

Abstract
Epilepsy is characterized by both neuronal and astroglial dysfunction. The endogenous anticonvulsant adenosine, the level of which is largely controlled by astrocytes, might provide a critical link between astrocyte and neuron dysfunction in epilepsy. Here we studied astrogliosis, a hallmark of the epileptic brain, adenosine dysfunction and the emergence of spontaneous seizures in a comprehensive approach including a new mouse model of focal epileptogenesis, mutant mice with altered brain levels of adenosine, and mice lacking adenosine A1 receptors. In wild type mice, following a focal epileptogenesis-precipitating injury, astrogliosis, upregulation of the adenosine-removing astrocytic enzyme adenosine kinase (ADK), and spontaneous seizures all coincided in a spatio-temporally restricted manner. Importantly, these spontaneous seizures were mimicked in untreated transgenic mice overexpressing ADK in brain or those lacking A1 receptors. Conversely, mice with reduced forebrain ADK did not develop astrogliosis or spontaneous seizures. Our studies define ADK as critical upstream regulator of A1 receptor mediated modulation of neuronal excitability and support the ADK hypothesis of epileptogenesis implicating that upregulation of ADK during astrogliosis provides a critical link between astrocyte and neuron dysfunction in epilepsy. These findings define ADK as rational target for therapeutic intervention.
Keywords: Epileptogenesis, kainic acid, status epilepticus, A1 receptor, astrocyte
Introduction
Despite the development of new antiepileptic drugs, pharmacotherapy of epilepsy remains unsatisfactory (Vajda, 2007) and up to 30% of patients with epilepsy do not enter remission with treatment (Sisodiya, 2007). Adenosine has been identified as an endogenous anticonvulsant of the brain more than 20 years ago (Dunwiddie, 1980; Lee et al., 1984; Dragunow et al., 1985). Its levels rise rapidly during seizures and are thought to be responsible for seizure termination and postictal refractoriness (Dragunow, 1991; During and Spencer, 1992). Indeed, augmentation of adenosine using brain implants of adenosine-releasing cells is an effective strategy to suppress seizures (Boison, 2007a). Adenosine exerts a wide range of effects within the brain by activation of the G protein coupled adenosine A1, A2A, A2B, and A3 receptors (ARs) (Fredholm et al., 2001; Fredholm et al., 2005a; Fredholm et al., 2005b; Boison, 2007b;, 2008a). Under physiological conditions adenosine modulates neuronal excitability largely via activation of A1 and A2ARs, due to their high affinity for adenosine and their high abundance within the brain; these receptors display reciprocal activity and distribution and thus are uniquely suited to integrate and fine-tune neuromodulation (Sebastiao and Ribeiro, 2000; Boison, 2007b). A1Rs, coupled to inhibitory G proteins, display high abundance in seizure-susceptible brain regions such as hippocampus. Consequently, A1R agonists are powerful anticonvulsants (Dunwiddie, 1999), effective even in pharmacoresistant epilepsy (Gouder et al., 2003), however associated with prominent cardiovascular and sedative side effects (Dunwiddie and Worth, 1982; Dunwiddie and Masino, 2001; Güttinger et al., 2005). As demonstrated in A1R knockout mice, activation of the receptor is necessary to prevent the spread of seizures and seizure-associated cell death; following either kainic acid (Fedele et al., 2006) or traumatic brain injury (Kochanek et al., 2006) induced status epilepticus (SE) all A1R knockout animals died within 5 days.
Extracellular, and thus synaptic, adenosine appears to be largely under the control of an astrocyte-based adenosine cycle (Boison, 2008a) that involves regulated release of ATP (Pascual et al., 2005), extracellular degradation of ATP to adenosine (Zimmermann, 2000), equilibration of extra- and intracellular pools of adenosine via nucleoside transporters (Baldwin et al., 2004; Gray et al., 2004), and intracellular phosphorylation of adenosine to AMP via adenosine kinase (ADK) (Boison, 2006). ADK functions largely as a metabolic reuptake system for adenosine, driving the influx of adenosine into the cell (Boison, 2006). It is important to note that in adult brain ADK expression is largely confined to astrocytes (Studer et al., 2006). Due to the existence of an active substrate cycle between adenosine and AMP involving ADK and 5′-nucleotidase, small changes in ADK activity are sufficient to translate into rapid and major changes in ambient adenosine (Arch and Newsholme, 1978; Bontemps et al., 1983). Thus, genetic disruption or RNAi mediated downregulation of ADK is an efficient means to induce cellular adenosine release (Fedele et al., 2004; Ren et al., 2007) and rapid downregulation of ADK following stroke has been proposed to be an endogenous neuroprotective mechanism of the brain (Pignataro et al., 2008).
Due to the preeminent role of astrocytes in regulating the ambient “tone” of adenosine, any pathological disturbance in astrocyte homeostasis is expected to translate into adenosine dysfunction. This is of particular importance since astrogliosis, a homotypic chronic response of the brain to injury, is a pathological hallmark of the epileptic brain (Rothstein et al., 1996; Duffy and MacVicar, 1999; Tashiro et al., 2002). The involvement of astrocytes in the pathogenesis of epileptogenesis is based on findings that astrocytes control synaptic transmission by the vesicular release of glutamate, ATP, and by the release of the NMDA receptor co-agonist D-serine (Haydon and Carmignoto, 2006). Recently, it was demonstrated that prolonged episodes of neuronal depolarization evoked by astrocytic release of glutamate contributed to epileptiform discharges (Tian et al., 2005). Thus, astrocyte dysfunction appears to be a major contributing factor to epileptogenesis.
Objective
Experiments were designed to address the “adenosine kinase hypothesis of epileptogenesis” which entails that astrogliosis, a homotypic response of the brain to injury, involves upregulation of the adenosine-removing enzyme adenosine kinase (ADK). This in turn causes adenosine deficiency, increased neuronal excitability and seizure generation. This hypothesis (Boison, 2008b) is based on previous work correlating astrogliosis and upregulation of ADK with the emergence of spontaneous seizures (Gouder et al., 2004; Fedele et al., 2005; Li et al., 2008). Here we expand these previous studies by providing a comprehensive analysis of the spatio-temporal requirements of ADK dysregulation in a new mouse model of focal CA3-selective epileptogenesis (Li et al., 2008). To provide mechanistic insight of adenosine dysfunction in epileptogenesis, we will study astrogliosis and seizure generation in transgenic mice (1) with overexpressed ADK in brain (having an astrogliosis-independent adenosine deficiency), (2) with reduced levels of forebrain ADK (having increased levels of forebrain adenosine), and (3) lacking A1 receptors to assess the role of this downstream effector of an altered brain adenosine tone.
Methods
Animals
All animal procedures were performed in an AAALAC-accredited facility in accordance with protocols approved by the Legacy IACUC and the principles outlined in the NIH Guide for the Care and Use of Laboratory Animals. Animals were kept in a 12-hour light/dark cycle with lights on from 7 am to 7 pm in individually ventilated isolator cages and food and water were supplied ad libitum. Four different genotypes of mice were used in this study, all maintained on a C57BL/6 background: (1) wild type; (2) homozygous Adk-tg mice with brain wide overexpression of a conditional Adk-transgene, but lack of the endogenous Adk gene; these animals are characterized by high ADK expression in CA3 neurons and have been characterized previously (Fedele et al., 2005; Pignataro et al., 2007; Wu and Boison, 2007; Yee et al., 2007; Li et al., 2008); (3) fb-Adk-def mice with reduced levels of forebrain ADK were created based on Emx1-Cre driven recombination of the loxP-flanked Adk-transgene in Adk-tg mice (Li et al., 2008); (4) homozygous and heterozygous A1 receptor knockout mice (Johansson et al., 2001; Fedele et al., 2006).
Epileptogenesis Model
Adult male mice weighing 25-30g underwent seizures induced by unilateral stereotaxic microinjection of KA into the basolateral amygdaloidal nucleus based on stereotactic coordinates relative to bregma: AP = -0.94 mm, L = -2.85 mm, V = -3.75 mm as described (Li et al., 2008). Briefly, under anesthesia with 68.5% N2O, 30% O2, and 1.5% isoflurane, mice were affixed with a 26-gauge steel guide cannula over the intact dura using dental cement, two cortical recording electrodes and a bipolar, coated, stainless steel electrode (0.2 mm in diameter, Plastics One, Roanoke, VA), which was implanted into the ipsilateral CA3 region and fixed with a pedestal of dental acrylate; this setup was used for chronic video-EEG monitoring (over up to three weeks, Fig. 2). Alternatively – for acute EEG recordings – the CA3 electrodes were implanted 3 weeks after the injection of KA into wild type mice or were implanted into untreated Adk-tg, A1R-/-, or A1R+/- mice (Fig. 1, Fig. 3). Coordinates for the CA3 electrodes were (toothbar at 0): 2.18 mm caudal to bregma, 2.5 mm lateral to midline, and 2.25 mm ventral to dura. Additional animals received CA1 or cortical recording electrodes. Anesthesia was discontinued, EEG recordings commenced, and then a 31-gauge internal cannula was inserted into the lumen of the guide to inject 0.3 μg KA in a volume of 0.2 μl PBS, pH 7.4, into the amygdala. The EEG was monitored for 30 min using a Nervus video-EEG recording device until lorazepam (6 mg/kg) was administered intravenously to terminate seizures. The EEG was further monitored for up to 30 min to ensure seizure cessation. An observer unaware of the experimental treatment performed quantification of EEG records. Selected animals were injected once 30 min prior to intraamygdaloid KA-injection with 1 mg/kg i.p. 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, A1R antagonist, Sigma). 24h after KA injection, animals with chronic CA3 electrodes were subjected to three weeks of continuous EEG-monitoring.
Figure 2. Temporal coincidence of ADK overexpression and development of spontaneous seizures.
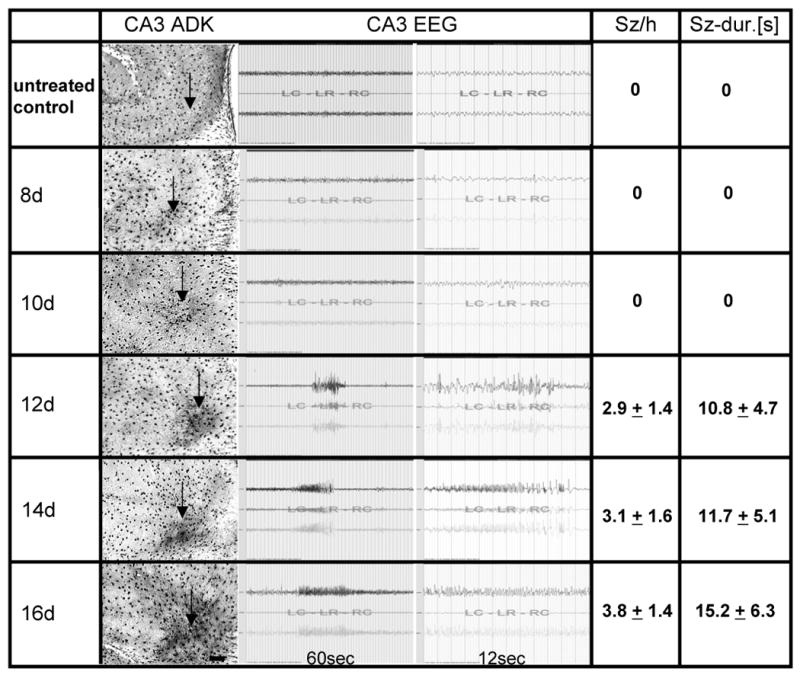
Ten wild type mice received intraamygdaloid injections of KA and received CA3 recording electrodes at the day of KA-injection. Five wild type control animals not injected with KA were included as control (top row: “untreated control”). All animals were EEG-monitored daily to detect the first onset of spontaneous seizures. Two animals each were processed for ADK immunohistochemistry at 8, 10, 12, 14, and 16 days (8d – 16d) after KA-injection. The column “CA3 ADK” shows representative brain sections (electrode insertion level not shown, but approximated by arrows) with ADK immunoreactivity visualized with DAB. Note the first emergence of significant ADK upregulation at day 12; scale bar 100 μm. Representative CA3 EEG traces are shown, demonstrating lack of seizures at day 8 and 10, but emergence of spontaneous CA3 seizures from day 12 onwards. Quantitative data are given as average number of seizures per hour (Sz/h) and average duration of each seizure in seconds (Sz-dur.[s]). Seizure data are based on n = 10 for day 8, n = 8 for day 10, n = 6 for day 12, n = 4 for day 14, and n = 2 for day 16.
Figure 1. Characterization of spontaneously epileptic mice 3 weeks after intraamygdaloid injection of kainic acid.
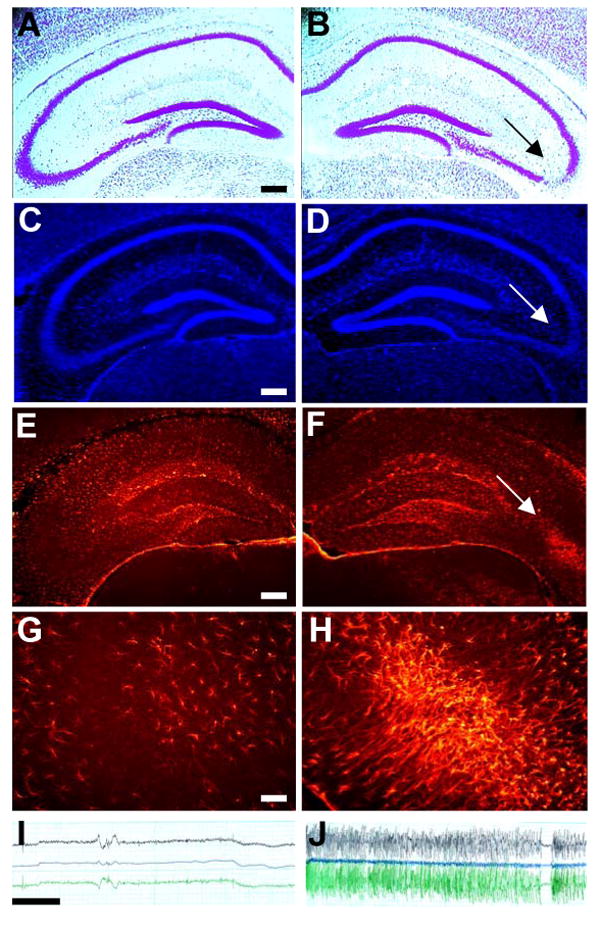
(A, B) Representative cresyl violet stained coronal brain section showing the hippocampal formation contralateral (A) or ipsilateral (B) to the KA-injection. Note the CA3-selective cell loss in the ipsilateral hippocampus (B, arrow). (C, D) Adjacent coronal brain section from the same animal stained with the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI) showing an increase in the number of cell nuclei in the injured CA3 (arrow). (E-H) The same section stained for ADK immunofluorescence (red) contralateral (E, G) or ipsilateral (F, H) to the KA-injection. Note the focal overexpression of ADK in the ipsilateral CA3 (F arrow, H). (I, J) Representative EEG recordings obtained from electrodes inserted into the contralateral (I) or ipsilateral CA3 (J). Scale bars: A-F: 300 μm; G, H: 37.5 μm; EEG: 10 sec; top lead: one tip of intra-CA3 bipolar electrode versus cortical electrode; central lead: two tips of bipolar CA3-electrode; bottom lead: “other” tip of bipolar CA3-electrode versus cortical electrode.
Figure 3. Compilation of ADK expression and seizure data from mice with induced or engineered alterations in the adenosine system.
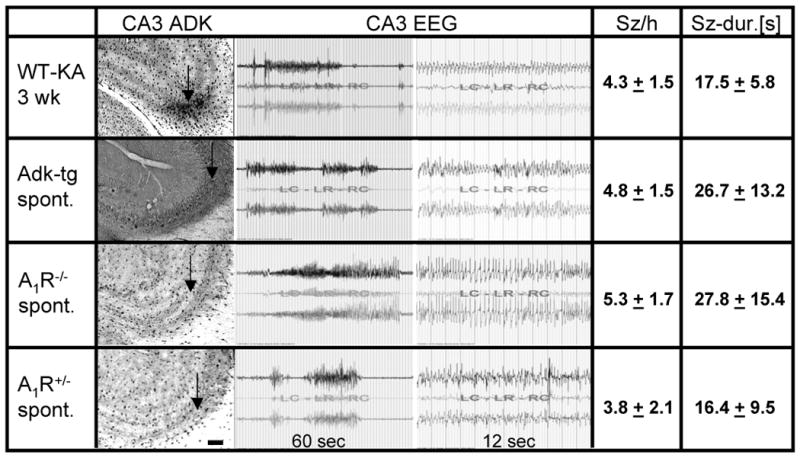
Wild type mice 3 weeks after intraamygdaloid KA-injection (WT-KA 3wk), untreated Adk-tg mice (Adk-tg spont.), untreated homozygous A1R knockout mice (A1R-/- spont.), and untreated heterozygous A1R knockout mice (A1R+/- spont.) were implanted with CA3 recording electrodes and subjected to 8 hours of continuous EEG monitoring (n = 5 animals, each). Subsequently, animals were processed for ADK immunohistochemistry. The column “CA3 ADK” shows representative brain sections (electrode insertion level not shown, but approximated by arrows) with ADK immunoreactivity visualized with DAB. Note the normal astrocytic expression pattern of ADK in A1R-/- spont. and A1R+/- spont. mice, but elevated levels of ADK in the CA3 of WT-KA 3wk and Adk-tg spont. mice; scale bar 100 μm. Representative CA3 EEG traces are shown, demonstrating similar seizure patterns in all mice. Quantitative data are given as average number of seizures per hour (Sz/h) and average duration of each seizure in seconds (Sz-dur.[s]).
EEG monitoring
Mice were placed in Plexiglas cages where they can move freely (one animal per cage). Electrical brain activity was monitored using a Nervus EEG Recording System connected with a Nervus magnus 32/8 Amplifier, and filtered (high-pass filter 0.3 Hz cutoff, low-pass 100 Hz). Each EEG file was analyzed individually by browsing through the EEG recording on the computer screen (30 mm/s chart speed). EEG-seizure activity was defined as high-amplitude rhythmic discharges that clearly represented a new pattern of tracing (repetitive spikes, spike-and-wave discharges, and slow waves) that lasted at least 5 s. Epileptic events occurring with an interval less than 5 s without the EEG returning to baseline were defined as belonging to the same seizure. Electrographic seizures were quantified as number of seizures per hour and according to the average duration of each seizure. Data were averaged across total recording time from all animals from each experimental group.
Histology
Brains obtained 24h following the KA injection were immediately frozen in 2-methylbutane (-30°C) and sectioned at 12 μm on a cryostat. Coronal sections at the level of bregma −1.7 mm were air dried, postfixed in 10% formalin (15 min), washed twice in PBS, and then processed for histopathology (cresyl violet staining) or for detection of DNA fragmentation (TUNEL) as described (Shinoda et al., 2004). The average number of TUNEL positive cells per brain slice was quantified by counting TUNEL positive cells from 3 adjacent brain sections, encompassing the entire CA1/3 field, derived from 3 animals from each genotype.
At different time points (day 8, 10, 12, 14, 16, 21) following KA-injection selected mice (n = 2 per time point) were transcardially perfused with 4% paraformaldehyde in phosphate buffer (0.15M, pH 7.4). The brains were post fixed in the same fixative at 4°C for 6 h and cryoprotected in 10% DMSO in PBS (v/v) before being cut into 40 μm coronal sections. 6 sections from each brain representing different levels of the hippocampal formation encompassing the complete rostro-caudal extent of the hippocampus were mounted onto gelatin-coated slides and stained with Cresyl violet. For the immunohistochemical detection of GFAP and ADK we followed our previously published procedures and stained sections adjacent to the Cresyl violet stained sections (Studer et al., 2006). ADK expression was quantified by analyzing fields of 200 × 300 μm encompassing the entire CA3a region. Corresponding fields from two sections from each animal were analyzed by scanning ADK immunofluorescence on DAB stained slices using a Kodak imaging device. For data analysis only those sections were chosen that were not affected by electrode insertion. Given a rostro-caudal extent of the CA3 of around 1 mm and an electrode diameter of 0.2 mm, 7 out of 10 brain sections were not affected by electrode insertion. Data were analyzed by ANOVA with Student-Newman-Keuls test.
Results
Spatial colocalization of upregulated ADK and spontaneous seizures in epileptic wild type mice
Non-convulsive electrographic status epilepticus (SE) was triggered in six adult male C57BL/6 mice by intraamygdaloid injection of kainic acid (KA) as described previously (Li et al., 2008). Three weeks after KA-injection bipolar electrodes were implanted into the ipsilateral CA3 region. Additional KA-injected animals were equipped with electrodes placed into the ipsilateral CA1, into the contralateral CA3 or onto the cortex. Although no seizure activity, based on a total recording time of 48 hours, was ever detected from ipsilateral CA1, contralateral CA3 (Fig. 1I) or from cortical electrodes, spontaneous focal seizures became evident from recordings of the ipsilateral CA3 at a frequency of 4.3 ± 1.5 seizures per hour with an average duration of 17.5 ± 5.8 seconds (based on 48 hours total recording time). These findings indicated focal epileptogenesis restricted to the ipsilateral CA3 region. CA3-selective spontaneous seizures were observed in every KA-injected animal and were never associated with convulsions.
Three weeks after KA-injection all animals were sacrificed for histological analysis. A Cresyl violet staining revealed CA3-selective neuronal cell loss in the ipsilateral hippocampus (Fig. 1B), whereas the contralateral hippocampus (Fig. 1A) was spared from injury. A nuclear stain (DAPI) (Fig. 1C,D) demonstrated the replacement of lost neuronal cell bodies with astrocytes as determined previously (Li et al., 2008). Most importantly, the CA3-selective injury (Fig. 1B) colocalized with increased expression of the astrocyte marker (Studer et al., 2006; Li et al., 2008) adenosine kinase (ADK) (Fig. 1F,H) demonstrating focal astrogliosis and upregulation of ADK. In contrast, the contralateral hippocampus was not affected by increased expression of ADK. These findings demonstrate spatial colocalization of CA3-selective neuronal cell loss (acute brain injury), astrogliosis (replacement of lost neurons by astrocytes), upregulation of ADK (reduction of the local adenosine “tone”), and focal spontaneous electrographic seizures. Restriction of focal seizures to the injured CA3 indicates that seizures might be due to astrogliosis and/or increased expression of ADK.
Temporal colocalization of upregulated ADK and development of spontaneous seizures in wild type mice
If astrogliotic overexpression of ADK is a cause for seizures, then – during epileptogenesis – upregulation of ADK should precede the first onset of spontaneous seizures. A separate set of animals (n = 10) received CA3 electrodes at the day of KA-injection and was compared to untreated wild type mice equipped with CA3 electrodes (n = 5). All animals were recorded daily to detect the first onset of spontaneous seizures. Two animals each were sacrificed at 8, 10, 12, 14, and 16 days following KA-injection for histological analysis. Electrographic seizures emerged first 12 days after KA-injection at a frequency of 2.9 ± 1.4 seizures per hour with an average duration of 10.8 ± 4.7 seconds (n = 6) (Fig. 2) and intensified with time with 3.1 ± 1.6 seizures per hour at 11.7 ± 5.1 seconds per seizure at day 14 (n = 4), and 3.8 ± 1.4 seizures per hour at 15.2 ± 6.3 seconds per seizure at day 16 (n = 2). In agreement with the gradual development of spontaneous CA3-selective seizures, histological analysis of brain sections revealed a parallel progressive upregulation of ADK. ADK-specific immuno-densities of scanned CA3 fields from diaminobenzidine (DAB) stained brain sections taken at days 8, 10, 12, 14, 16, and 21 (Fig. 2 & 3) after KA injection were normalized to ADK-specific immuno-densities in the CA3 of untreated wild type animals and analyzed by ANOVA. Compared to untreated wild type samples, ADK immuno densities were increased by 7.3 ± 7.5% at day 8 (p > 0.05), 10.3 ± 7.8% at day 10 (p < 0.05), 15.2 ± 6.3% at day 12 (p < 0.01), 20.0 ± 9.2% at day 14 (p < 0.01), 28.8 ± 10.9% at day 16 (p < 0.01), and 31.1 ± 12.3% at day 21 (p < 0.01). Most notably, seizures were not detected at earlier time points (day 8, 10) with moderate upregulation of ADK (Fig. 2). Remarkably, ADK immuno-densities at days 16 and 21 were similar (p > 0.05), but increased compared to those quantified at days 8 through 14 (p < 0.01) (Fig. 2 & 3). Likewise, seizure parameters at day 16 (3.8 ± 1.4 seizures per hour with an average duration of 15.2 ± 6.3 seconds) approached those observed 21 days after KA injection (Fig. 3).
In conclusion, the data presented here demonstrate that upregulation of ADK precedes the onset of epilepsy in mice indicating that upregulated ADK cannot be a consequence of seizures.
Spontaneous seizures in ADK overexpressing transgenic mice
Our data obtained in wild type mice (Fig. 1 & 2) suggest that either focal astrogliosis and/or focal upregulation of ADK are sufficient to cause seizures. To discriminate between astrogliosis and ADK dependent effects and to provide a mechanistic explanation for seizure generation we generated a transgenic mouse line (Adk-tg mice) overexpressing an Adk-transgene in astrocytes and neurons within an Adk-knockout background (Fedele et al., 2005; Pignataro et al., 2007; Li et al., 2008). These animals are characterized by a reduced tone of endogenous adenosine (Fedele et al., 2005) and display increased susceptibility to seizures and to seizure- and stroke-induced cell death (Pignataro et al., 2007; Li et al., 2008). Most importantly, Adk-tg mice display an ectopic overexpression of ADK within the hippocampal CA3 region (Fig. 3). If diffuse overexpression of ADK in CA3 is sufficient to generate spontaneous seizures in the absence of astrogliosis, then Adk-tg mice should have spontaneous seizures in CA3. To investigate the possibility for spontaneous seizures in Adk-tg mice, 6 naïve animals were equipped with bipolar recording electrodes implanted into the CA3 region. In parallel, untreated homozygous and heterozygous A1 receptor knockout mice (Johansson et al., 2001), as well as KA injected wild type mice 3 weeks after SE received intra-CA3 recording electrodes (n = 5 per group). One day after electrode implantation each animal was subjected to 8 hours of continuous EEG recordings. Strikingly, Adk-tg mice displayed spontaneous non-convulsive CA3 seizures at a frequency (4.8 ± 1.5 seizures per hour) and duration (26.7 ± 13.2 seconds) similar (p > 0.05) to CA3 seizures recorded from the astrogliotic region of KA-injected wild type mice (4.3 ± 1.5 seizures per hour, at 17.5 ± 5.8 seconds) (Fig. 3). These results suggest that overexpression of ADK in the absence of astrogliosis or any other epileptogenetic process (untreated Adk-tg mice) is sufficient for seizure generation. We have previously demonstrated that A1 receptor activation by endogenous adenosine is needed to prevent spread of seizure activity (Fedele et al., 2006). Thus, we hypothesized that seizure activity in Adk-tg mice (reduced adenosine tone) and in A1 receptor knockout mice should be essentially similar. Indeed, untreated homozygous A1R knockout mice displayed spontaneous CA3 seizures at a similar frequency and duration (p > 0.05) (5.3 ± 1.7 seizures per hour at 27.8 ± 15.4 seconds) compared to Adk-tg mice, indicating that the adenosine/ADK system acts as an upstream regulator of A1R mediated seizure control. Even heterozygous A1 receptor knockout mice, which have 50% of normal A1Rs display spontaneous seizures, however at a reduced frequency and duration (3.8 ± 2.1 seizures per hour at 16.4 ± 9.5 seconds) indicating that a reduced tonic activation of A1R by endogenous adenosine (corresponding to moderate overexpression of ADK) might be sufficient to induce spontaneous seizures. It is important to note that the inborn spontaneous electrographic seizures in A1R-/- and A1R+/- mice do not trigger astrogliosis or upregulation of ADK, nor do they induce neuronal cell loss (Fedele et al., 2006). These non-injurious seizures are in contrast to KA-induced injurious seizures that cause neuronal cell loss and subsequent astrogliosis with upregulation of ADK. In summary, our experiments demonstrate that overexpression of ADK, but not astrogliosis is responsible for the induction of spontaneous seizures, likely by reduced activation of A1Rs.
Adk-tg and A1R knockout mice display increased KA-induced acute brain injury
To investigate the influence of different levels of ADK on the development of acute seizure-induced hippocampal injury, additional Adk-tg, A1R knockout, and wild type mice received intraamygdaloid injections of KA (n = 6 per group). 24 h after KA injection all animals were analyzed histologically by Cresyl violet and TUNEL staining (Fig. 4). KA injected wt mice developed a well-defined lesion in the ipsilateral CA3 of the hippocampus, which was characterized by prominent TUNEL-positive cells (73.8 ± 17.1 TUNEL positive cells per brain section as averaged across 3 brain sections from 3 animals, each), being indicative of ongoing cell death (Fig. 4B,C). Neither the contralateral hippocampus nor any other brain region was affected by KA-induced seizures (Fig. 4A). In contrast, ADK overexpression in Adk-tg mice led to a much larger cell loss in the ipsilateral CA3 region (117.6 ± 31.1 TUNEL positive cells, p < 0.05) (Fig. 4E,F), to an extension of cell loss to the ipsilateral CA1 region (38.6 ± 4.0 TUNEL positive cells, p < 0.001) (Fig. 4H) and dentate gyrus (Fig. 4H), and, most remarkably, to significant cell loss in the contralateral hippocampus (28.3 ± 10.4 TUNEL positive cells in the contralateral CA3 and 25.0 ± 5.6 TUNEL positive cells in the contralateral CA1, p < 0.01, each) (Fig. 4D,G,I). As expected, A1R-KO mice displayed a similar pattern of cell loss (129.0 ± 33.0 TUNEL positive cells in the ipsilateral CA1, p < 0.05; 315.6 ± 62.7 TUNEL positive cells in the ipsilateral CA1, p < 0.001; 40.3 ± 23.5 TUNEL positive cells in the contralateral CA3, p < 0.05; 272.0 ± 39.9 TUNEL positive cells in the contralateral CA1, p < 0.001, compared to wt by t-test), but of further increased magnitude, particularly in the contralateral CA1 region (Fig. 4 J-O). All KA-injected Adk-tg and A1R knockout mice died within the first three days after KA-injection and displayed excessive SE (Li et al., 2008). Again, Adk-tg and A1R knockout mice displayed striking similarity with regard to increased vulnerability to acute brain injury, consistent with earlier reports of lethal status epilepticus in A1R knockout mice induced by intrahippocampal kainate (Fedele et al., 2006) or traumatic brain injury (Kochanek et al., 2006).
Figure 4. Epilepsy associated cell loss.
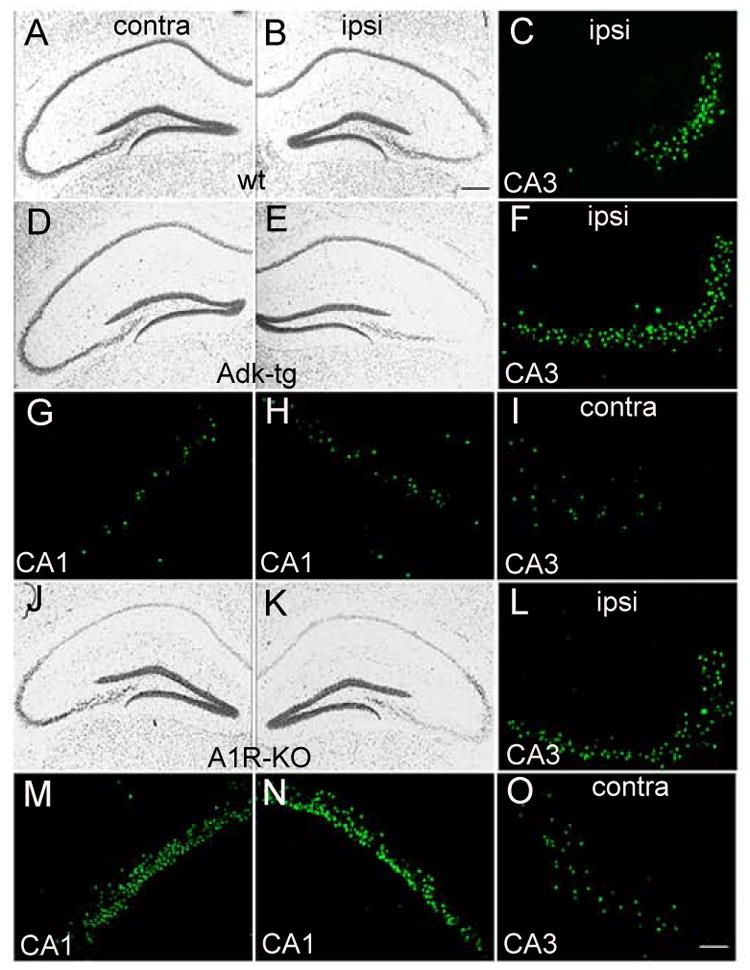
Representative micrographs of the hippocampal formation from coronal brain sections taken from wild type mice (wt) (A-C), Adk-tg mice (D-I), and A1R-KO mice (J-O) 24 h after intraamygdaloid injection of 0.3 μg KA. Sections were stained either with cresyl violet or with TUNEL (green). (A-C) Typical apoptotic cell death in the CA3 region of the hippocampus of wt mice ipsilateral (ipsi) to the KA-injected amygdala. (D-I) Adk-tg mice show aggravated cell loss in the ipsilateral CA3 region (F), and novel cell loss in the ipsilateral CA1 region (H), and the contralateral CA1 (G) and CA3 (I) regions. (J-O) A1R-KO mice show aggravated cell loss in the ipsilateral CA3 region (L), and novel cell loss in the ipsilateral CA1 region (N), and the contralateral CA1 (M) and CA3 (O) regions. Scale bars: A,B,D,E,G,H,J,K,M,N: 100 μm; C,F,I,L,O: 25 μm.
Fb-Adk-def mice are protected from seizure development
The experiments described above suggest the following: (i) increased ADK triggers seizures by reduction of the level of ambient adenosine, thereby reducing the tonic activation of A1Rs; (ii) activation of the A1R prevents spread of seizures and lethality. To investigate the net effect of increased adenosine on the brains' susceptibility to seizures we generated transgenic mice with a forebrain selective reduction of ADK to 62% of normal levels (fb-Adk-def mice) (Li et al., 2008). This was achieved by breeding Adk-tg mice with Emx1-tg3-Cre mice (Iwasato et al., 2004) leading to a forebrain selective recombination of the loxP-flanked Adk-transgene within the context of an Adk(endogenous) null background (Li et al., 2008). It is important to note that in these animals ADK is completely lacking in both astrocytes and neurons, with the exception of ADK remaining present in GABAergic interneurons (Li et al., 2008). As demonstrated pharmacologically (Li et al., 2008) these animals are characterized by reduced levels of forebrain adenosine.
In the first set of experiments 10 fb-Adk-def mice were subjected to intraamygdaloid injection of KA. None of the animals developed SE and type IV seizure activity. 24 hours after KA injection five of the animals were sacrificed for histological analysis. Nissl, as well as TUNEL stained brain sections demonstrated a complete lack of acute brain injury. Indeed, not a single TUNEL positive cell was found (Fig. 5A). These findings demonstrate strong anticonvulsant and neuroprotective functions as a consequence of reduced forebrain ADK. The remaining five animals received CA3 recording electrodes and were subjected to histological analysis 3 weeks after KA-injection. As expected, due to the lack of acute brain injury, these animals neither developed spontaneous seizures, nor did they show any signs of astrogliosis or upregulation of ADK (Fig. 5A).
Figure 5. Assessment of epileptogenesis 3 weeks after intraamygdaloid KA.
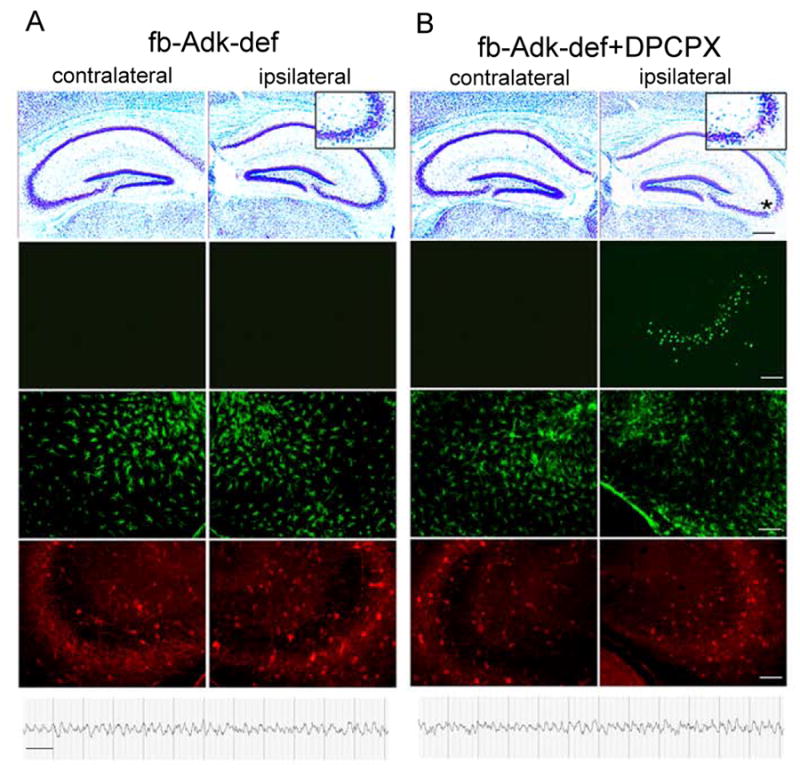
Representative micrographs and EEGs taken from the hippocampal formation of fb-Adk-def mice (A), and fb-Adk-def mice pretreated with 1mg/kg i.p. DPCPX (B), 3 weeks after intraamygdaloid injection KA. 1st row: Cresyl violet stained section showing the characteristic ipsilateral SE induced CA3 lesion in fb-Adk-def/DPCPX mice (B, asterisk), but not in fb-Adk-def mice (A). Insets show enlarged ipsilateral CA3. 2nd row: TUNEL staining showing ipsilateral CA3-selective cell loss only in fb-Adk-def/DPCPX mice. 3rd row: GFAP immunofluorescence staining showing lack of astrogliosis in fb-Adk-def and fb-Adk-def/DPCPX mice. 4th row: ADK immunofluorescence staining of the same specimen. 5th row: Representative intrahippocampal EEG recordings taken from CA3 recordings showing protection from the development of spontaneous seizures in fb-Adk-def and fb-Adk-def/DPCPX mice. Scales: black bar: 300 μm; white bar: 75 μm; EEG: 10 sec.
In the second experiment, performed with additional fb-Adk-def mice (n = 10), we paired KA injection with DPCPX (1 mg/kg, i.p. 30 min before KA) to remove the A1R-mediated acute seizure- and neuroprotection. As a result of intraamygdaloid KA paired with DPCPX all animals developed SE, which was terminated after 30 min with lorazepam. The duration of cumulative type IV seizures (= high-voltage, polyspike paroxysmal discharges of > 1Hz frequency) was 318.0 ± 100.6 sec and was similar (p > 0.05) in intensity and duration to type IV seizures recorded in KA-treated wild type mice (443.3 ± 147.2). Thus, DPCPX paired with KA restored wild type-like SE in fb-Adk-def mice. This experiment also demonstrates an increased tone of ambient adenosine in the forebrain of fb-Adk-def mice, preventing the development of SE via activation of A1Rs. 24 hours after KA-injection five animals were subjected to histological analysis. All showed a prominent CA3 injury with 51.8 ± 17.7 TUNEL positive cells per brain section (Fig. 5B), with the number of TUNEL positive cells similar (p > 0.05) to those counted in respective wild type controls (73.8 ± 17.1 cells per section). Again, DPCPX in KA-injected fb-Adk-def mice restored wild type-like brain injury, which, in wild type mice is considered to be a sufficient acute epileptogenesis-triggering injury.
To demonstrate that DPCPX (1 mg/kg, i.p.) blocks the effects of increased adenosine (as in forebrains of fb-Adk-def mice) rather than interfering with normal adenosine levels in wild type mice, a group of wild type mice (n = 6) was injected with DPCPX 30 min prior to the injection of KA. Pairing of DPCPX with KA in wild type mice did not influence acute seizure parameters. In concordance with “normal” acute seizure patterns in these animals, counts of TUNEL positive cells in the ipsilateral CA3 performed 24 hours after KA (78.2±20.3) were comparable (p > 0.05) to those obtained from KA-injected wild type mice not treated with DPCPX (73.8±17.1). These data suggest that 1 mg/kg DPCPX normalized the effects of increased adenosine on A1Rs in fb-Adk-def mice and thereby recreated a wild type-like acute injury in these mutants.
To determine whether fb-Adk-def mice, in which KA was paired with DPCPX, are protected from the subsequent expression of spontaneous seizures after the initial precipitating injury (IPI) (see above), the remaining five animals were studied at 3 weeks after KA-injection. Most importantly, EEG recording electrodes implanted into the ipsilateral CA3 of these animals did not reveal any seizure activity during 40 hours of cumulative recordings (Fig. 5B) indicating that the expression of spontaneous seizures was prevented in these animals. Remarkably, and despite the presence of the IPI, histological assessment of brain sections did not reveal any signs of astrogliosis or upregulation of ADK (Fig. 5B) indicating the prevention of the development of two histopathological parameters associated with epileptogenesis.
Conclusions
Upregulation of ADK coincides at high spatial and temporal resolution with the expression of spontaneous seizures.
Upregulation of ADK, which can occur as a consequence of astrogliosis, is sufficient to cause spontaneous seizures.
Upregulation of ADK and loss of A1Rs cause an equally large increase in the susceptibility of the brain to seizures and neuronal cell death.
Levels of ADK, as an upstream regulator of A1R-mediated responses, are critical to determine the brain's susceptibility to seizures.
Discussion
Purinergic signalling plays an important role in mediating neuron-glia interactions (Fields and Burnstock, 2006; Halassa et al., 2007). In particular, astrocytes play a key role in regulating synaptic levels of adenosine – and thus in the modulation of neuronal activity – based on astrocyte-mediated release of the adenosine precursor ATP (Pascual et al., 2005; Haydon and Carmignoto, 2006), and based on ADK-driven reuptake of adenosine into astrocytes (Boison, 2006;, 2007b; 2007a). Thus, an astrocyte-based “adenosine-cycle” has been proposed as an upstream regulator and integrator of glutamatergic and dopaminergic neurotransmission (Yee et al., 2007; Boison, 2008a). Based on the capacity of astrocytes to regulate and control neuronal transmission, it is not surprising that astrocyte dysfunction contributes to epileptogenesis. Recently an “astrocytic basis for epilepsy” was proposed based on findings that Ca2+ homeostasis in astrocytes plays a key role in the hyper-synchronization of epileptic bursts (Tian et al., 2005). Here we provide experimental evidence in support of the “ADK hypothesis of epileptogenesis” (Boison, 2008b), which is based on astrogliotic upregulation of the key adenosine-removing enzyme ADK.
The ADK hypothesis of epileptogenesis
In this and in a previous study (Li et al., 2008) we provide experimental evidence in support of the ADK hypothesis of epileptogenesis on four different levels:
Spatio-temporal colocalization of upregulated ADK and the emergence of spontaneous seizures: In our new mouse model of CA3-selective focal epileptogenesis (Li et al., 2008), we demonstrate at high spatial resolution colocalization of astrogliosis, upregulated ADK, and spontaneous electrographic seizures (Fig. 1). Epileptogenesis in our model is triggered by SE, which is secondary to intraamygdaloid injection of KA. Thus, KA will have no direct effects on the ipsilateral hippocampus. It is important to note that ADK levels in the vicinity of the astrogliotic scar are normal. Thus, normal adenosine levels in the surrounding tissue are likely to prevent the spread of seizures via an A1R dependent mechanism (Fedele et al., 2006). The spatial correlation of upregulated ADK with spontaneous seizures (Fig. 1) however, did not allow us to discriminate whether upregulated ADK was cause or consequence of spontaneous chronic seizures. One remaining possibility was that upregulated ADK might be a compensatory reaction of the brain to remove excessive adenosine released during spontaneous chronic seizures. We now provide evidence that upregulation of ADK temporally precedes the emergence of spontaneous chronic seizures (Fig. 2). These are important findings suggesting that upregulated ADK cannot be a consequence of spontaneous chronic seizures. These findings – as will further be discussed below – also suggest that in our model astrocytes react differently to acute injury/SE (triggering astrogliosis and upregulation of ADK) whereas spontaneous chronic electrographic seizures do not appear to trigger astrogliosis and upregulation of ADK as suggested by the lack of astrogliosis and upregulated ADK in A1R knockout mice that experience spontaneous chronic electrographic seizures in the absence of KA-triggered brain injury (see also discussion below).
Spontaneous seizures in Adk-tg mice: To address the question whether astrogliosis or upregulation of ADK is the primary cause for ictogenesis, transgenic mice were created with altered levels of ambient adenosine due to genetic modification of the Adk gene. Global genetic disruption of ADK leads to hepatic steatosis and perinatal lethality (Boison et al., 2002) precluding any further studies of epileptogenesis. However, mutant mice with a transgenic overexpression of ADK within an ADK-deficient background (Adk-tg) are viable (Fedele et al., 2005). Due to the lack of endogenous ADK, adenosine levels in these mice are largely under the control of an Adk transgene, which is ubiquitously overexpressed in brain (in both glia and neurons) including an ectopic overexpression of ADK in pyramidal neurons, which was most prominent in the hippocampal CA3 region (Fedele et al., 2005; Li et al., 2008). It is important to note that the endogenous ADK, which is subject to regulation, is deleted and replaced by the ubiquitously expressed transgene. As determined by GFAP immunofluorescence analysis, this genetic modification did not lead to overt changes in astrocyte number or distribution (Fedele et al., 2005). Since intracellular ATP levels are about 100,000 times higher than respective adenosine levels (Fredholm et al., 2005a), modulation of ADK activity will translate into changes in adenosine, but will not influence levels of ATP. Thus, a reduced tone of ambient adenosine was demonstrated in brain slices derived from Adk-tg mice: increased pictrotoxin-induced seizure activity, as well as the lack of changes in the EPSP response after pharmacological blockade of the A1R in mossy fiber CA3 synapses, was consistent with a reduction in the hippocampal adenosine tone. In agreement with these findings, A1R dependent adenosine-mediated inhibition of the EPSP amplitude could be restored by inhibition of ADK (Fedele et al., 2005). As a result of the Adk-tg mutation and reduced levels of adenosine, the animals exhibit aggravated cell death in ischemia (Pignataro et al., 2007) and following KA-induced acute seizures (Fig. 4). In addition, transgenic overexpression of ADK in CA3 was associated with spontaneous electrographic CA3 seizures – and this in the absence of astrogliosis – at a frequency of 4.8 ± 1.5 seizures per hour with each seizure lasting 26.7 ± 13.2 seconds (Fig. 3). Consistent with the global diffuse overexpression of ADK in brain, occasional spontaneous seizures were also recorded in CA1 and cortex (Fedele et al., 2005), however it was not further investigated whether these seizures were primary or resulted from spread of the regular CA3 seizures. Remarkably, the CA3 seizures in Adk-tg mice were highly similar to CA3 seizures in KA treated epileptic mice (4.3 ± 1.5 seizures per hour, each lasting 17.5 ± 5.8 seconds) (Fig. 3). It is important to note that in mutant animals only modest increases in forebrain ADK (147% of normal) were sufficient to elicit spontaneous seizures (Li et al., 2008). This correlates well with the 1.8-fold increase of ADK activity in hippocampi of kainic acid treated epileptic mice (Gouder et al., 2004). Although Adk-tg mice overexpress ADK from birth, they do not show any histopathological alterations in brain morphology. In particular, untreated naïve adult Adk-tg mice, despite having regular spontaneous seizure activity, do not show any signs of astrogliosis, mossy fiber sprouting, granule cell dispersion, or neuronal cell death [(Fedele et al., 2005) and unpublished observations]. These observations indicate that naïve Adk-tg mice do not underlie the process of epileptogenesis. Most importantly, spontaneous seizures in Adk-tg mice appear to be non-injurious, indicating that the increased susceptibility to neuronal cell death (Fig. 4) is related to the spread of injurious KA-induced acute type IV seizures (Shinoda et al., 2004; Henshall and Simon, 2005). Our findings further imply that tight regulation of adenosine levels by ADK is a necessity and that moderate overexpression of ADK in the absence of astrogliosis or epileptogenesis is sufficient to induce spontaneous seizures (i.e. to trigger ictogenesis). One limitation of the Adk-tg mice used here is the fact that they do not completely mimic epileptic overexpression of ADK that is confined largely to astrocytes. Ectopic expression of ADK in CA3 neurons might also have some direct consequences for neuronal excitability that are independent of the general tone of adenosine within the CA3. However, ADK in epileptic CA3 not only shows upregulation, but also redistribution (from nucleus to periphery and processes of cells) (Li et al., 2008) thus leading to a “diffuse” overexpression of ADK not un-similar to what is seen in Adk-tg mice. In addition, as pointed out above, global ADK levels are similar between forebrain of Adk-tg mice (Li et al., 2008) and epileptic hippocampus (Gouder et al., 2004), and Adk-tg mice clearly have a reduced tone of adenosine in CA3 (Fedele et al., 2005). The incidence of spontaneous CA3 seizures in A1R KO mice is a direct explanation that a reduced tone of adenosine caused by overexpressed ADK (whether in astrocytes of the epileptic CA3 or globally in Adk-tg mice) is sufficient to trigger ictogenesis.
Prevention of astrogliosis, upregulation of ADK, and spontaneous seizures in fb-Adk-def mice: In contrast to the enhanced brain vulnerability seen in Adk-tg mice (Fig. 4), a reduction of ADK in forebrain (62% of normal) of fb-Adk-def mice provides profound protection from acute seizures and neuronal injury. Wild type-like brain injury can be restored in fb-Adk-def mice upon pairing KA-injection with A1R blockade, indicating that the observed protection is due to increased forebrain adenosine. Most remarkably, under these conditions (i.e. despite the presence of an initial epileptogenesis-precipitating injury) the animals fail to develop astrogliosis, fail to overexpress ADK, and fail to develop spontaneous CA3-selective seizures. As described previously (Li et al., 2008), despite the lack of ADK in pyramidal neurons and astrocytes, fb-Adk-def mice retain ADK expression in GABAergic interneurons (complete disruption of ADK in forebrain is lethal, unpublished data). This could theoretically lead to decreased adenosinergic inhibition of these neurons and thus to increased GABA-release, which might contribute to seizure resistance in these animals. However, this possibility is unlikely, since wild type-like acute seizures and wild type-like acute injury can be restored in KA-injected fb-Adk-def mice pre-treated with DPCPX. Overall, our current findings suggest, that fb-Adk-def mice might be resistant to epileptogenesis. However, strictly speaking, the data presented here do not allow discrimination between the suppression of epileptogenesis and the suppression of ictogenesis due to increased levels of forebrain adenosine. Thus, epileptogenesis might be masked by continued seizure suppression by elevated adenosine. However, the lack of astrogliosis – a pathologic hallmark of the epileptic brain – in fb-Adk-def mice suggests that at least one pathological parameter of epileptogenesis is prevented. Future studies are warranted to study different parameters of epileptogenesis in fb-Adk-def mice, such as mossy fiber sprouting or seizure susceptibility under DPCPX, delivered during different time windows, in more detail.
Prevention of seizures by cell transplantation: Cell therapy for temporal lobe epilepsy has emerged as a novel and promising prospect to achieve seizure control by focal cell-based intervention (Boison, 2007c; Raedt et al., 2007; Shetty and Hattiangady, 2007; Loscher et al., 2008). Thus, cells have been engineered to release adenosine as a local source to augment endogenous adenosinergic functions (Boison, 2007a). The therapeutic potential of stem cells engineered to release adenosine was recently tested in two cell transplantation experiments (Li et al., 2007; Li et al., 2008). In these studies mouse embryonic stem (ES) cells engineered to lack both alleles of ADK (Fedele et al., 2004) were differentiated in vitro into adenosine-releasing neural precursor cells (NPs). ES cell derived NPs, when grafted into the infrahippocampal cleft, can form densely clustered infrahippocampal implants thought to provide a reservoir for paracrine adenosine release (Li et al., 2007). In addition, grafted cells migrate into the ipsilateral CA1 region, assume neuronal morphology including the formation of long processes and the expression of the neuronal marker NeuN (Li et al., 2007). These implants display excellent survival for at least 26 days (Li et al., 2007). When tested in the rat kindling model of epilepsy, infrahippocampal implants of Adk-/- ES-derived NPs provide profound suppression of kindling epileptogenesis, characterized by the lack of secondarily generalized seizures after 48 kindling stimulations, conditions under which all sham and wild type cell-treated control animals regularly displayed generalized seizures (Li et al., 2007). Similarly, Adk-/- ES-derived NPs transplanted into the infrahippocampal cleft of mice 24 hours after the intraamygdaloid injection of KA (i.e. after establishment of the initial precipitating injury) significantly reduced the degree of CA3-selective astrogliosis, prevented upregulation of ADK, and – most importantly – prevented the occurrence of CA3-selective seizures 3 weeks after acute brain injury (Li et al., 2008). The beneficial mechanisms how chronic augmentation of brain adenosine in the nanomolar range as achieved by cell-based brain implants (Huber et al., 2001) lead to a reduction of astrogliosis and to lack of ADK upregulation need to be distinguished from the mechanisms of acute rises in adenosine to micromolar levels (Fredholm et al., 2005a) as a response to injury that are thought to trigger astrogliosis (Boison, 2008b). More work is needed to fully understand the mechanistic differences of opposing downstream-effects of chronic low-level increases in adenosine versus acute high-level increases in adenosine. Several hypothetical mechanisms may be involved: (i) A moderate increase in adenosine levels might not be sufficient to trigger receptor expression changes on astrocytes, might preferentially activate astrocytic A1 receptors, and thus reduce astrocyte proliferation, astrogliosis, and consequential upregulation of ADK. (ii) In contrast, high levels of adenosine inhibited ADK (Mimouni et al., 1994); thus high levels of adenosine could trigger upregulation of ADK as a compensatory mechanism. (iii) A sustained rise in adenosine to micromolar changes is expected to lead to changes in astrocytic adenosine receptors, most notably downregulation of A1 receptors that are involved in regulating astrocyte proliferation; changes in astrocytic adenosine receptors could then trigger astrogliosis. In summary, our data suggest that focal augmentation of adenosine by cellular brain implants has the potential to prevent at least some aspects of epileptogenesis. The lack of upregulated ADK in the recipients of adenosine-releasing cells (Li et al., 2007; Li et al., 2008) also demonstrates that upregulation of ADK is not a compensatory response to increased adenosine, but rather the cause for an adenosine deficit.
Downstream effectors
Seizure susceptibility is largely controlled by the relative availability of two receptors with antagonistic activity: A1R vs. A2AR. Since adenosine exerts most of its epileptogenesis-relevant effects by activation of A1 and A2A receptors, astrocytes expressing ADK as the key regulator of ambient adenosine can be considered as an upstream regulatory control system for adenosine receptor-mediated downstream effects. To further elucidate the downstream components of the adenosine / epileptogenesis pathways, in the present study we subjected A1 receptor knockout mice to our paradigm of CA3-selective epileptogenesis and provide the following findings: We previously demonstrated that A1R activation is necessary to keep an epileptic focus localized (Fedele et al., 2006). Likewise, traumatic brain injury in A1R knockout mice led to increased seizure spread and lethal status epilepticus (Kochanek et al., 2006). Here we demonstrate for the first time that A1R knockout mice develop focal spontaneous seizures in the absence of an epileptogenesis-precipitating injury. Remarkably, in CA3 these seizures are highly similar, in terms of frequency and duration, to seizures recorded from the CA3 of Adk-tg mice, or spontaneously epileptic mice with upregulated ADK in the CA3 seizure focus (Fig. 3). After intraamygdaloid injection with KA, A1R knockout mice developed increased SE, a spread of neuronal cell death within the hippocampal formation, and lethality within 3 days, consistent with our previous study subjecting A1R knockout mice to intrahippocampal KA-induced SE. The capacity of A1Rs to prevent the spread of seizures and neuronal cell death described here is of importance to the understanding of our mouse model of CA3-selective epileptogenesis. While we find upregulated ADK restricted to the ipsilateral CA3, ADK expression in the vicinity – and thus, presumably, levels of ambient adenosine – are normal. Thus, normal A1R activation and therefore prevention of seizure spread is expected in areas outside the seizure focus. In contrast to the predominant inhibitory effects of the A1R, the A2AR has pro-excitatory functions that could theoretically limit the effectiveness of adenosine-augmentation therapies (AATs). However, AATs increase the global inhibitory tone of “paracrine” adenosine acting as a “heterosynaptic modulator” via activation of A1Rs, thus exerting broad inhibitory/protective therapeutic effects; in contrast, A2ARs are activated more specifically by “autocrine” adenosine as a “synaptic modulator (Cunha, 2008). Thus, the pro-excitatory functions of A2ARs seem to be limited to high frequency stimulation of selected synapses within a globally inhibited network (Cunha, 2008). Thus, the net-effect of AATs (e.g. transplantation of adenosine-releasing cells) appears to be inhibitory, as has been found in a variety of experimental AAT approaches (Boison, 2007a).
Clinical findings
Several clinical data support the ADK hypothesis of epileptogenesis: Microdialysis studies performed in patients with intractable temporal lobe epilepsy have demonstrated a seizure-induced 6- to 31-fold increase in extracellular adenosine levels (During and Spencer, 1992). Likewise, adenosine metabolites were increased in cerebrospinal fluid following SE (Chin et al., 1995). Based on these findings adenosine was described as “endogenous mediator of seizure arrest and postictal refractoriness” (During and Spencer, 1992). Remarkably, and in agreement with upregulated ADK in epileptogenic hippocampus, in these studies basal adenosine levels sampled before the onset of seizures were lower in the epileptogenic hippocampus compared to the non-epileptic hippocampus (During and Spencer, 1992). Findings on A1R expression in human temporal lobe epilepsy are controversial; both down- and upregulation of A1R have been described (Angelatou et al., 1993; Glass et al., 1996). Indeed, the incidence of spontaneous seizures in heterozygous A1R knockout mice described here (Fig. 3) indicates that downregulation of A1Rs in temporal lobe epilepsy may directly be linked to seizures. Loss of A1R during aging as analyzed in a recent positron emission tomography (PET) study may also be a contributing factor to the increased incidence of epilepsy in the elderly (Meyer et al., 2007). Recent findings from human studies suggest energetic dysfunction and mitochondrial dysfunction to be implicated in epileptogenesis (Kunz, 2002; Pan et al., 2005; Williamson et al., 2005). Therapeutically, the purinergic drug allopurinol was used as adjunctive therapy in a double-blind and placebo-controlled trial in intractable epilepsy (Togha et al., 2007). Remarkably, seizures were decreased in the allopurinol group compared to control, however allopurinol-induced side effects were common. Nevertheless, this study demonstrates that modulation of the purinergic system might be beneficial in intractable epilepsy.
Acknowledgments
The work of the corresponding author is supported by grants RO1NS058780-01, R21NS057475-01, and R21NS057538-01 from the National Institute of Neurological Disorders and Stroke (NINDS), the Good Samaritan Hospital Foundation, the Epilepsy Research Foundation through the generous support of Arlene & Arnold Goldstein Family Foundation, and Citizens United in Research against Epilepsy (CURE) in collaboration with the Department of Defense (DoD).
Footnotes
Statement of Interest: None
References
- Angelatou F, Pagonopoulou O, Maraziotis T, Olivier A, Villemeure JG, Avoli M, Kostopoulos G. Upregulation of A1 adenosine receptors in human temporal lobe epilepsy: a quantitative autoradiographic study. Neuroscience Letters. 1993;163:11–14. doi: 10.1016/0304-3940(93)90217-9. [DOI] [PubMed] [Google Scholar]
- Arch JR, Newsholme EA. Activities and some properties of 5′-nucleotidase, adenosine kinase and adenosine deaminase in tissues from vertebrates and invertebrates in relation to the control of the concentration and the physiological role of adenosine. Biochemical Journal. 1978;174:965–977. doi: 10.1042/bj1740965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Archive European Journal of Physiology. 2004;447:735–743. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends in Pharmacological Sciences. 2006;27:652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine-based cell therapy approaches for pharmacoresistant epilepsies. Neurodegenerative Diseases. 2007a;4:28–33. doi: 10.1159/000100356. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine as a modulator of brain activity. Drug News and Perspectives. 2007b;20:607–611. doi: 10.1358/dnp.2007.20.10.1181353. [DOI] [PubMed] [Google Scholar]
- Boison D. Cell and gene therapies for refractory epilepsy. Current Neuropharmacology. 2007c;5:115–125. doi: 10.2174/157015907780866938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine as a neuromodulator in neurological diseases. Current Opinion in Pharmacology. 2008a;8:2–7. doi: 10.1016/j.coph.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. The adenosine kinase hypothesis of epileptogenesis. Progress in Neurobiology. 2008b;84:249–262. doi: 10.1016/j.pneurobio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rülicke T, Litynski P, Fowler B, Brandner S, Mohler H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proceedings of the National Academy of Sciences USA. 2002;99:6985–6990. doi: 10.1073/pnas.092642899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontemps F, Van den Berghe G, Hers HG. Evidence for a substrate cycle between AMP and adenosine in isolated hepatocytes. Proceedings of the National Academy of Sciences USA. 1983;80:2829–2833. doi: 10.1073/pnas.80.10.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JH, Wiesner JB, Fujitaki J. Increase in adenosine metabolites in human cerebrospinal fluid after status epilepticus. Journal of Neurology, Neurosurgery, and Psychiatry. 1995;58:513–514. doi: 10.1136/jnnp.58.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA. Different cellular sources and different roles of adenosine: A(1) receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A(2A) receptor-mediated facilitation of plasticity. Neurochemistry International. 2008;52:65–72. doi: 10.1016/j.neuint.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Dragunow M. Adenosine and seizure termination. Annals of Neurology. 1991;29:575. doi: 10.1002/ana.410290524. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Goddard GV, Laverty R. Is adenosine an endogenous anticonvulsant? Epilepsia. 1985;26:480–487. doi: 10.1111/j.1528-1157.1985.tb05684.x. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. Modulation of neuronal excitability by astrocytes. Advances in Neurology. 1999;79:573–581. [PubMed] [Google Scholar]
- Dunwiddie TV. Endogenously released adenosine regulates excitability in the in vitro hippocampus. Epilepsia. 1980;21:541–548. doi: 10.1111/j.1528-1157.1980.tb04305.x. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV. Adenosine and suppression of seizures. Advances in Neurology. 1999;79:1001–1010. [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annual Review in Neuroscience. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Worth T. Sedative and anticonvulsant effects of adenosine analogs in mouse and rat. Journal of Pharmacology and Experimental Therapeutics. 1982;220:70–76. [PubMed] [Google Scholar]
- During MJ, Spencer DD. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Annals of Neurology. 1992;32:618–624. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Güttinger M, Gabernet L, Scheurer L, Rulicke T, Crestani F, Boison D. Astrogliosis in epilepsy leads to overexpression of adenosine kinase resulting in seizure aggravation. Brain. 2005;128:2383–2395. doi: 10.1093/brain/awh555. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Brüstle O, Scheurer L, Simpson EM, Mohler H, Boison D. Engineering embryonic stem cell derived glia for adenosine delivery. Neuroscience Letters. 2004;370:160–165. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Experimental Neurology. 2006;200:184–190. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nature Review Neuroscience. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. International Review in Neurobiology. 2005a;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annual Review in Pharmacology and Toxicology. 2005b;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Ijzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacological Reviews. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Glass M, Faull RL, Bullock JY, Jansen K, Mee EW, Walker EB, Synek BJ, Dragunow M. Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Research. 1996;710:56–68. doi: 10.1016/0006-8993(95)01313-x. [DOI] [PubMed] [Google Scholar]
- Gouder N, Fritschy JM, Boison D. Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia. 2003;44:877–885. doi: 10.1046/j.1528-1157.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. Journal of Neuroscience. 2004;24:692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JH, Owen RP, Giacomini KM. The concentrative nucleoside transporter family, SLC28. Pflugers Archive European Journal of Physiology. 2004;447:728–734. doi: 10.1007/s00424-003-1107-y. [DOI] [PubMed] [Google Scholar]
- Güttinger M, Padrun V, Pralong W, Boison D. Seizure suppression and lack of adenosine A1 receptor desensitization after focal long-term delivery of adenosine by encapsulated myoblasts. Experimental Neurology. 2005;193:53–64. doi: 10.1016/j.expneurol.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends in Molecular Medicine. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiological Reviews. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Simon RP. Epilepsy and apoptosis pathways. Journal of Cerebral Blood Flow and Metabolism. 2005;25:1557–1572. doi: 10.1038/sj.jcbfm.9600149. [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D. Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proceedings of the National Academy of Sciences USA. 2001;98:7611–7616. doi: 10.1073/pnas.131102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Nomura R, Ando R, Ikeda T, Tanaka M, Itohara S. Dorsal telencephalon-specific expression of Cre recombinase in PAC transgenic mice. Genesis. 2004;38:130–138. doi: 10.1002/gene.20009. [DOI] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proceedings of the National Academy of Sciences USA. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, Jenkins LW, Clark RS, Homanics GE, Dixon CE, Schnermann J, Jackson EK. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. Journal of Cerebral Blood Flow and Metabolism. 2006;26:565–575. doi: 10.1038/sj.jcbfm.9600218. [DOI] [PubMed] [Google Scholar]
- Kunz WS. The role of mitochondria in epileptogenesis. Current Opinion in Neurology. 2002;15:179–184. doi: 10.1097/00019052-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Lee KS, Schubert P, Heinemann U. The anticonvulsive action of adenosine: a postsynaptic, dendritic action by a possible endogenous anticonvulsant. Brain Research. 1984;321:160–164. doi: 10.1016/0006-8993(84)90694-2. [DOI] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. Journal of Clinical Investigation. 2008;118:571–582. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain. 2007;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Loscher W, Gernert M, Heinemann U. Cell and gene therapies in epilepsy - promising avenues or blind alleys. Trends in Neurosciences. 2008 doi: 10.1016/j.tins.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Meyer PT, Elmenhorst D, Boy C, Winz O, Matusch A, Zilles K, Bauer A. Effect of aging on cerebral A(1) adenosine receptors: A [F-18] CPFPX PET study in humans. Neurobiology of Aging. 2007;28:1914–1924. doi: 10.1016/j.neurobiolaging.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Mimouni M, Bontemps F, Van den Berghe G. Kinetic studies of rat liver adenosine kinase. Journal of Biological Chemistry. 1994;269:17820–17825. [PubMed] [Google Scholar]
- Pan JW, Kim JH, Cohen-Gadol A, Pan C, Spencer DD, Hetherington HP. Regional energetic dysfunction in hippocampal epilepsy. Acta Neurologica Scandinavica. 2005;111:218–224. doi: 10.1111/j.1600-0404.2005.00398.x. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Maysami S, Studer FE, Wilz A, Simon RP, Boison D. Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. Journal of Cerebral Blood Flow and Metabolism. 2008;28:17–23. doi: 10.1038/sj.jcbfm.9600499. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Simon RP, Boison D. Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. Journal of Cerebral Blood Flow and Metabolism. 2007;27:1–5. doi: 10.1038/sj.jcbfm.9600334. [DOI] [PubMed] [Google Scholar]
- Raedt R, Van Dycke A, Vonck K, Boon P. Cell therapy in models for temporal lobe epilepsy. Seizure-European Journal of Epilepsy. 2007;16:565–578. doi: 10.1016/j.seizure.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Ren G, Li T, Lan JQ, Wilz A, Simon RP, Boison D. Lentiviral RNAi-induced downregulation of adenosine kinase in human mesenchymal stem cell grafts: a novel perspective for seizure control. Experimental Neurology. 2007;208:26–37. doi: 10.1016/j.expneurol.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sebastiao AM, Ribeiro JA. Fine-tuning neuromodulation by adenosine. Trends in Pharmacological Sciences. 2000;21:341–346. doi: 10.1016/s0165-6147(00)01517-0. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B. Concise review: Prospects of stem cell therapy for temporal lobe epilepsy. Stem Cells. 2007;25:2396–2407. doi: 10.1634/stemcells.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, Lan JQ, Taki W, Simon RP, Henshall DC. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. Journal of Clinical Investigation. 2004;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodiya S. Etiology and management of refractory epilepsies. Nature Clinical Practice Neurology. 2007;3:320–330. doi: 10.1038/ncpneuro0521. [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience. 2006;142:125–137. doi: 10.1016/j.neuroscience.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Goldberg J, Yuste R. Calcium oscillations in neocortical astrocytes under epileptiform conditions. Journal of Neurobiology. 2002;50:45–55. doi: 10.1002/neu.10019. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu QW, Peng WG, Lin J, Oberheim N, Lou NH, Wang XH, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nature Medicine. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togha M, Akhondzadeh S, Motamedi M, Ahmadi B, Razeghi S. Allopurinol as adjunctive therapy in intractable epilepsy: a double-blind and placebo-controlled trial. Archives of Medical Research. 2007;38:313–316. doi: 10.1016/j.arcmed.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Vajda FJE. Pharmacotherapy of epilepsy: New armamentarium, new issues. Journal of Clinical Neuroscience. 2007;14:813–823. doi: 10.1016/j.jocn.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Pan J, Spencer DD, Hetherington H. Correlations between granule cell physiology and bioenergetics in human temporal lobe epilepsy. Brain. 2005;128:1199–1208. doi: 10.1093/brain/awh444. [DOI] [PubMed] [Google Scholar]
- Wu NL, Boison D. Adenosine kinase expression modulates expression of myelin proteolipid protein. The Open Neuroscience Journal. 2007;1:15–19. [Google Scholar]
- Yee BK, Singer P, Chen JF, Feldon J, Boison D. Transgenic overexpression of adenosine kinase in brain leads to multiple learning impairments and altered sensitivity to psychomimetic drugs. European Journal of Neuroscience. 2007;26:3237–3252. doi: 10.1111/j.1460-9568.2007.05897.x. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Archives of Pharmacology. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]