

Abstract
We present a novel computational method for predicting which proteins from highly and abnormally expressed genes in diseased human tissues, such as cancers, can be secreted into the bloodstream, suggesting possible marker proteins for follow-up serum proteomic studies. A main challenging issue in tackling this problem is that our understanding about the downstream localization after proteins are secreted outside the cells is very limited and not sufficient to provide useful hints about secretion to the bloodstream. To bypass this difficulty, we have taken a data mining approach by first collecting, through extensive literature searches, human proteins that are known to be secreted into the bloodstream due to various pathological conditions as detected by previous proteomic studies, and then asking the question: ‘what do these secreted proteins have in common in terms of their physical and chemical properties, amino acid sequence and structural features that can be used to predict them?’ We have identified a list of features, such as signal peptides, transmembrane domains, glycosylation sites, disordered regions, secondary structural content, hydrophobicity and polarity measures that show relevance to protein secretion. Using these features, we have trained a support vector machine-based classifier to predict protein secretion to the bloodstream. On a large test set containing 98 secretory proteins and 6601 non-secretory proteins of human, our classifier achieved ∼90% prediction sensitivity and ∼98% prediction specificity. Several additional datasets are used to further assess the performance of our classifier. On a set of 122 proteins that were found to be of abnormally high abundance in human blood due to various cancers, our program predicted 62 as blood-secreted proteins. By applying our program to abnormally highly expressed genes in gastric cancer and lung cancer tissues detected through microarray gene expression studies, we predicted 13 and 31 as blood secreted, respectively, suggesting that they could serve as potential biomarkers for these two cancers, respectively. Our study demonstrated that our method can provide highly useful information to link genomic and proteomic studies for disease biomarker discovery. Our software can be accessed at http://csbl1.bmb.uga.edu/cgi-bin/Secretion/secretion.cgi.
Contact: xyn@bmb.uga.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Alterations in gene and protein expression provide important clues about the physiological states of a tissue or an organ. During malignant transformation, genetic alterations in tumor cells can disrupt autocrine and paracrine signaling networks, leading to the over-expression of some classes of proteins such as growth factors, cytokines and hormones that may be secreted outside the cancerous cells (Hanahan and Weinberg, 2000; Sporn and Roberts, 1985). These secreted proteins may get into blood, urine or other body fluids through various complex secretion pathways and can potentially be used as marker proteins for blood or urine tests. Recent genomic studies on various cancer specimens have identified numerous genes that are consistently over-expressed and some of these genes encode secreted proteins (Buckhaults et al., 2001; Welsh et al., 2001). For example, prostasin and osteopontin genes have elevated expression levels in ovarian cancer while MIC1 gene is over-expressed in colorectal, breast and prostate cancers. The increased abundance of these secretory proteins has been detected in the serum of patients harboring these cancers compared to the healthy individuals (Kim et al., 2002; Mok et al., 2001; Welsh et al., 2003). It has also been found that some of the secreted proteins have shown varying levels of concentration increases in serum associated with different developmental stages of cancers, suggesting that they could possibly be used as markers of both cancer typing and staging (Huang et al., 2006).
The human serum proteome is a very complex mixture of highly abundant native serum proteins such as albumin, immunoglobulins, transferrin, haptoglobin and lipoproteins as well as proteins and peptides that are secreted from different tissues, diseased or normal or leak from cells throughout the human body (Adkins et al., 2002; Schrader and Schulz-Knappe, 2001). A challenging issue when working with the human serum proteome is that most of the circulating native blood proteins are orders of magnitude more abundant than those of the putative proteins of interest. Hence, it is very difficult to experimentally detect such secreted proteins, though with increased relative abundance in blood, among thousands or possibly more native blood proteins without knowing what proteins we are looking for in blood a priori. It is thus desirable to investigate computational approaches to predict proteins that are both abnormally highly expressed in cancer tissues and can get secreted into the bloodstream, providing a target list for targeted proteomic work of human blood serum and making the identification problem of such marker proteins in blood more realistically solvable.
Numerous studies have been carried out to predict proteins that can get secreted to cell surface or into the extracellular environments in both eukaryotes and prokaryotes, and several public prediction servers are available (Guda, 2006; Horton et al., 2007; Menne et al., 2000; Nair and Rost, 2005). Most of these methods have been developed based on our general understanding about protein subcellular localization—localization of most proteins is done through a cascade of sorting events that are directed by short (signal) peptides or motifs that enable site-specific uptake, retention and transport (Doudna and Batey, 2004; Tjalsma et al., 2000). These programs have been developed using various statistical learning methods, based on information such as amino acid composition, co-occurrence of protein domains and annotated protein functions (Guda, 2006; Mott et al., 2002). A fundamental difference between our work and the previous work is that while they all are concerned about whether a protein gets secreted outside of a cell, previous studies are not concerned about where the proteins will end up but ours is.
In order to predict proteins that can get secreted into the bloodstream, we have collected 305 non-native blood proteins that have been experimentally found in the blood under various physiological conditions from the published literature. We have analyzed these proteins carefully to derive various sequence and structural features commonly shared by these proteins or by some subsets of these proteins, including signal peptides, transmembrane domains, glycosylation sites, disordered regions, secondary structural content, radius of gyration of a protein tertiary structure, hydrophobicity and polarity measures that show relevance to protein secretion. Using these features, we have trained support vector machine (SVM)-based classifier to distinguish these experimentally verified blood-secreted proteins from the rest of the human proteins. We found that the prediction results of this classifier are highly promising, providing very useful information that could bridge the information about abnormally and highly expressed genes detected through microarray experiments and proteomic studies of blood serum for effective marker–protein identification.
2 METHODS
2.1 Collection of blood-secreted proteins and non-blood-secreted proteins
We have collected a total of 1620 human proteins that are annotated as secretory proteins from the Swissprot and SPD database (Chen et al., 2005), and then determined if any of these proteins have been detected experimentally in blood by previous studies. We have done this by checking the 1620 proteins against the known serum protein dataset compiled by the Plasma Proteome Project (PPP) (Omenn et al., 2005) and a few additional datasets generated by other serum proteomic studies (Adkins et al., 2002; Pieper et al., 2003), which consist of a total of ∼16 000 proteins. We found that 305 of the 1620 proteins match at least two peptides with the ∼16 000 proteins, and hence we consider that these 305 proteins are secreted into blood—a common practice for protein identification based on mass spectrometry data. To ensure the good quality of our dataset, it is should be noted that we only chose these 305 proteins which meet two criteria (both secreted and serum/plasma detected), as the positive dataset and did not include proteins that leak into the blood as a result of cell damage (e.g. cardiac myoglobin released into plasma after a heart attack).
To generate a negative dataset of proteins for the classification, we selected representatives from non-blood-secreted proteins, which should include both proteins unrelated to secretory pathway and secreted proteins not involved in the circulatory system. We have selected three representatives from each of the Pfam protein families (Bateman et al., 2002) that contain no previously mentioned blood-secreted proteins as the negative set.
In order to obtain a non-redundant dataset for a final independent validation step, we used BLAST (Altschul et al., 1997) to remove the redundant proteins using 20% sequence identity as the cutoff, giving rise to 56 positive and 13 716 negative proteins. We then divided the remains, 249 positive and 13 246 negative proteins into separate training and testing sets, respectively, using the following procedure. All the proteins in the positive set were divided into clusters based on the similarity of our selected features (see Section 2.2), measured by the Euclidean distance, using a hierarchical clustering method. A total of 151 clusters were obtained with the ratio between the maximum intra-cluster distance and the minimum inter-cluster distance for each cluster, ranging from 0.27 to 0.51. From each cluster, one representative protein was chosen randomly to form the positive training set. We do the same for the negative training set. The training set was selected in this way to ensure it is sufficiently diverse and broadly distributed in the feature space. The remaining proteins are used as the test set. We repeated this process to construct five different datasets to train the classifier, which can be used to assess the stability of our data generation strategy.
2.2 Feature construction
We have examined a number of features computed based on protein sequences and secondary structures that are possibly relevant to the classification of proteins being blood secreted or not. Some features are included because they are known to be relevant to protein secretion, while others are included because of their statistical relevance to our classification problem. For example, signal peptides and transmembrane domains are known to be important factors to prediction of extracellularly secreted proteins. Twin-arginine (TAT) signal peptides, only observed in prokaryotes so far, are known to be used to export proteins into the periplasmic compartment or extracellular environment independent of the well-studied sec-dependent translocation pathway (Bendtsen et al., 2005; Taylor et al., 2006). We included this motif information in our study to check if it might be relevant to transporting folded proteins across the human cell membrane. In addition, it is known that the structures of the capillaries determine that only proteins under a certain size can diffuse through their walls and get into the bloodstream. For example, blood proteins are expected to be larger than 45 kDa, the kidney filtration cutoff, and not smaller than the capillary leakage size that is up to 400 nm in diameter (under some tumor conditions), for their retention in blood (Anderson and Anderson, 2002; Brown and Giaccia, 1998). Hence, we have included information about the protein size and shape in our initial feature list. Another important feature is the glycosylation sites. It has been observed that most blood-secreted proteins are glycosylated (Bosques et al., 2006), including important tumor biomarkers such as prostate-specific antigen (PSA) and the ovarian cancer marker CA125.
In addition, we have included a number of general features in our initial feature list, derived from protein sequence, secondary structural and physicochemical properties widely used in various protein classification studies, such as protein function prediction and protein–protein interaction prediction, as reviewed in Cui et al. (2007b), which might be relevant to our prediction of blood-secreted proteins. Supplementary Table 1 summarizes the features discussed above. The actual relevance of these features to our classification problem is assessed using a feature-selection algorithm presented in the following section.
Features in Supplementary Table 1 can be roughly grouped into four categories: (i) general sequence features such as amino acid composition, sequence length and di-peptide composition (Bhasin and Raghava, 2004; Reczko and Bohr, 1994); (ii) physicochemical properties such as solubility, unfoldability, disordered regions, hydrophobicity, normalized Van der Waals volume, polarity, polarizability and charges, (iii) structural properties such as secondary structural content, solvent accessibility and radius of gyration and (iv) domains/motifs such as signal peptides, transmembrane domains and twin-arginine signal peptides motif (TAT). In total, 25 properties are included in the initial list, which give rise to a 1521-dimensional feature vector for each protein sequence. Note that for each included property, different amount of information is needed to encode it in our feature vector representation of the properties. For example, amino acid composition and dipeptide composition are represented as a 20- and 400(20×20)-dimensional feature vector, respectively. The feature vector of the secondary structural content is a four-dimensional vector, including alpha-helix content, beta-strand content, coil content and the assigned class by the SSCP program (Eisenhaber et al., 1996). Our encoding of physicochemical properties is illustrated by the example of hydrophobicity feature vector: amino acids can be divided into hydrophobic (CVLIMFW), neutral (GASTPHY) and polar (RKEDQN) groups. Then three descriptors, composition (C), transition (T) and distribution (D), are used to describe the global composition with C being the number of amino acids of a particular group (such as hydrophobic) divided by the total number of amino acids in the protein sequence (Cai et al., 2003; Cui et al., 2007; Dubchak et al., 1995); T being the relative frequency in changing amino acid groups along the protein sequence, and D denoting the chain length within which the first, 25%, 50%, 75% and 100% of the amino acids of a particular group is located, respectively. Overall, 21 elements are used to represent these three descriptors: 3 for C, 3 for T and 15 for D. By following these procedures, the feature vector of a protein is constructed using a total of 1521 feature elements.
2.3 Classification and feature selection
We have trained an SVM-based classifier to distinguish the positive from the negative training data, using a Gaussian kernel (Keerthi et al., 2001; Platt, 1999). SVM has been successfully applied to a wide range of pattern recognition problems in data mining and bioinformatics, such as protein function prediction (Cui et al., 2007), protein–protein interaction prediction (Ben-Hur and Noble, 2005) and protein subcellular location prediction (Su et al., 2007). The Gaussian radial basis function kernel has been extensively used in those studies with good results, which consistently shows superior performance to other kernels used in SVM such as linear and polynomial kernels (Ben-Hur and Noble, 2005; Burbidge et al., 2001; Su et al., 2007). Thus, Gaussian kernel SVM is used in our classification study. The inputs to the SVM are the aforementioned 1521 features for each protein in the training set, and the output of the classifier is an assignment of the input protein to be blood secreted or not. An independent evaluation set is used to estimate the accuracy of the overall protein assignment for the whole dataset. The classification performance is measured using the prediction sensitivity SE=TP/(TP+FN), prediction specificity SP=TN/(TN+FP), the overall prediction accuracy Q=(TP+TN)/N, precision=TP/(TP+FP), AUC (Mason and Graham, 2002) and Matthews correlation coefficient 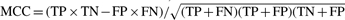 . Here TP, TN, FP and FN are the number of true positive, true negative, false positive and false negative, respectively, and N=TP+FN+TN+FP is the total number of proteins in the training set. We have used a reliability score, R-value, to assess the reliability for each of our predictions, shown as follows:
. Here TP, TN, FP and FN are the number of true positive, true negative, false positive and false negative, respectively, and N=TP+FN+TN+FP is the total number of proteins in the training set. We have used a reliability score, R-value, to assess the reliability for each of our predictions, shown as follows:
whered is the distance between the position of a target protein in the feature space and the optimal separating hyperplane derived through our SVM training. There is a strong correlation between the R-value and the classification accuracy (probability of correct classification) (Hua and Sun, 2001). Thus, a P-value is introduced to indicate the expected classification accuracy, derived from the statistical relationship between the R-value and the actual classification accuracy based on the analysis of 305 positive and 26 962 negative proteins, as shown in Figure 1.
Fig. 1.

Statistical relationship between the R-value and P-value (probability of correct classification) derived from the analysis of 305 positive and 26 962 negative samples of proteins.
Based on the performance of each initially trained SVM, a feature selection process, named recursive feature elimination (RFE) (Tang et al., 2007), is used to remove features irrelevant or negligible to our classification goal. For example, Moreau–Broto autocorrelation descriptors defined as:
were reported to be useful to the prediction of membrane proteins based on the hydrophobic index of amino acids (Feng and Zhang, 2000), but our classification study shows that it does not contribute to the accuracy of our classification, where d is the lag of the autocorrelation, and Pi and Pi+d are the hydrophobicity of the amino acids at position i and i+d, respectively. Hence, it is removed from our initial feature list, by the RFE procedure. The feature selection process iteratively removes such irrelevant features based on a consensus-scoring scheme and gene-ranking consistency evaluation (Tang et al., 2007). Specifically, in each iteration, features with the lowest score (least ranked) given by RFE based on randomly sampled training data are eliminated from the feature list. Essentially a majority-rule voting scheme is used to overcome possible discrepancies among different randomly chosen samples. This process continues until a minimal set of features, without losing the classification performance, is obtained.
3 RESULTS
Using the initial list of 1521 features, we trained an SVM classifier based on the provided positive and negative training sets. We then evaluated the performance of the best classifier, measured by the overall accuracy as defined in Section 2.3, using an independent evaluation set containing 47 positive and 3296 negative samples. We found that the prediction performance of this classifier gives only ∼40% accuracy, a clearly undesirable result. We believe that this is mostly due to the reason that the classifier used a number of features that are irrelevant to our classification and only add noises to the training of the SVM classifier; in addition, over-fitting the data by this large classifier with many parameters may be another cause for the underpar performance. Hence, it is desirable to remove some of the less relevant features by carrying out feature selection to optimize the performance of the SVM-based classifier.
Using the feature selection procedure outlined in Section 2.3, we have selected a total of 85 features, which gives the best cross-validation performance of our SVM classifier (Tang et al., 2007), as shown in Supplementary Table 2. We found that the following features are the most important ones for our classification, transmembrane domains, charges, TatP motif, solubility, polarity, signal peptides, hydrophobicity, O-linked glycosylation motif and secondary structural content, which rank among the top 20 features. This observation is consistent with our general understanding of secretory proteins, except that we found the TatP motif contributes substantially to our prediction result, which ranks among the top three features in our prediction, where TatP is known to be used to export proteins into the periplasmic compartment or extracellular environment in prokaryotes (Bendtsen et al., 2005; Taylor et al., 2006). To the best of our knowledge, this represents a novel finding linking the TatP motifs to protein secretion in eukaryotes.
Based on the 85 selected features, we have trained five new SVM-based classifiers and tested their performance using the reduced feature list on the same independent evaluation set. We found that the level of performance by these five classifiers is generally consistent, ranging from 87.2% to 93.7% for the blood-secreted proteins and from 98.2% to 98.6% for non-blood-secreted proteins, which is detailed in Supplementary Table 3. We have also calculated the precision, MCC and AUC values of our prediction performance, which have average values 44.6%, 0.63 and 0.94, respectively. Clearly, the AUC value is consistent with the earlier performance measures. Interestingly, the precision and MCC seem to be relatively low. It should be noted that MCC may fluctuate substantially on comparable evaluation sets, a general problem that has been reported previously (Klee and Sosa, 2007; Smialowski et al., 2007). In our case, the relatively low precision and MCC value are partially due to the skewed sizes between the positive and negative evaluation sets, which cause underestimation of the system performance. This could possibly be improved by increasing the size of positive set. Our goal here is to include as many previously unknown blood-secreted proteins as possible, while keeping the specificity high, so we have chosen one of the classifiers with the best sensitivity, as shown in Table 1.
Table 1.
Performance statistics of our classifier on prediction of blood-secreted protein and non-blood-secreted proteins in the training, testing and independent evaluation sets
| Dataset | Blood-secreted |
Non-blood-secreted |
Prediction accuracy |
||||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FN | TN | FP | SE (%) | SP (%) | Q (%) | MCC | AUC | |
| Training | 151 | 0 | 6545 | 0 | 100 | 100 | 100 | 1.00 | 1.00 |
| Testing | 46 | 5 | 3253 | 52 | 90.2 | 98.4 | 98.3 | 0.64 | 0.94 |
| Evaluation | 44 | 3 | 3237 | 59 | 93.6 | 98.2 | 98.1 | 0.63 | 0.95 |
When applying WolF PSORT (Horton et al., 2007) to the same evaluation set, the most cited method for protein extracellular secretion prediction, it gives 81.0% prediction accuracy with MCC value 0.37. This is not surprising since all the previous protein-secretion prediction methods, including WolF PSORT, are not designed for solving our problem as we are interested in both extracellular secretion and secretion to the bloodstream.
Our classifier has been further evaluated through a screening test against all human proteins in the Swissprot database, which can provide a more realistic estimate of our prediction performance when applied to large datasets. For this test, we collected 20 832 human proteins, among which 1563 are annotated as secreted proteins and additional ∼750 proteins are considered to be relevant to secretion based on their signal peptides and annotated subcellular locations (Welsh et al., 2003). As shown in Table 2, our classifier predicted 4063 proteins, 19.5% of the 20 832 as blood-secreted proteins, which largely agrees with the total (estimated and reported) numbers of secreted proteins and blood proteins (Welsh et al., 2003). All these results suggest that our initial set of 249 positive and 13 244 negative proteins shows good representation of the relevant proteins across the whole protein space.
Table 2.
Results of screening all human proteins in Swissprot for blood-secreted proteins
| No.of human proteins in Swissprot | 20 832 |
| No. of proteins annotated as secreted | 1563 |
| No. of potentially secreted proteins based on signal | 2 308 |
| peptide and location (Welsh et al., 2003) | |
| No. of blood-proteins | |
| All reported | 15 710 |
| High confidence | 3020 |
| No. of SVM predicted blood-secreted proteins | 4063 |
In addition to the above tests, we have done an extensive literature search of published proteomics studies and compiled a list of 240 differentially expressed proteins in human blood due to various diseases. These studies cover multiple cancers in 14 types of human tissues such as pancreas, ovary, melanoma, lung, prostate, stomach, liver, colon, nasopharynx, kidney, uterine cervix, brain, breast and bladder. Among the 240 proteins, 122 are not included in our initial collection of the 305 blood-secreted proteins, whose names are listed in Supplementary Table 4. The main reasons for not including these 122 proteins in our initial collection of blood-secreted proteins are (i) mis-annotation of these proteins in Swissprot and (ii) failing to detect them by the proteomics studies, from which we collected this initial list of proteins. As indicated in their respective studies, all these 122 proteins can be used as potential biomarkers in blood of a particular cancer to discriminate the normal from the tumor tissues or distinguish different developmental stages of a particular cancer, e.g. heat shock protein beta-1 for breast cancer (Rui et al., 2003), cathepsin D for melanoma (Pardo et al., 2007), l-lactate dehydrogenase for renal cancer (Unwin et al., 2003) and PSA for prostate cancer (Bradford et al., 2006). We predicted 97 out of 122 (79.5%) proteins correctly, while the remaining 25 proteins have prediction results inconsistent with the published literature (the names of these 122 proteins are given in Supplementary Table 4).
The following gives a few examples of our predictions on the 122 proteins and relevant evidence as reported in the literature. Among the correct predictions with supporting evidence from the literature, the tumor necrosis factor, tenascin, C–C motif chemokine 3 and the insulin-like growth factor-binding protein 7 are detected with elevated gene expression levels in cancer patients' serum and are annotated as secreted proteins in Swissprot and SPD database (Chen et al., 2005). Some membrane proteins like calsyntenin-1, immunoglobulin alpha chain C and hepatocyte growth factor receptor are predicted as secreted proteins but these predictions can only be considered having partial supporting evidence in the published literature since there is evidence that these proteins are found outside the cells, through secretion or other means, e.g. proteolytic cleavage of membrane-associated proteins. Besides, some predictions can also be partially supported by the annotated protein functions. For example, thrombospondin 1 precursor is described as an adhesive glycoprotein that mediates cell-to-cell and cell-to-matrix interactions, thus it is expected to function outside the cells. We consider those proteins annotated as secreted proteins but predicted as non-blood-secreted or as blood-secreted proteins but without any evidence showing relevance to secretion as ‘not consistent with the literature’, such as profilin-1 and carbonic anhydrase 1.
One key planned application of our SVM-based classifier is to predict if abnormally and highly expressed genes, detected by microarray gene expression experiments, will have their proteins secreted into the bloodstream. Previous studies have identified a number of such genes that show abnormally high expression levels in patients of various cancers. For examples, a total of 26 and 57 genes were found to have abnormal expression levels, including both up-regulated and down-regulated in comparison with the normal cells (Supplementary Table 5), from studies on gastric cancer (Kim et al., 2005) and lung cancer (Lo et al., 2008). All these genes have been considered as potential markers for cancer diagnosis or for distinguishing different cancer stages, as shown in Supplementary Figure 1 from Lo et al. (2008). We have run our classifier on each of these genes to check if its encoded protein is predicted to be blood-secreted and thus can possibly serve as biomarkers for the corresponding cancer. Our prediction results show that 13 and 31 proteins out of the 26 and 57 proteins, respectively, can be secreted into the bloodstream. For example, complement factor D is encoded by the CFD gene. According to a quantitative analysis of factor D secretion by gastric cancer cells (Kitano and Kitamura, 2002), factor D secreted by gastric tissues is considered to likely contribute to the factor D level in blood circulation, which is consistent with our prediction. Another example is the multi-drug and toxin extrusion protein 2, encoded by gene MATE1 with elevated expression in gastric cancer patients. It is a solute transporter for tetraethylammonium (TEA), 1-methyl-4-phenylpyridinium (MPP), cimetidine and ganciclovir, and directly transports toxic organic cations OCs into urine and bile (Otsuka et al., 2005). Members of the MATE families are observed on the surface of various tissue cells including endothelial cells of blood vessels (Pardo et al., 2007). Thus, our prediction of this protein as being blood secreted is consistent with the previous knowledge.
Based on the results on multiple datasets presented above, we can see that the overall prediction accuracy of our SVM-based classifier ranges from 79.5% to 98.1%, with at least 80% of known blood-secreted proteins correctly predicted for both independent evaluation test and the extra blood proteins test. From the independent negative evaluation test, the false positive rate is found to be ∼10%, a reasonable percentage of misclassified non-blood-secreted proteins, which is helpful in alleviating the doubts of the low precision. The prediction accuracies show a good level of consistency across different datasets.
It should be noted that several factors may affect the accuracy of our prediction. One is the diversity of protein samples used for training the SVM-based classifier. It is likely that not all possible types of blood-secreted proteins are adequately represented in our training set. The current limitations in the proteomic technologies for precise separation, detection and identification of relevant proteins might explain why some of the proteins with relatively low abundance (lower than ng/ml in serum) are not detected when in the presence of the high-abundance native blood proteins (greater than mg/ml in serum). This apparent discrepancy can overcome with the accumulation of more proteins identified through more cancer studies focusing on proteins with low abundance in blood. Another problem is that the protein secretion mechanisms may not be sufficiently represented by the structural and physicochemical descriptors currently used in our classifier, leading to false predictions. Additional more informative descriptors (features) may be needed to alleviate this problem.
4 CONCLUDING REMARKS
We have developed a novel sequence-based approach to the classification of proteins into ‘blood-secreted’ and ‘non-blood-secreted’ proteins. Global and local characteristics of sequence-derived properties have been studied for their dominance and usefulness in predicting proteins that are likely to be possibly secreted into the bloodstream. A number of proteins encoded by abnormally and highly expressed genes in tumor cells are predicted to be blood-secreted proteins, and further investigation by blood tests can evaluate the potential of those proteins as serum biomarkers. We expect that this strategy will prove to be highly useful for marker protein identification for various human diseases including cancers.
Funding
National Science Foundation (DBI-0354771, ITR-IIS-0407204, CCF-0621700, DBI-0542119); National Institutes of Health (1R01GM075331); ‘Distinguished Scholar’ grant from the Georgia Cancer Coalition.
Conflict of Interest: none declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Yunmei Lu, Zhongbo Cao from Jilin University of China and Zhiyi Tong from CSBL (UGA) for their helpful discussions and their help in data collection.
REFERENCES
- Adkins JN, et al. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Mol. Cell Proteomics. 2002;1:947–955. doi: 10.1074/mcp.m200066-mcp200. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- Bateman A, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, et al. Prediction of twin-arginine signal peptides. BMC Bioinformatics. 2005;6:167. doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Hur A, Noble WS. Kernel methods for predicting protein-protein interactions. Bioinformatics. 2005;21(Suppl. 1):i38–i46. doi: 10.1093/bioinformatics/bti1016. [DOI] [PubMed] [Google Scholar]
- Bhasin M, Raghava GP. Classification of nuclear receptors based on amino acid composition and dipeptide composition. J. Biol. Chem. 2004;279:23262–23266. doi: 10.1074/jbc.M401932200. [DOI] [PubMed] [Google Scholar]
- Bosques CJ, et al. The sweet side of biomarker discovery. Nat. Biotechnol. 2006;24:1100–1101. doi: 10.1038/nbt0906-1100. [DOI] [PubMed] [Google Scholar]
- Bradford TJ, et al. Molecular markers of prostate cancer. Urol. Oncol. 2006;24:538–551. doi: 10.1016/j.urolonc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–1416. [PubMed] [Google Scholar]
- Buckhaults P, et al. Secreted and cell surface genes expressed in benign and malignant colorectal tumors. Cancer Res. 2001;61:6996–7001. [PubMed] [Google Scholar]
- Burbidge R, et al. Drug design by machine learning: support vector machines for pharmaceutical data analysis. Comput. Chem. 2001;26:5–14. doi: 10.1016/s0097-8485(01)00094-8. [DOI] [PubMed] [Google Scholar]
- Cai CZ, et al. SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003;31:3692–3697. doi: 10.1093/nar/gkg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, et al. SPD – a web-based secreted protein database. Nucleic Acids Res. 2005;33:D169–D173. doi: 10.1093/nar/gki093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, et al. Computer prediction of allergen proteins from sequence-derived protein structural and physicochemical properties. Mol. Immunol. 2007a;44:514–520. doi: 10.1016/j.molimm.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Cui J, et al. Advances in exploration of machine learning methods for predicting functional class and interaction profiles of proteins and peptides irrespective of sequence homology. Curr. Bioinformatics. 2007b;2:95–112. [Google Scholar]
- Doudna JA, Batey RT. Structural insights into the signal recognition particle. Annu. Rev. Biochem. 2004;73:539–557. doi: 10.1146/annurev.biochem.73.011303.074048. [DOI] [PubMed] [Google Scholar]
- Dubchak I, et al. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl Acad. Sci. USA. 1995;92:8700–8704. doi: 10.1073/pnas.92.19.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber F, et al. Prediction of secondary structural content of proteins from their amino acid composition alone. I. New analytic vector decomposition methods. Proteins. 1996;25:157–168. doi: 10.1002/(SICI)1097-0134(199606)25:2<157::AID-PROT2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Zhang CT. Prediction of membrane protein types based on the hydrophobic index of amino acids. J. Protein Chem. 2000;19:269–275. doi: 10.1023/a:1007091128394. [DOI] [PubMed] [Google Scholar]
- Guda C. pTARGET: a web server for predicting protein subcellular localization. Nucleic Acids Res. 2006;34:W210–W213. doi: 10.1093/nar/gkl093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Horton P, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S, Sun Z. A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach. J. Mol. Biol. 2001;308:397–407. doi: 10.1006/jmbi.2001.4580. [DOI] [PubMed] [Google Scholar]
- Huang LJ, et al. Proteomics-based identification of secreted protein dihydrodiol dehydrogenase as a novel serum markers of non-small cell lung cancer. Lung Cancer. 2006;54:87–94. doi: 10.1016/j.lungcan.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Keerthi SS, et al. Improvements to Platt's SMO algorithm for SVM classifier design. Neural Comput. 2001;13:637–649. [Google Scholar]
- Kim JH, et al. Osteopontin as a potential diagnostic biomarker for ovarian cancer. J. Am. Med. Assoc. 2002;287:1671–1679. doi: 10.1001/jama.287.13.1671. [DOI] [PubMed] [Google Scholar]
- Kim JM, et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin. Cancer Res. 2005;11:473–482. [PubMed] [Google Scholar]
- Kitano E, Kitamura H. Synthesis of factor D by gastric cancer-derived cell lines. Int. Immunopharmacol. 2002;2:843–848. doi: 10.1016/s1567-5769(02)00028-0. [DOI] [PubMed] [Google Scholar]
- Klee EW, Sosa CP. Computational classification of classically secreted proteins. Drug Discov. Today. 2007;12:234–240. doi: 10.1016/j.drudis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Lo KC, et al. Identification of genes involved in squamous cell carcinoma of the lung using synchronized data from DNA copy number and transcript expression profiling analysis. Lung Cancer. 2008;59:315–331. doi: 10.1016/j.lungcan.2007.08.037. [DOI] [PubMed] [Google Scholar]
- Mason SJ, Graham NE. Areas beneath the relative operating characteristics (ROC) and levels (ROL) curves: statistical significance and interpretation. Q. J. Roy. Meteorol. Soc. 2002;128:2145–2166. [Google Scholar]
- Menne KM, et al. A comparison of signal sequence prediction methods using a test set of signal peptides. Bioinformatics. 2000;16:741–742. doi: 10.1093/bioinformatics/16.8.741. [DOI] [PubMed] [Google Scholar]
- Mok SC, et al. Prostasin, a potential serum marker for ovarian cancer: identification through microarray technology. J. Natl Cancer Inst. 2001;93:1458–1464. doi: 10.1093/jnci/93.19.1458. [DOI] [PubMed] [Google Scholar]
- Mott R, et al. Predicting protein cellular localization using a domain projection method. Genome Res. 2002;12:1168–1174. doi: 10.1101/gr.96802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R, Rost B. Mimicking cellular sorting improves prediction of subcellular localization. J. Mol. Biol. 2005;348:85–100. doi: 10.1016/j.jmb.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Omenn GS, et al. Overview of the HUPO plasma proteome project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- Otsuka M, et al. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl Acad. Sci. USA. 2005;102:17923–17928. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo M, et al. Biomarker discovery from uveal melanoma secretomes: identification of gp100 and cathepsin D in patient serum. J. Proteome Res. 2007;6:2802–2811. doi: 10.1021/pr070021t. [DOI] [PubMed] [Google Scholar]
- Pieper R, et al. The human serum proteome: display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics. 2003;3:1345–1364. doi: 10.1002/pmic.200300449. [DOI] [PubMed] [Google Scholar]
- Platt JC. Advances in Kernel Methods: Support Vector Learning. Cambridge, MA, USA: MIT Press; 1999. Fast training of support vector machines using sequential minimal optimization; pp. 185–208. [Google Scholar]
- Reczko M, Bohr H. The DEF data base of sequence based protein fold class predictions. Nucleic Acids Res. 1994;22:3616–3619. [PMC free article] [PubMed] [Google Scholar]
- Rui Z, et al. Use of serological proteomic methods to find biomarkers associated with breast cancer. Proteomics. 2003;3:433–439. doi: 10.1002/pmic.200390058. [DOI] [PubMed] [Google Scholar]
- Schrader M, Schulz-Knappe P. Peptidomics technologies for human body fluids. Trends Biotechnol. 2001;19:S55–S60. doi: 10.1016/S0167-7799(01)01800-5. [DOI] [PubMed] [Google Scholar]
- Smialowski P, et al. Protein solubility: sequence based prediction and experimental verification. Bioinformatics. 2007;23:2536–2542. doi: 10.1093/bioinformatics/btl623. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB. Autocrine growth factors and cancer. Nature. 1985;313:745–747. doi: 10.1038/313745a0. [DOI] [PubMed] [Google Scholar]
- Su EC, et al. Protein subcellular localization prediction based on compartment-specific features and structure conservation. BMC Bioinformatics. 2007;8:330. doi: 10.1186/1471-2105-8-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZQ, et al. Derivation of stable microarray cancer-differentiating signatures using consensus scoring of multiple random sampling and gene-ranking consistency evaluation. Cancer Res. 2007;67:9996–10003. doi: 10.1158/0008-5472.CAN-07-1601. [DOI] [PubMed] [Google Scholar]
- Taylor PD, et al. TATPred: a Bayesian method for the identification of twin arginine translocation pathway signal sequences. Bioinformation. 2006;1:184–187. doi: 10.6026/97320630001184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H, et al. Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol. Mol. Biol. Rev. 2000;64:515–547. doi: 10.1128/mmbr.64.3.515-547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin RD, et al. Serological and proteomic evaluation of antibody responses in the identification of tumor antigens in renal cell carcinoma. Proteomics. 2003;3:45–55. doi: 10.1002/pmic.200390008. [DOI] [PubMed] [Google Scholar]
- Welsh JB, et al. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc. Natl Acad. Sci. USA. 2001;98:1176–1181. doi: 10.1073/pnas.98.3.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JB, et al. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc. Natl Acad. Sci. USA. 2003;100:3410–3415. doi: 10.1073/pnas.0530278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.