

Abstract
A concise synthesis of (+)-rishirilide B (2) is described. This is the first synthesis to be reported for the (+)-enantiomer of rishirilide B (2) as found in nature. The strategy accentuates the valuable combination of a method for o-quinone methide coupling with a method for enantioselective resorcinol dearomatization, which provides a densely functionalized chiral building block. The convergent synthesis illustrates several improvements and refinements to these methods and their supporting chemistries. Among these is the in situ generation of PhI[OTMS]OTf. Combination of this oxidant with phenol 31 constitutes the first example of a diastereoselective oxidative dearomatization of a resorcinol displaying a 2-alkyl substituent. In addition, the preparation of the cyclic sulfone 34 is reported. As a new dimethide precursor expressing a readily cleavable O-benzyl residue, sulfone 34 should prove useful in future endeavors. A protocol using the aluminum amide of dimethylhydrazine for opening and cleavage of a [1,4]-dioxan-2-one is also described. This procedure unmasks the hydroxy dione 36 by jettisoning the chiral directing group. Regioselective O-carbamylation of the 1,3-dione 36 enables the transformation of the remaining carbonyl into the α-hydroxy carboxylic acid found in 2. The total synthesis of (+)-rishirilide B (2) requires 15 pots from benzaldehyde 17 and 13 pots from benzaldehyde 32. The final product emerges in yields of 12.5% and 20.3% from compounds 17 and 32, respectively. The longest linear sequence requires eight chromatographies. Important observations leading to the development of the principle asymmetric method are described within the context of the total synthesis.
Introduction
(+)-Rishirilide A (1) and (+)-rishirilide B (2) were isolated from Streptomyces rishiriensis OFR-1056 by Takeuchi and found to contain structurally related oxygenated anthracene skeletons (Figure 1). These natural products were found to inhibit α2-macroglobulin with respective IC50’s of 100 μg/mL for 1 and 35 μg/mL for 2.1 α2-Macroglobulin is a tetrameric serum glycoprotein that provides irreversible protease inhibition through a unique trapping mechanism. Inhibitors of α2-macro-globulin are useful in treating thrombosis caused by fibrinolytic accentuation. (+)-Rishirilide B (2) has also been shown to inhibit glutathione S-transferase with an IC50 of 26.9 μM.2 Glutathione S-transferase catalyzes the addition of the tripeptide glutathione to electrophilic substrates, resulting in their increased water solubility and excretion. Glutathione S-transferase activity also causes increased resistance toward anticancer drugs; therefore, an inhibitor of it may serve to potentiate certain anticancer therapies.
Figure 1.

The natural products (+)-rishirilides A (1) and B (2), and some known derivatives (±)-3, (±)-4, and (±)-5.
In 1999, Hauser and Xu claimed a racemic synthesis of an analogue of 2, (±)-methyl-rishirilide B (3).4 Their strategy employed a cycloaddition reaction between a well-known isobenzofuranone enolate and a novel enone. The remainder of their efforts was spent in an attempt to remove undesired oxygen substituents from the resulting naphthazarin intermediate.
In 2001, Danishefsky and Allen completed the first racemic synthesis of (±)-2.3 Their convergent two-pronged strategy includes a naphthalene disconnection similar to the earlier Hauser approach. However, there were some crucial distinctions. The Danishefsky synthesis employs an elaborate o-quinone dimethide intermediate for combination with a simpler cyclic enedione. Each component displays a reverse regioselective preference as compared to their Hauser counterparts. A chiral hydroxyl residue serves as an internal Lewis acid and activates a portion of the enedione. Because the o-quinone dimethide exists at a lower oxidation state than Hauser’s isobenzofuranone enolate, the Danishefsky synthesis is almost completed by the cycloaddition reaction. The synthesis concludes after just two additional steps. The chiral hydroxyl residue directs the addition of an iso-pentyl organometallic reagent to the adjacent carbonyl affording (±)-4, which succumbs to fluoride-mediated deprotection of the ester and phenol protecting groups to afford (±)-2. Furthermore, by conversion of rishirilide B (±)-2 into its methyl analogue (±)-3 and performing an X-ray analysis, Danishefsky and co-workers proved that the Hauser–Xu effort had failed to reach methyl rishirilide (±)-3 as earlier claimed.4 The Danishefsky work also illuminates some pitfalls associated with the rishirilide skeleton. Their choice of fluoride labile protecting groups arose from appreciation of the instability of (±)-3 and related alkyl esters toward basic saponification conditions. In 2003, the Danishefsky team reported an enantioselective addendum to the shorter spur of their two-pronged racemic strategy.5 The crucial chiral enedione dienophile is prepared in six steps and a 1.9% yield from 3-methylcyclohex-anone. At that time, only the (R)-antipode of this starting material was commercially available. It was used to arrive at ent-(−)-rishirilide B in 0.4% yield, thereby leading to the assignment of the absolute configuration for the dextrorotatory natural product.
The difficult enantioselective synthesis of the Danishefsky enedione intermediate, which might be viewed as a chiral o-quinol when enolized, underscores our belief that more methods for the enantioselective construction of highly oxidized six-membered rings are needed by the synthetic community. We, in turn, have investigated general methods leading to the oxidative dearomatization of resorcinols such as 6 (Figure 2). Few reactions can be imagined to yield such a densely functionalized chiral product with such readily distinguished functionality. Derivatives of 7 would seem to be reasonable synthetic precursors to many medically relevant natural products.6 Sometime ago, we imagined that a lactonization resulting in the formation of a [1,4]-dioxan-2-one fused onto the 3,4-positions of the cyclohexa-2,5-dienone product might overcome some of the problems surrounding oxidative dearomatizations and their products.7 Oxidative dearomatization is well known to be a capricious and fickle transformation.8 Side reactions often occur, and yields of greater than 50% are an exception rather than the norm. We postulated that by proper selection of substituents we could improve the performance of the reaction and stabilize the product. For example, the orthogonal attachment of the tertiary oxygen substituent relative to the π system of the product discourages reductive rearomatization, which is well known to afflict p-quinols. Moreover, the ring fusion enables the angular alkyl residue to assert diastereocontrol in subsequent reactions, which normally suffer because of the inherently flat structure of a p-quinol. In addition, the newly created alkoxy stereocenter emerges in a protected state. Our thoughts regarding convenience, stability, and diastereoselectivity were confirmed by a six-pot synthesis of (±)-epoxysor-bicillinol,9 and later by a racemic synthesis of (±)-5, a close relative of (±)-2 (Figure 2).10
Figure 2.
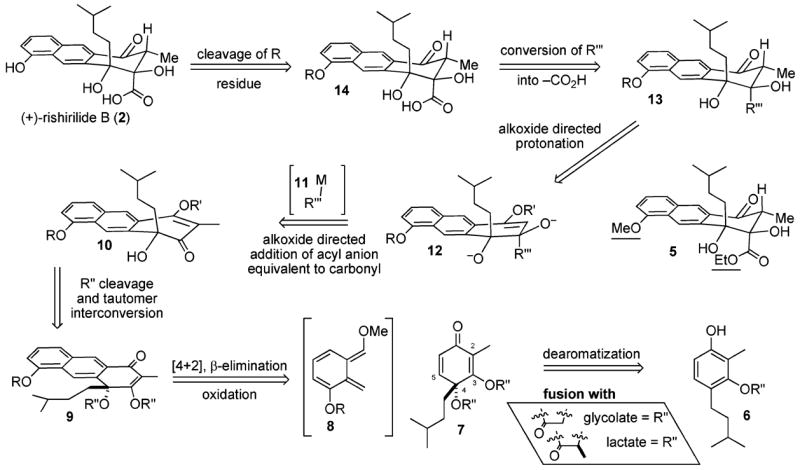
Our strategy for rishirilide B (2).
One pleasing series of reactions in the latter project was the addition of the carboxylic acid equivalent 11 directed by the alkoxide of 10 to produce the bisalkoxide 12, whereupon cleavage of the enol protecting group (R′) and diastereoselective protonation affords ketone 13. This cascade establishes two new stereocenters in a single pot. The transformation also proves that the conformation of rishirilide B (2) prevents β-elimination as seen in similar systems.11 However, the remainder of our strategy was seriously flawed. The saponification of the ethyl ester in 5 proves impossible. In addition, the naphthol protecting group (R = –Me) fails to cleave. Moreover, the yields for the perruthenate cleavage of enol ether 13 (R‴= –C(OEt)=CH2) into the ethyl ester 5 are meager at best (less than 35%). These problems had to be resolved, and our method for the enantioselective dearomatization of C2 brominated resorcinol derivatives had to be expanded,12 if we were to complete the synthesis of the natural product (+)-rishirilide B (2).
Early experiments had suggested our chiral technology with C2 bromo resorcinols would transfer to C2 methyl derivatives such as 6. However, a new problem surfaced. Whereas the corresponding R″ glycolate derivatives of 9 cleaved, the corresponding lactate derivatives proved too robust. Herein, we report the observations that led to the development of this enantioselective dearomatization procedure and the solutions that consequently emerged to these problems that enabled an enantioselective synthesis of (+)-rishirilide B (2).
Results and Discussion
To develop an enantioselective dearomatization process applicable to rishirilide B as well as future prospective targets, we required easy access to the appropriately substituted phenols that would serve as the platform for dearomatization. Building upon the observations of McLoughlin,13 we developed a cascade process for their synthesis beginning with the benzaldehydes (15–17). This new o-quinone methide generation and consumption technology subsequently enables easy access to many different prochiral resorcinols, such as 18–23, which display an assortment of C4-substituents (Scheme 1).14
Scheme 1.
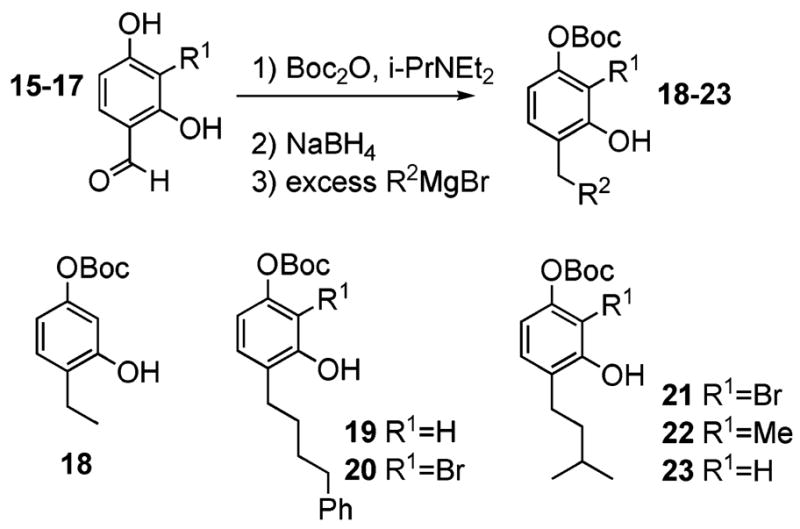
Synthesis of Differentially Protected 4-Alkyl Resorcinols
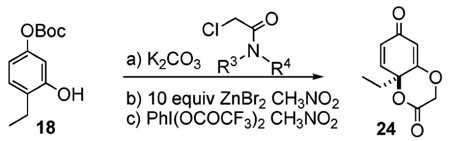
The oxidative dearomatization involving substrates 18–23 is unusual because it involves an ortho cyclization as opposed to the usual ipso cyclization most often found among the intramolecular reactions triggered by oxidative dearomatization. We thought that this ortho cyclization could be modified to an enantioselective format by incorporation of a chiral amine. However, this proved more difficult than we anticipated. The achiral glycolate-derived pyrrolidine amide already affords low yields of the desired lactone 24 (15–36%, Table 1, entry 1). Chiral amides derived from more encumbered amines proceed to 24 in even lower yields than their pyrrolidine counterpart.
Table 1.
Auxilliary Approach for Chiral Cyclohexa-2,5-dienones
 | ||||
|---|---|---|---|---|
| entry | chiral amide | sign for α | ee for 24 | yield of 24 three steps |
| 1 |
|
0 | 15–36% | |
| 2 |
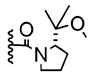
|
+ | 59% | 5–18% |
| 3 |
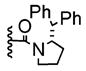
|
+ | 24% | 10–25% |
| 4 |
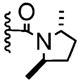
|
− | 7% | 0–5% |
The poor efficiency of the sequence means that the nonracemic lactone 24 and its analogues are useless from the standpoint of enantioselective syntheses even though these adducts easily undergo a variety of chemoselective and diastereoselective reactions.7 We therefore investigated structural modifications designed to lower the energy barrier for the desired ortho-lactonization and improve the prospects for these cyclohexadienone building blocks. The phenoxonium cation derived from 19 and 20 undergoes two types of cyclization, the ortho cyclization leading to lactone-A or the more traditional ipso cyclization leading to the spirocycle-B (Table 2). Modifications to the R1, R3, and R4 substituents, which improve the A/B ratio, result in more efficient and higher yielding reactions. The data shown (Table 2, entries 1–6) testify to the importance of tether length, amide nucleophilicity, and the steric impact of the [R1] aryl substituent on the efficiency of the dearomatization process.
Table 2.
Survey of Substituents and Their Effects on the Ratio of Ortho versus Ipso Cyclization
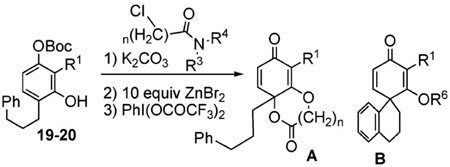 | |||||
|---|---|---|---|---|---|
| entry | n | R1 | amide | A/B ratio | yield of A three steps |
| 1 | 0 | -H |
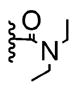
|
<1:99 | 0% |
| 2 | 1 | -H |
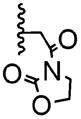
|
18:82 | 7% |
| 3 | 1 | -H |
|
43:57 | 28% |
| 4 | 1 | -Br |
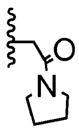
|
92:8 | 49% |
| 5 | 1 | -Br |
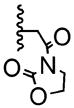
|
66:33 | 40% |
| 6 | 1 | -Br |

|
46:54 | 14% |
These findings illuminate several key features affecting the course of the transition into products as well as determining the ultimate success of the reaction. The results suggested further electronic and structural amendments to improve yields and enantioselectivites (Table 3). For example, exchange of the amine functionality [–NR3R4] for an alkoxyamine [–N(OR3)-R4] increases amide carbonyl nucleophilicity and improves the yield of the ortho cyclization. However, the effort required to build and to survey the new complex chiral amines suggested by these studies seemed wasteful. Instead, we opted for simpler modifications that would enable an enantioselective synthesis of the chiral cyclohexadienone building block. The simplest was replacement of the achiral glycolate-derived amide with an amide derived from lactate or leucate, thereby introducing the chiral center R5 onto a carbon of the [1,4]-dioxan-2-one that was fused to the cyclohexadienone. This method assumes that the reaction for coupling the chiral amide occurs with 100% inversion and that the chiral starting material would be jettisoned at some later point rather than recycled. However, the availability and costs associated with the R and S antipodes of methyl lactate and leucate justified this sacrificial approach, particularly in view of the scarcity of methods for the enantioselective synthesis of cyclohexadienones. Furthermore, unlike the steric interactions of amine auxiliaries, the interaction between the R5 and R1 substituents appears to improve the likelihood of a successful cyclization. Our studies further recommended that the carbonyl moiety, which is involved in the cyclization, carries sufficient steric encumbrance to reinforce the steric effects of the R5 stereocenter so that the overall reaction proves diastereoselective. These findings eventually led us to adopt Paterson’s procedure for preparation of the chiral methoxyamide derived from methyl lactate.15
Table 3.
Synthesis of a Chiral [1,4]-Dioxan-2-one Fused at the 3,4-Positions with a Cyclohexa-2,5-dienone
 | ||||
|---|---|---|---|---|
| entry | R1 | carbonyl species | dr | yield over three steps |
| 1 | -Br |
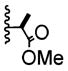
|
3:1 | 22% of 26 |
| 2 | -H |
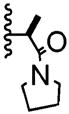
|
2:1 | 24% of 27 |
| 3 | -Br |
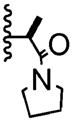
|
7:1 | 50% of 26 |
| 4 | -Br |
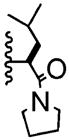
|
8:1 | 53% of 28 |
| 5 | -Br |
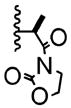
|
10:1 | 59% of 26 |
| 6 | -Br |
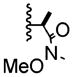
|
>20:1 | 65% of 26 |
| 7 | -Me |
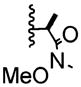
|
13:1 | 61% of 29 |
Given the results (Tables 1–3), some of the important structural features of this reaction are evident. The amide substituent R3 forces the R5 substituent into an axial orientation in the transition states preceding the major and minor products. Because of the number of contributing sp2 atoms, the boat experiences few destabilizing effects. Its dipole (μ) is smaller than the dipole for the corresponding chair transition state. Amides with substituents on the sides of the nitrogen atom favor the chairax transition state, but the yield suffers. Nevertheless, the six-step protocol that has emerged from these studies enables an efficient method for preparing enantiomerically enriched cyclohexa-2,5-dienones carrying an alkyl or bromine C2 substituent. The process begins with the three-step conversion of the benzaldehyde 17 into a differentially protected resorcinol22 (Scheme 1). This material is then coupled with the (R)-enantiomer of 30 to yield the inverted corresponding product (Scheme 2). The remaining Boc-residue is cleaved by dissolving the crude product in nitromethane (0.1 M) and adding zinc bromide (10 equiv). The phenol 31 emerges in an 84% yield after chromatography, a 61% yield over the three steps from 22. The stereointegrity of this SN2 inversion process has been confirmed by chiral HPLC analysis and comparison of 31 with a racemic standard. Oxidative dearomatization of 31 (0.2 M in CH2Cl2, −78 °C) with PhIO/TMSOTf (1.1 equiv) affords a 73% yield of (−)-29. We find this combination of reagents superior to PhI(OCOCF3)2 or PhI(OCOCH3)2. The carboxylate ligandsof the latter reagents often intercept the phenoxonuim cation and lead to appreciable amounts of side products. The major product 29 arises in a readily separable 13:1 mixture with the minor diastereomer. The relative and absolute stereochemistry of 29 and its analogs has been established by NOE experiments, X-ray analysis, and relay synthesis. The optimized six-step procedure easily provides multigram quantities of (−)-29 for application toward (+)-rishirilide B.
Scheme 2.

While Comins’s ortho lithiation process was again employed,16 an adjustment was made to the precursor of the dimethide 8 used in our earlier racemic strategy.10 In this instance, the O-benzylated benzaldehyde 32 is converted into the C-methylated product 33 (Scheme 3). The subsequent Durst and Charlton’s procedure (substrate, 0.6 M benzene, 2.5 equiv of SO2, 450 W Hg lamp, 23 °C, 4 h) provides the new sulfone 34 in 75% yield over the three steps.17 Upon warming 34 to 155 °C as compared to 110 °C used in our earlier racemic strategy,10 the cycloaddition proceeds with the nonracemic cyclohexadienone (−)-29 in both a regioselective and a diastereoselective manner. At this elevated temperature, however, the cycloadduct suffers immediate β-elimination. Subsequent oxidation of the cyclohexenone intermediate with DDQ provides the new anthracene (−)-35 in a 68% overall yield.
Scheme 3.

In our earlier racemic approach that had used glycolate derivatives (R5 = H, Figure 3), the cleavage and removal of the lactone functionality had proven straightforward.10 The 1,4-dioxan-2-one opens upon addition of an aqueous solution of metal hydroxide to give the metalated carboxylate. Subsequent addition of acid affords the spirocycle. The successive additions of aqueous lithium hydroxide and hydrochloric acid afford the β-diketone in good yields. Alternatively, prolonged exposure of the glycolate-derived lactone to aqueous hydroxide followed by the addition of acid provides the corresponding β-diketone. However, the lactate derivative (R5 = Me) proves much more robust than its glycolate counterpart. Saponification is very slow. It can be accompanied by decomposition and even epimerization of R5. Acidification of the carboxylate leads to a complex diastereomeric mixture of spirocycles. Upon addition of aqueous hydroxide, one of these adducts transforms into the 1,3-dione and the other reverts to the metal carboxylate by β-elimination.
Figure 3.

Lactate and glycolate derivatives behave differently.
Faced with poor yields of 36 (21–41%), other options for jettisoning the chiral directing group were investigated. We had found that treatment of a dimethyl hydrazone expressing an α-alkoxy substituent with a Lewis acid causes the hydrazone to expel the alkoxy fragment,18 and therefore we were curious to see if this protocol could be employed with the corresponding 1,4-dioxan-2-one. Application of Benderly’s conditions for lactone opening using the aluminum amide derived from dimethyl hydrazine causes the [1,4]-dioxan-2-one 35 to open as expected. However, in this case, the hydroxy dione 36 is also unmasked by cleavage of the chiral directing group that had fulfilled its role as a stabilizing and protecting group (69% yield, a mixture of tautomers, Scheme 4).19 We explored the generality of this cleavage reaction and found it limited to the expulsion of stabilized leaving groups.
Scheme 4.

a Other conditions for lactate cleavage: Ba(OH)2/EtOH yields 21% of 36, LiOH/THF yields 41% of 36.
Addition of diethylcarbamyl chloride (1.5 equiv) and Hünig’s base (4.0 equiv) to the dione 36 gives the O-carbamyl product 37 after 4 h (Scheme 5). None of the tautomeric product is evident. To the best of our knowledge, these are the only conditions that enable regioselective production of the thermodynamically preferred tautomer. Compound 37 proves very unstable toward chromatography. Evaporation (0.01 Torr, 1 day) supplies crude material for the subsequent series of reactions. Upon addition of an excess of lithiated ethyl vinyl ether (30 equiv, at −78 °C), we suspect that the first equivalent causes deprotonation of the tertiary alcohol. The resulting metal alkoxide then directs the addition of a subsequent equivalent to the vicinal ketone. This chelation manifold affords the corresponding anti dialkoxide. The carbonyl of the vinyl carbamate then slowly undergoes reaction with additional equivalents of the lithiated vinyl ether to afford the enolate 38. Workup with aqueous NaCl produces a single diastereomer. This intermediate enol ether is submitted in crude form to epoxidation with DMDO (3.0 equiv). Upon workup with 1 N HCl, the robust α-hydroxy ketone (−)-39 emerges in a very gratifying 87% overall yield from the β-dione 36.
Scheme 5.
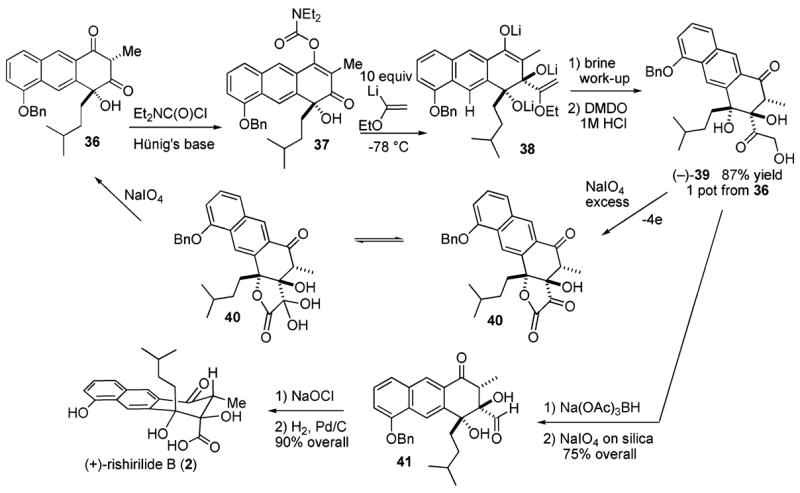
Much to our surprise given several reports employing hydroxycortisone,20 the α,α′-dihydroxy ketone 39 fails to cleave on the least substituted side with sodium periodate to yield the desired acid. Instead, these conditions afford the dione 36, which seems to indicate cleavage between the carbonyl and more hindered tertiary alcohol. However, a comprehensive investigation performed with a simple tetralone analogue reveals an unusual sequence of events.21 The primary alcohol first undergoes oxidation to yield the corresponding aldehyde. However, in this instance, a cyclization occurs to afford the corresponding γ-lactol that succumbs to a rapid oxidation to produce lactone 40, whereupon the ketone hydrates and periodate cleavage occurs between the hydrate and the tertiary alcohol (Scheme 5).
Once the origins of this unexpected problem were grasped, a solution became evident. The oxidation state of the α,α′-dihydroxy ketone in 39 was lowered so that the primary and secondary alcohols could both interact with the periodate oxidant. The α,α′-dihydroxy carbonyl reduces with a freshly prepared solution of sodium triacetoxyborohydride in acetic acid.22 The tetrol that emerges is a single diastereomer of unassigned configuration. This crude material (0.08 M in CH2-Cl2:H2O/5:1, 25 °C) undergoes cleavage with NaIO4 on silica (2.0 equiv) to cleanly afford the aldehyde (−)-41 in 75% yield (Scheme 5).23 The aldehyde 41 is then transformed into the desired carboxylic acid using Kraus’s modification of the Lindgren oxidation.24 Subsequent hydrogenolysis of the benzyl residue in 95% EtOH occurs over a catalytic amount of 5% palladium on carbon under a hydrogen atmosphere and yields (+)-rishirilide B (2) in 90% from aldehyde 41. This synthetic material proves identical in all respects with natural (+)-rishirilide B and displays a rotation of (+) 12.6° for c = 0.5.
Conclusion
In conclusion, the natural product (+)-rishirilide B (2) has been prepared for the first time. Its synthesis requires 15 steps from benzaldehyde 7 and 13 steps from aldehyde 32 and proceeds in overall yields of 12.5% and 20.3%, respectively. The synthesis requires seven chromatographies. It demonstrates the powerful combination of a novel method for the enantioselective dearomatization of cyclohexadienones with a process for the synthesis of differentially protected resorcinols for accessing highly functionalized chiral building blocks. In addition, an unusual process for the cleavage of α-alkoxy esters and [1,4]-dioxan-2-ones is presented. In the near future, we hope to report syntheses of several other natural products using this combination of procedures.
Acknowledgments
Research grants from the National Science Foundation (Career-0135031) for support of the o-quinone methide chemistry and the National Institutes of Health (GM-64831) for support of the cyclohexadienone chemistry are greatly appreciated.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for compounds 19–36, 39, 41, and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Komagata D, Sawa R, Kinoshita N, Imada C, Saw T, Naganawa H, Hamada M, Okami Y, Takeuchi T. J Antibiot. 1984;37:1681. doi: 10.7164/antibiotics.45.1681. [DOI] [PubMed] [Google Scholar]
- 2.Iwaki H, Nakaya Y, Takahashi M, Uetsuki S, Kido M, Fukuyama Y. J Antibiot. 1992;45:1091. doi: 10.7164/antibiotics.37.1091. [DOI] [PubMed] [Google Scholar]
- 3.Allen JG, Danishefsky SJ. J Am Chem Soc. 2001;123:351. doi: 10.1021/ja003272a. [DOI] [PubMed] [Google Scholar]
- 4.Hauser FM, Xu YJ. Org Lett. 1999;1:335. doi: 10.1021/ol9906561. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto K, Hertemann MF, Allen JG, Danishefsky SJ. Chem-Eur J. 2003;9:3242. doi: 10.1002/chem.200304931. [DOI] [PubMed] [Google Scholar]
- 6.Magdziak D, Meek S, Pettus TRR. Chem Rev. 2004;104:1383. doi: 10.1021/cr0306900. [DOI] [PubMed] [Google Scholar]
- 7.Van De Water RW, Hoarau C, Pettus TRR. Tetrahedron Lett. 2003;44:5109. [Google Scholar]
- 8.Wipf P, Kim Y, Jahn H. Synthesis. 1995:1549. [Google Scholar]
- 9.Pettus LH, Van de Water RW, Pettus TRR. Org Lett. 2001;3:905. doi: 10.1021/ol0155438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Wang JH, Pettus TRR. Tetrahedron Lett. 2004;45:5895. [Google Scholar]; (b) Wang JH, Pettus TRR. Tetrahedron Lett. 2004;45:1793. [Google Scholar]
- 11.Stork G, Danheiser R, Ganem B. J Am Chem Soc. 1973;95:3414. [Google Scholar]
- 12.Mejorado L, Hoarau C, Pettus TRR. Org Lett. 2004;6:1535–1538. doi: 10.1021/ol0498592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLoughlin BJ. J Chem Soc Chem Commun. 1969:540. [Google Scholar]
- 14.(a) Van De Water R, Magdziak D, Chau J, Pettus TRR. J Am Chem Soc. 2000;122:6502. [Google Scholar]; (b) Jones RW, Van De Water R, Lindsey CL, Hoarau C, Ung T, Pettus TRR. J Org Chem. 2001;122:3435. doi: 10.1021/jo001752e. [DOI] [PubMed] [Google Scholar]
- 15.Paterson I, Wallace DJ, Cowden CJ. Synthesis. 1998:639. [Google Scholar]
- 16.Comins DL, Brown JD. J Org Chem. 1989;54:3730. [Google Scholar]
- 17.Durst T, Kozma EC, Charlton JL. J Org Chem. 1985;50:4829. [Google Scholar]
- 18.Hoarau C, Pettus TRR. Org Lett. 2006;8:2843–2846. doi: 10.1021/ol061000s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benderly A, Stavchansky S. Tetrahedron Lett. 1988;29:739–740. [Google Scholar]
- 20.Kertesz DJ, Marx MJ. J Org Chem. 1986;51:2315. [Google Scholar]
- 21.Mejorado L, Pettus TRR. Synthesis. 2006:3209. doi: 10.1055/s-2006-950188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans DA, Chapman KT. Tetrahedron Lett. 1986;27:5939. [Google Scholar]
- 23.(a) Daumas M, Vo-Quang Y, Vo-Quang L, Le Goffic F. Synthesis. 1989:64–65. [Google Scholar]; (b) Zhong YL, Shing TKM. J Org Chem. 1997;62:2622. doi: 10.1021/jo9621581. [DOI] [PubMed] [Google Scholar]
- 24.Kraus GA, Taschner MJ. J Org Chem. 1980;45:1175. [Google Scholar]