their loss. Likewise, as a cautionary caveat to inferring conservation of function from genetic interaction data, Tischler et al. demonstrated that synthetic lethal gene interactions between yeast and worm genes are not significantly conserved (Tischler et al., 2008). In this context, as the march toward systems biology proceeds and integrated analyses of gene or protein activity continue to lead to an increasing number of complex datasets, our ability to eventually define causes and cures will depend on functional strategies for wading through the emerging ‘information overload’. Science will benefit from the efficient manner by which C. elegans research can contribute to the quest for translational and personalized medical breakthroughs, by boldly going where no worm has gone before.
Footnotes
COMPETING INTERESTS The authors declare no competing financial interests.
References
- Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. (2004). Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. New Engl. J. Med. 351, 1972–1977 [DOI] [PubMed] [Google Scholar]
- Augood S. J., Penney J. B., Jr, Friberg I. K., Breakefield X. O., Young A. B., Ozelius L. J., Standaert D. G. (1998). Expression of the early-onset torsion dystonia gene (DYT1) in human brain. Ann. Neurol. 43, 669–673 [DOI] [PubMed] [Google Scholar]
- Bargmann C. I. (1998). Neurobiology of the C. elegans. Genome Science 282, 2028–2033 [DOI] [PubMed] [Google Scholar]
- Braungart E., Gerlach M., Riederer P., Baumeister R., Hoener M. C. (2004). Caenorhabditis elegans MPP+ model of Parkinson’s disease for high-throughput drug screenings. Neurodegener Dis. 1, 175–183 [DOI] [PubMed] [Google Scholar]
- Caldwell G. A., Cao S., Sexton E. G., Gelwix C. C., Bevel J. P., Caldwell K. A. (2003). Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum. Molec. Genet. 12, 307–319 [DOI] [PubMed] [Google Scholar]
- Cao S., Gelwix C. C., Caldwell K. A., Caldwell G. A. (2005). Torsin-mediated protection from cellular stress in the domaminergic neurons of Caenorhabditis elegans. J. Neurosci. 12, 3801–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase D. L., Pepper J. S., Koelle M. R. (2004). Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat. Neurosci. 7, 1096–1103 [DOI] [PubMed] [Google Scholar]
- Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- Clark L. N., Ross B. M., Wang Y., Mejia-Santana H., Harris J., Louis E. D., Cote L. J., Andrews H., Fahn S., Waters C., et al. (2007). Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson’s disease. Neurology 69, 1270–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. A., Gitler A. D., Cashikar A., Haynes C. M., Hill K. J., Bhullar B., Liu K., Xu K., Strathearn KE., Liu F., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung H. C., Scholz S., Matarin M., Simon-Sanchez J., Hernandez D., Britton A., Gibbs J. R., Langefeld C., Stiegert M. L., Schymick J., et al. (2006). Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet 5, 911–916 [DOI] [PubMed] [Google Scholar]
- Gitler A. D., Bevis B. J., Shorter J., Strathearn K. E., Hamamichi S., Su L. J., Caldwell K. A., Caldwell G. A., Rochet J. C., McCaffery J. M., et al. (2008). The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 105, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamichi S., Rivas R. N., Knight A. L., Cao S., Caldwell K. A., Caldwell G. A. (2008). Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 105, 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath R. S., Ahringer J. (2003). Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–321 [DOI] [PubMed] [Google Scholar]
- Kenyon C. (2005). The plasticity of aging: insights from long-lived mutants. Cell 120, 449–460 [DOI] [PubMed] [Google Scholar]
- Kim D. H., Feinbaum R., Alloing G., Emerson F. E., Garsin D. A., Inoue H., Tanaka-Hino M., Hisamoto N., Matsumoto K., Tan M. W., et al. (2002). A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623–626 [DOI] [PubMed] [Google Scholar]
- Kim J., Inoue K., Ishii J., Vanti W. B., Voronov S. V., Murchison E., Hannon G., Abeliovich A. (2007). A microRNA feedback circuit in midbrain dopamine neurons. Science 317, 1220–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik K. S. (2006). The neuronal microRNA system. Nat. Rev. Neurosci. 7, 911–920 [DOI] [PubMed] [Google Scholar]
- Kuwahara T., Koyama A., Gengyo-Ando K., Masuda M., Kowa H., Tsunoda M., Mitani S., Iwatsubo T. (2006). Familial Parkinson mutant α-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J. Biol. Chem. 281, 334–340 [DOI] [PubMed] [Google Scholar]
- Lasko M., Vartiainen S., Moilanen A-M., Sirvio J., Thomas J. H., Nass R., Blakley R. D., Wong G. (2003). Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human α-synuclein. J. Neurochem. 86, 165–172 [DOI] [PubMed] [Google Scholar]
- Haugarvoll K., Rademakers R., Kachergus J. M., Nuytemans K., Ross O. A., Gibson J. M., Tan E. K., Gaig C., Tolosa E., Goldwurm S., et al. (2008). Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology 70, 1456–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez-Ventoso C., Yang M., Guo S., Robins H., Padgett R. W., Driscoll M. (2006). Modulated microRNA expression during adult lifespan in Caenorhabditis elegans. Aging Cell 5, 235–246 [DOI] [PubMed] [Google Scholar]
- Maranova M., Nichols C. D. (2007). Identification of neuroprotective compounds of Caenorhabditis elegans dopaminergic neurons against 6-OHDA. J. Mol. Neurosci. 31, 127–137 [DOI] [PubMed] [Google Scholar]
- Mohri-Shiomi A., Garsin D. A. (2008). Insulin signaling and the heat shock response modulate protein homeostasis in the Caenorhabditis elegans intestine during infection. J. Biol. Chem. 283, 194–201 [DOI] [PubMed] [Google Scholar]
- Murphy C. T., McCarroll S. A., Bargmann C. I., Fraser A., Kamath R. S., Ahringer J., Li H., Kenyon C. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283 [DOI] [PubMed] [Google Scholar]
- Nass R., Hahn M. K., Jessen T., McDonald PW., Carvelli L., Blakely R. D. (2005). A genetic screen in Caenorhabditis elegans for dopamine neuron insensitivity to 6-hydroxydopamine identifies dopamine transporter mutants impacting transporter biosynthesis and trafficking. J. Neurochem. 94, 774–785 [DOI] [PubMed] [Google Scholar]
- Nass R., Hall D. H., Miller D. M., 3rd, Blakely R. D. (2002). Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 99, 3264–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panowski S. H., Wolff S., Aguilaniu H., Durieux J., Dillin A. (2007). HA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature 447, 550–555 [DOI] [PubMed] [Google Scholar]
- Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J. M., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161 [DOI] [PubMed] [Google Scholar]
- Poole A. C., Thomas R. E., Andrews L. A., McBride H. M., Whitworth A. J., Pallanck L. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 105, 1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A., Heimbach A., Grundemann J., Stiller B., Hampshire D., Cid L. P., Goebel I., Mubaidin A. F., Wriekat A. L., Roeper J., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191 [DOI] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A., Meir J. Y., Jin Y., Matsumoto K., Hisamoto N. (2007). LRK-1, a C. elegans PARK8-related kinase, regulates axonaldendritic polarity of SV proteins. Curr. Biol. 7, 592–598 [DOI] [PubMed] [Google Scholar]
- Sawin E. R., Ranganathan R., Horvitz H. R. (2000). C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619–631 [DOI] [PubMed] [Google Scholar]
- Springer W., Hoppe T., Schmidt E., Baumeister R. (2005). A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet. 14, 3407–3423 [DOI] [PubMed] [Google Scholar]
- Tischler J., Lenher B., Fraser A. G. (2008). Evolutionary plasticity of gene networks. Nat. Genet. 4, 390–391 [DOI] [PubMed] [Google Scholar]
- van Ham T. J., Thijssen K. L., Breitling R., Hofstra R. M. W., Plasterk R. H. A., Nollen E. A. A. (2008). C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLoS Genet. 4, 10000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ved R., Saha S., Westlund B., Perier C., Burnam L., Sluder A., Hoener M., Rodrigues C. M., Alfonso A., Steer C., et al. (2005). Similar patterns, of, mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein parkin and DJ-1 in Caenorhabditis elegans. J. Biol. Chem. 280, 42655–42668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. B., Moore D. J., Choi C., Andrabi S. A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K. L., Dawson V. L., Dawson T. M. (2007). Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 16, 223–232 [DOI] [PubMed] [Google Scholar]
- Wolozin B., Saha S., Guillily M., Ferree A., Riley M. (2008). Investigating convergent actions of genes linked to familial Parkinson’s disease. Neurodegener Dis. 5, 182–185 [DOI] [PMC free article] [PubMed] [Google Scholar]