

Abstract
Motivation: Searching genomes for non-coding RNAs (ncRNAs) by their secondary structure has become an important goal for bioinformatics. For pseudoknot-free structures, ncRNA search can be effective based on the covariance model and CYK-type dynamic programming. However, the computational difficulty in aligning an RNA sequence to a pseudoknot has prohibited fast and accurate search of arbitrary RNA structures. Our previous work introduced a graph model for RNA pseudoknots and proposed to solve the structure–sequence alignment by graph optimization. Given k candidate regions in the target sequence for each of the n stems in the structure, we could compute a best alignment in time O(ktn) based upon a tree width t decomposition of the structure graph. However, to implement this method to programs that can routinely perform fast yet accurate RNA pseudoknot searches, we need novel heuristics to ensure that, without degrading the accuracy, only a small number of stem candidates need to be examined and a tree decomposition of a small tree width can always be found for the structure graph.
Results: The current work builds on the previous one with newly developed preprocessing algorithms to reduce the values for parameters k and t and to implement the search method into a practical program, called RNATOPS, for RNA pseudoknot search. In particular, we introduce techniques, based on probabilistic profiling and distance penalty functions, which can identify for every stem just a small number k (e.g. k ≤ 10) of plausible regions in the target sequence to which the stem needs to align. We also devised a specialized tree decomposition algorithm that can yield tree decomposition of small tree width t (e.g. t ≤ 4) for almost all RNA structure graphs. Our experiments show that with RNATOPS it is possible to routinely search prokaryotic and eukaryotic genomes for specific RNA structures of medium to large sizes, including pseudoknots, with high sensitivity and high specificity, and in a reasonable amount of time.
Availability: The source code in C++ for RNATOPS is available at www.uga.edu/RNA-Informatics/software/rnatops/
Contact: cai@cs.uga.edu
Supplementary information: The online Supplementary Material contains all illustrative figures and tables referenced by this article.
1 INTRODUCTION
Non-coding RNAs (ncRNAs) have been shown to be involved in many biological processes including gene regulation, chromosome replication and RNA modification (Frank and Pace, 1998; Nguyen et al., 2001; Yang et al., 2001). Searching genomes using computational methods has become important for annotation of ncRNAs (Griffiths-Jones, 2007; Hofacker, 2006; Lowe and Eddy, 1997; Rivas and Eddy, 2001; Rivas et al., 2001; Washietl et al., 2005). In general, to annotate an individual genome for a specific family of ncRNAs, a computational tool needs to scan through the genome and align its sequence segments to some structure model for the ncRNA family. Those segments with significant alignment scores are then reported as the results. An algorithm that can perform an accurate sequence–structure alignment is thus the core of such a searching tool.
A few programs (Brown and Wilson, 1996; Klein and Eddy, 2003; Liu et al., 2006; Lowe and Eddy, 1997) have been developed for genome annotation using the covariance model CM introduced by Eddy and Durbin (1994). Based on a CM, the optimal alignment between a sequence and a pseudoknot-free structure can be performed with a dynamic programming algorithm in O(W N3), where N is the size of the model and W is the length of the sequence. In particular, RSEARCH (Klein and Eddy, 2003) and Infernal (http://infernal.janelia.org/) are two programs that can perform such searches. CM-based methods can achieve high searching accuracy; however, due to the time complexity needed for sequence–structure alignment, a CM-based search may be inefficient on complex or large RNA structures. Further, pseudoknot structures, which contain at least two interweaving stems, cannot be modeled with CMs.
Searches on genomes can be speeded up with filtering methods (Bafna and Zhang, 2004; Lowe and Eddy, 1997; Weinberg and Ruzzo, 2004, 2006; Zhang et al., 2005). Sometimes it is possible to efficiently remove genome segments unlikely to contain the desired pattern. For example, in tRNAscan-SE (Lowe and Eddy, 1997), two efficient filters are used to preprocess a genome and remove the part that is unlikely to contain the searched tRNA structure; the remaining part of the genome is then scanned with a CM to identify the tRNA. FastR (Bafna and Zhang, 2004) considers the structural units of an RNA structure; it evaluates the specificity of each structural unit and construct filters based on the specificity of these structural units. In Weinberg and Ruzzo (2004), an algorithm is developed to safely break the base pairs in an RNA structure and automatically select filters from the resulting Hidden Markov Model (HMM). These approaches have significantly improved the computational efficiency of genome searches.
RNA structures that contain pseudoknots pose special problems. A number of creative approaches (Cai et al., 2003; Rivas and Eddy, 1999, 2000; Uemura et al., 1999) have been tried to model the crossing stems of pseudoknots; however, the time and space complexities for optimal sequence–structure alignment based on these models are O(N4) or O(N5). These models are not practical for efficient searching. Intersecting CMs have been proposed for pseudoknots (Brown and Wilson, 1996), and used to search small genomes (Liu et al., 2006), but these have the same efficiency problem. Several heuristic search methods have been developed that can work with RNAs containing pseudoknots; as heuristics, each has some limitations. For example, ERPIN (Gautheret and Lambert, 2001), considers the stem loops contained in a secondary structure. The genome is then scanned to find the possible hit locations for each stem loop. A hit for the overall structure is reported when there exists a combination of hit locations for different stem loops that conform with the overall structure. However, ERPIN does not allow gaps in the alignment and thus may have low sensitivity when the target is a remote homolog of the query structure model.
Our previous work introduced a graph modeling method that can profile the secondary structure of a family of RNAs including pseudoknots (Song et al., 2005, 2006). In this method, the topology of an RNA structure is specified with a mixed graph, with non-directed edges denoting stems and directed edges for loops. With this model, we proposed to efficiently solve the structure–sequence alignment problem, including pseudoknots, by exploiting the small tree width (Robertson and Seymour, 1986) demonstrated by the structure graphs of almost all existing RNA pseudoknots. Theoretically, given k (pairs of) regions as candidates for each of the n stems in the structure and given a tree decomposition of tree width t for the structure graph, the alignment can be computed in time O(ktn). However, to implement the algorithm into computer programs that can routinely perform fast, accurate RNA pseudoknot search, heuristics for the preprocessing steps need to be able to associate results with small values of parameters k and t while maximizing search accuracy.
In this article, we present our current work, built upon the previous one, to develop a practical program, called RNATOPS, for RNA pseudoknot search. In this work, we have introduced new, effective heuristic techniques for generating stem candidates and for tree decomposition of RNA structure graphs. In particular, parameter k can be chosen relatively small (e.g. k ≤ 10) to ensure both accuracy and efficiency of the search. The alignment algorithm (and thus the search algorithm) runs in time O(ktn), linear in the number n of stems in the profiled RNA structure. It is scalable with the complexity of the profiled structure because the yielded tree decompositions have small tree width t, t ≤ 4, for almost all RNA secondary structures (including pseudoknots). In this article, we evaluate RNATOPS with search tests conducted on several medium to large size RNAs (including pseudoknots) and make comparisons with existing RNA structure search programs such as Infernal.
2 APPROACH
We refer the reader to the publications (Song et al., 2005, 2006) for detailed discussions of our graph modeling method for RNA structures and on the solution to structure–sequence alignment based on tree decomposition of the structure graph. In this section, we give a brief recap of the necessary notions and techniques relevant to the current article. We then present the new heuristic techniques for stem candidate identification and for tree decomposition designated for RNA structure graphs. These heuristic techniques aim at achieving a fast structure–sequence alignment without degrading the accuracy.
2.1 A graph model for structure search
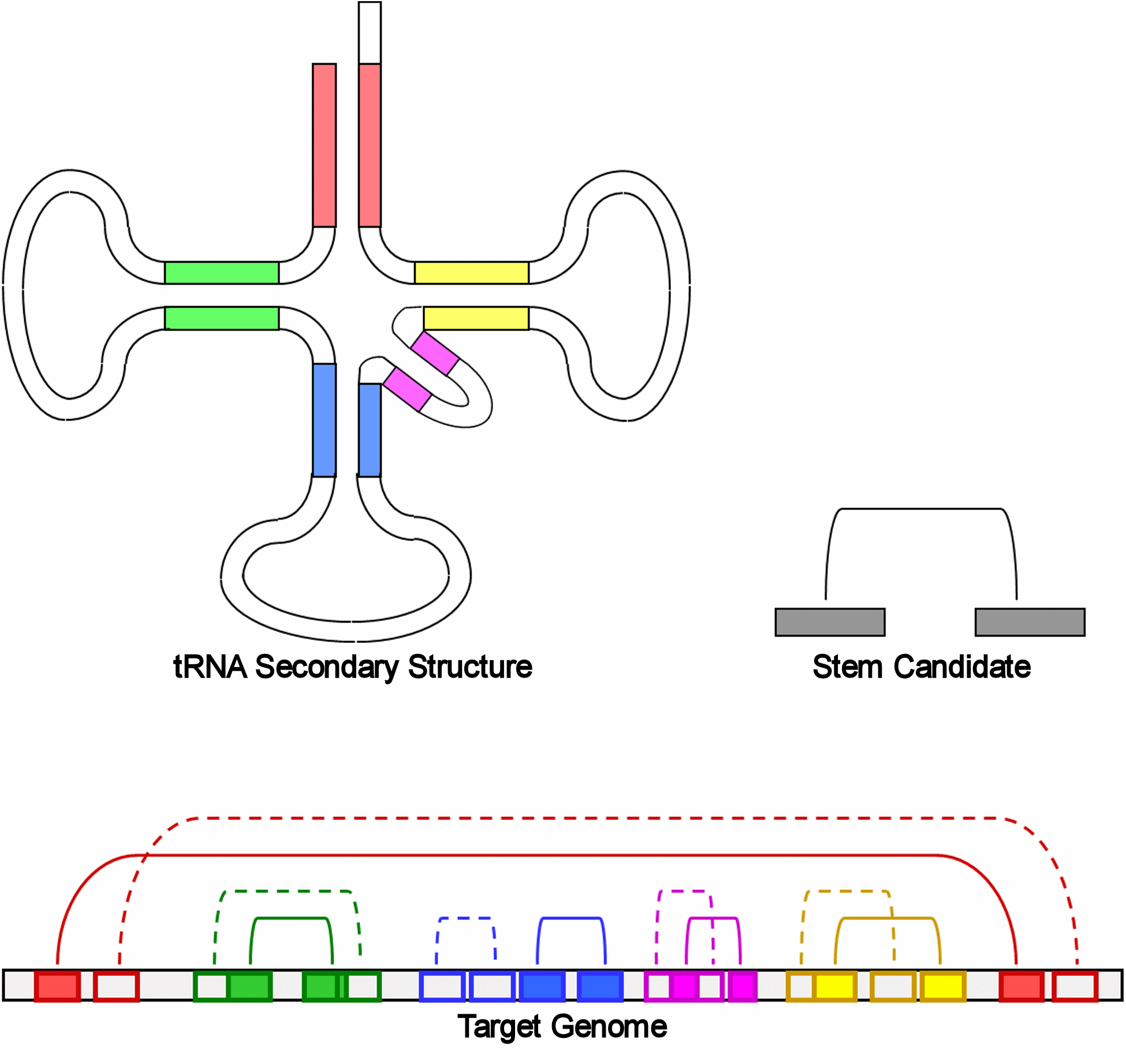
Our structure model based on a mixed graph specifies the consensus structure of an RNA family as a relation among all involved structural units: stems and loops. In this graph, each vertex defines either base pairing regions of a stem; two vertices representing two complementary regions (forming a stem) are connected with a non-directed edge. Two vertices defining two regions that are physically next to each other (forming a loop) are connected with a directed edge (from 5′ to 3′). The individual structural units are stochastically modeled; every stem is associated with a simplified CM and every loop with a profile HMM. The structure graph is capable of modeling RNA structures resulting from multi-body interactions of nucleotides, such as triple helices, as well as pseudoknots. Figure 1 in Supplementary Material shows the structure graph of a typical bacterial tmRNA.
Searching in a target genome consists of sliding a window of appropriate size along the target genome, then testing for a possible alignment of the structural model with the sequence segment within the current window. With the graph model, the structure–sequence alignment is identical to the task of finding the optimal subgraph of a graph G isomorphic to another graph H, where H is the RNA structure graph and G is constructed from the target sequence in a preprocessing step. We proposed two methods to cope with the computational intractability of the subgraph isomorphic problem. One method was to pre-identify in the target sequence top k candidates for every stem in the structure. The other method was to tree decompose the structure graph. Based upon a tree decomposition, a dynamic programming algorithm could solve the subgraph isomorphic (thus the structure–sequence) problem in theoretical time O(ktn), where n is the number of stems in the structure and t is the tree width of the graph tree decomposition (Song et al., 2005, 2006). This article presents new heuristic techniques to support these two methods.
2.2 Model training
Model training involves defining the structure graph, individual CMs and profile HMMs from a set of training RNA sequences given in a pasta file. The pasta format (pairing plus fasta) is a representation we developed for multiple structural alignment and consensus structure of RNA sequences (Fig. 2 in Supplementary Material). It labels stem positions with an upper case letter for one side, the corresponding lower case letter for the other side. The first line of the file denotes the consensus structure using matching (upper and lower case) letters for conserved base pairs and ‘.’s for unpaired nucleotides or possibly consensus insertions. Representation with pairing letters has the advantage of being able to denote arbitrary RNA structures, including pseudoknots and triple helices. A structure graph is produced from the consensus structure, where one vertex is for one letter, one non-directed edge connects the two vertices of matched letters and one directed edge connects two neighboring letters (from 5′ to 3′, Fig. 1 in Supplementary Material).
The rest of the lines in the pasta file are RNA sequences structurally aligned to the consensus structure, possibly containing ‘-’s for deletions. Individual CMs and profiles HMMs are constructed from the multiple structure alignment as follows. Every stem of base-paired regions (with matching letters) produces one simplified CM that does not contain bifurcation rules or rules for the sequence connecting the two base-paired regions. One profile HMM is generated from every two neighboring base regions. The profile HMM allows possible match, insertion and deletion states in every column of the multiple alignment. The parameters of these stochastic models are computed from the multiple structural alignment using the maximum likelihood method. To avoid over-fitting the models, we incorporate background statistics. In particular, we allow pseudocounts for nucleotides in the match, insertion and deletion states of the profile HMM. For the simplified CM, a 4 × 4 prior probability matrix Pp for base pairs and a weighting parameter w are introduced so that the probability of a base pair P(x, y) is defined as the weighted sum wPt (x, y)+(1−w) Pp(x, y), where Pt is the base pair probability matrix obtained from the training data.
2.3 Identifying stem candidates
The sequence segment within the sliding window is preprocessed to identify top k candidates for the CM of every stem. Given a CM modeling some consensus stem, the score of every possible structural motif within the window aligned to the model is computed (Fig. 3 in Supplementary Material). Candidates can be found by a simple dynamic programming algorithm; we describe here four heuristic techniques developed to ensure that the correct motif structure for the CM, if it does exist in the sequence, is highly likely to be among the selected top k candidates for some small value of k.
Regions from which candidates can be selected are constrained according to the statistical distribution of the consensus stem in the sample (training sequences). In particular, we assume a Gaussian distribution for the position of the consensus stem in the RNA structure. The constrained region for the correct motif of the consensus stem is within a certain number (e.g. 3) of the SD of the average position.
For training sequences that demonstrate a large SD for the position of some consensus stem, training sequences are partitioned into clusters, each with a small SD for the stem position. Therefore, more than one (constrained) region may be derived for the correct motif of the consensus stem.
The candidates so identified are then ranked again according to statistical distributions of various length parameters associated with a consensus stem, including the length of the stem, the distance between the two stem arms and the head and tail offsets. The scores of every possible motif candidate c of the CM M are recalculated according to the formula: S(c, M)=uA(c, M)+(1−u)P(c, M), where A(c, M) is the logodds score from the alignment, P(c, M) is the penalty function for the deviations of all lengths list above from their means and u, 0≤u≤1, is a weighting parameter. In particular, P(c, M) is computed based on the log score log(1/cK2), where K=|l−μ|/σ ≥ 1 for the length l deviating from mean μ (with a SD σ) and c is a selected constant.
Finally, since it is possible that several structural motifs, heavily overlapping in their positions, may all have decent alignment scores with respect to a stem model, it suffices to record only one representative for them. Strategies have been used to select representatives and to ensure a low value for k, the number of top candidates.
2.4 Tree decomposition for structure graphs
With our model, almost all ncRNAs have structure graphs of small tree width. However, finding the optimal tree decomposition (one with the smallest tree width) is NP-hard. Available efficient tree decomposition algorithms are for general graphs and usually do not guarantee the optimal tree width. For RNA structure graphs, we develop a linear-time greedy algorithm that can yield tree decomposition of tree width almost always bounded by 4. An earlier version of this algorithm was given in (Song et al., 2005), but it used the idea of minimum fill-in and may produce decompositions of unnecessarily larger tree widths. We present a self-contained version of the algorithm here.
First, the algorithm removes arcs (i.e. non-directed edges) in the structure graph that cross with other arcs. It does this by greedily removing the arc crossing the most other arcs and repeating the step on the remaining graph until there is no crossing arc in the graph (Fig. 4a and b in Supplementary Material). This step actually removes stems involved in pseudoknots in the corresponding RNA structure; a crossing arc-free structure graph corresponds to a pseudoknot-free RNA structure. Such a graph is an outer-planar graph that has tree width 2, whose optimal tree decomposition can be found as follows.
Note that in a structure graph, the vertices are arranged in the direction of from 5′ to 3′ (left to right in the figures) based on the directed edge relation. We also add the source s and sink t as the left most and the right most vertices, respectively. We use notation Hab to represent the subgraph induced by the set of vertices ‘from’ vertex a ‘to’ vertex b (inclusive, from 5′ to 3′). Then to decompose the subgraph Hst, the algorithm handles the following three major scenarios recursively (and the recursive process terminates when the considered subgraph is empty).
If (s, X) is a directed edge but (x, t) is not, where (X, x) is an arc (Fig. 5a in Supplementary Material), then the root node {s, t} has child node {s, x, t}, which in turn has child node {s, X, x} (Fig. 5b in Supplementary Material). Node {s, X, x} will be the root for the subtree generated from subgraph HXx and node {s, x, t} will be the root for the subtree generated from subgraph Hxt.
If (s, X) and (x, t) both are directed edge, where (X, x) is an arc (Fig. 5c in Supplementary Material), then the root {s, t} has child node {s, X, t}, which in turn has child node {X, x, t}. Node {X, x, t} will be the root for the subtree generated from subgraph HXx (Fig. 5d in Supplementary Material).
If (s, X) is a directed edge but (X, x) is not an arc (Fig. 5e in Supplementary Material), then the root {s, t} has a child node {s, X, t}, which in turn will be the root for the subtree generated from subgraph HXt (Fig. 5f in Supplementary Material).
The algorithm modifies the resulting tree decomposition as follows. For every removed arc (v, v′), the algorithm identifies two nodes, one containing vertex v and another containing its counterpart v′. For every tree node on the path from the former node to the latter, the algorithm adds v to it (Fig. 6 in Supplementary Material). This gives a tree decomposition for the original structure graph.
3 IMPLEMENTATION
RNATOPS, implemented in language C++, has been compiled and tested on several systems, including Desktop Linux computers, a Linux cluster and a SUN workstation running SunOS 5.1.
4 EVALUATION
To evaluate the search program and the effective of the heuristics, we tested RNATOPS using four types of RNAs of medium to large sizes: bacterial tmRNA, bacterial RNaseP type B RNA, yeast telomerase RNA and bacterial 16S rRNA. We compare both search accuracy and efficiency of RNATOPS with those of Infernal and FastR, two of the best known general-purpose programs for RNA structure search.
4.1 Data preparation and tests conducted
Bacterial tmRNAs (Moore and Sauer, 2007; Nameki et al., 1999) have a complex structure containing four pseudoknots; there are 178 molecules in the Rfam (Griffiths-Jones et al., 2005) seed alignment with an average length of 364 bases (Fig. 1 in Supplementary Material). The tmRNA sequences have variations in structure with certain stem loops present in some sequences and absent in others. We extracted a subset of 43 tmRNA sequences from the 178 molecules in the alignment, which did not differ from each other in the presence or absence of any stem loops, and for which the entire bacterial genome sequence was available; columns consisting entirely of gaps were then removed from the alignment.
RNaseP, bacterial type B, RNAs have multiple stem loops and one sophisticated pseudoknot (Brown, 1999; Harris et al., 2001; Fig. 7 in Supplementary Material). There are 31 sequences of average length 367 in the Rfam seed alignment. We extracted a subset of 10 sequences which did not differ from each other in the presence or absence of any stem loops; the full genome sequence was available for 7 of the 10.
Yeast telomerase RNAs contain a conserved, essential, pseudoknot within a large stem loop (Chen and Greider, 2004). We used an alignment, of length 834, for this region (Dandjinou et al., 2004) of six Saccharomyces species telomerase RNAs. While the genome of S.cerevisiae has been completely sequenced, those of the other Saccharomyces species are available in varying degrees of completeness and assembly. We were able to collect four Saccharomyces genomes total, three in addition to S. cerevisiae, to search.
The bacterial 16S rRNA is a conserved molecule which has been extensively used for phylogenetic studies of bacteria. We obtained an alignment (of 1570 bp) of the 16S rRNA for gammaproteobacteria from the ribosomal database (Cole et al., 2007); from this we selected those sequences which contained an identical match in a fully sequenced bacterial genome. Although many gammaproteobacteria genomes have been sequenced, for only 12 was there an exact match between the database sequence and a genomic sequence, which we required to take advantage of the expert alignment from the database. These sequences were used as the training set.
For all the genomic searches, we followed a cross-validation approach in which the RNA found in a genomic sequence was removed from the alignment, and the remaining sequences were used as a training set for a search on that genome.
To search genomes of a considerable length, we identified highly conserved motifs of the RNA molecules, then searched the genomes with these, after which we examined the region around a potential hit for a structural match to the whole molecule. We note that a program that can automatically identify a conserved motif as the optimal filter is currently being developed for RNATOPS.
4.2 Comparison to other search programs
To compare with Infernal (infernal.janelia.org), we downloaded Infernal from its website, compiled it and installed it, and compared its performance on one of the same Linux computers we used for testing of RNATOPS. Both Infernal and RNATOPS use multiple structural alignments for model training and use filters to speed up the search.
We used FastR (Bafna and Zhang, 2004; Zhang et al., 2005) through job submission at a website. As such, it is difficult for us to compare the performance of FastR on a server of unknown configuration and numbers of cpus with the performance of RNATOPS. During the times we tested it, our analyses were the only ones listed in the job queue. We estimated the time of the run from the time of submission and the time at which the job finished e-mail was sent. The user can pick from pre-defined profiles for searching. It is unknown to us if these profiles included the tmRNAs for the genomes we tested. Hence these FastR tests may or may not correspond to the training sets we used, in which we left out the RNAs for the genome targeted for searching.
4.3 Search accuracy
4.3.1 Bacterial tmRNAs
We searched 43 bacterial genomes with RNATOPS for tmRNAs using a leave-one-out cross-validation approach. Table 1 in Supplementary Material gives a comparison of the results achieved with RNATOPS with those of Infernal. RNATOPS was evaluated with varying parameter k, the number of candidate regions examined for each stem in the structure. Increasing k from 10 to 15 to 25 increased the sensitivity of the whole structure search, but also increased the time taken. For example, at k=10, the bacterial genome searches gained 88% sensitivity and 100% specificity; at k=25, the sensitivity increased to 98%. Infernal had 100% sensitivity and specificity for these searches with comparable times spent.
We observed that the tmRNAs missed by RNATOPS at the low k values generally had one or more portions in the structure, which significantly deviated from the consensus structure. In particular, several stems in these sequences consisted of mainly rare, non-canonical base pairs, which may have been placed in pairing positions during the multiple alignment process.
We also compared the alignments of the tmRNA structures found by Infernal and RNATOPS. Four structures identified by RNATOPS have stem alignments off their correct positions for more than a few nucleotides in their alignments; Infernal identified seven such structures. There are in total nine such stem misalignments in the structures identified by RNATOPS; there were total 17 in those structures identified by Infernal. In addition, because Infernal is based on the pseudonot-free CM, in a structure alignment, regions ‘belonging to’ a pseudoknot may be mistakenly aligned to pseudoknot-free substructures. In particular, in this set of search tests, there were totally five such incorrect assignments found in the search results of Infernal while the issue was not raised on RNATOPS (Table 6 in Supplementary Material).
We also tried the search with FastR web server, which includes tmRNAs as a profile. We selected one bacterial genome on which RNATOPS successfully found the tmRNA, and one genome on which RNATOPS failed to find the tmRNA, then submitted these to the FastR server. FastR gave the same results as RNATOPS with these two sequences, finding the structure in one sequence and missing it in the other (Table 2 in Supplementary Material), again suggesting there is something unusual about the tmRNA that both programs missed. Several additional bacterial genomes were submitted to the FastR server, but no results were returned.
4.3.2 Bacterial RNaseP (Bact. B) RNAs
The bacterial RNaseP (Bact. B) RNA is similar in size to the tmRNAs, but has a more complex pseudoknot structure. Both the RNATOPS and Infernal programs had 100% sensitivity and 100% specificity in finding the RNaseP RNAs in the seven genomes tested (Table 3 in Supplementary Material); RNATOPS identified two structures whose alignments put in total four stems off their correct positions by more than a few nucleotides (Table 6 in Supplementary Material). A comparison with the tmRNA results suggests that the more complex pseudoknot structure in RNaseP (Bact. B) was handled well by RNATOPS, with less than a doubling in time taken for similar sized genomes, while Infernal took about nine times as long.
4.3.3 Saccharomyces telomerase RNAs
The conserved core region of Saccharomyces telomerase RNAs is more than twice as long as the bacterial tmRNAs or RNaseP RNAs, and the Saccharomyces genomes are 2 to 10 times larger than the bacterial genomes tested. The pseudoknot structure itself is not complex, but it is contained within a stem-loop and some additional stem-loops are present. Both programs found the four Saccharomyces fungal telomerase RNAs perfectly in their genomes; RNATOPS took from 5.5 to 6.4 min, while Infernal took from 295 to 654 min for the same searches (Table 4 in Supplementary Material).
4.3.4 Bacterial 16S rRNAs
The bacterial 16S rRNAs are the longest molecule we tested with lengths around 1500 bp. The results were similar to the telomerase and RNAseP RNAs, with both RNATOPS and Infernal finding the target with perfect specificity and sensitivity, but with RNATOPS performing the search in an average of 14.1 min as opposed to 88 min for Infernal (Table 5 in Supplementary Material).
4.4 Efficiency
The theoretical time of the search method can be expressed as O(TaN), where Ta is the time needed for the structure alignment between the structure model and the sequence segment within the window sliding through the genome of N nucleotides. Ta actually consists of two parts: the time for the preprocessing step and the time for the dynamic programming step for the subgraph isomorphism based upon a tree decomposition. The latter takes O(ktn) time, where t, usually not >4, is the tree width of the tree decomposition and n is the number of stems in the structure. Recall that k is the number of candidates selected for the simplified CM model of a stem during the preprocessing; it is a relatively small parameter that can be used to tune the accuracy of the alignment. The time for the preprocessing step is O(R2Mn), where M is the maximum size of a CM and R is the maximum length of the sequence regions from which candidates are selected. These regions are fairly restricted by the preprocessing techniques we introduced here (Section 2.3). Our experiments showed that the preprocessing time O(R2Mn) is roughly the same as the time O(ktn) needed for the dynamic programming step when k is around 10 and that it is dominated by the latter for larger values of k or t. So the time for searching a whole genome is very much scalable with the size and complexity of the RNA structure searched.
Overall, our results indicate that the RNA graph model plus tree decomposition method incorporated into RNATOPS performed very well in efficiency while maintaining high search accuracy. The advantage of RNATOPS in speed, compared to other programs, increased as the length of the modeled molecule increased. This is because its search time depends on the number of stems, not the number of nucleotides, in the structure. Thus, the efficiency advantage becomes even more significant for RNATOPS to search for the larger yeast telomerase RNA and bacterial 16S rRNA (Tables 4 and 5 in Supplementary Material). Note that RNATOPS search accuracy can be tuned by the user through parameter k, to balance search sensitivity versus running time. The problems that RNATOPS had, where target RNAs were not found, were in stem-loop regions of tmRNAs where individual molecules deviated from the consensus structure; increasing the k value allowed RNATOPS to resolve most of these, at the cost of a slight decrease in speed.
5 DISCUSSION
Heuristic techniques have been presented in this article with the aim to develop a fast and accurate RNA pseudoknot search program based on our previous work in an RNA graph modeling method. Through search tests on the implemented program RNATOPS, we have shown its performance comparable with or better than that of Infernal and FastR in identifying large or complex RNA structures including pseudoknots. We discuss in the following the strengths and weaknesses of RNATOPS.
One apparent advantage of RNATOPS is its ability to detect pseudoknots accurately without compromising computation time. Theoretically, RNATOPS can feasibly consider all combinations of stems for pseudoknot alignment through a non-conventional, tree decomposition-based dynamic programming. Detecting a pseudoknot as a whole structure avoids the difficulty with pseudoknot-free models that the predicted alignment sometimes incorrectly forms pseudoknot-free substructures in a ‘pseudoknot territory’.
Another advantage of RNATOPS is its search speed. The theoretical time O(ktn) for structure–sequence alignment with RNATOPS has been effectively speeded up by the introduced heuristic techniques that can yield small values for k and t. Another important factor contributing to the efficient time is parameter n, the number of stems, not the number of nucleotides in the structure which would otherwise be at least one magnitude larger. As shown in the test results, RNATOPS has essentially broken the inefficiency barrier that might have heldback other pseudoknot detection models, reducing the computation time from hours to minutes.
Nevertheless, since the introduced heuristics produce only k pairs of candidate regions for each individual stem in the structure to align to, for a small k, they may not include the real candidate of the stem and may bring inaccuracy to the search result. In particular, when a stem in the RNA contains non-canonical base pairings, for which candidates may not be accurately identified, it is possible that all pairs of candidates between this stem and another are ‘incompatible’, resulting in a invalid alignment and lower sensitivity. This issue does not exists in the CM–CYK-based programs like Infernal as its stem candidates are found globally instead of locally.
Another issue with the current version of RNATOPS is the computation of the structure–sequence alignment without reusing the data from the previous scanning window frame. In fact, the CM–CYK-based search method can save a factor of O(M) computation time by reusing data between two consecutive window frames (Durbin et al., 1998), where M is the CM model length. This issue might have cost RNATOPS some speed in the search tests; however, we believe that it is possible to make technical improvements for RNATOPS in reusing the data between scanning window frames to further speed up the search.
We consider two future developments for RNATOPS. First, the graph model can also easily profile structures caused by nucleotide interactions beyond the binary base pairing. For example, the graph model makes it easy to profile tertiary interactions or triple helices recently found in the telomerase RNA genes of human and yeast genomes (Chen and Greider, 2004; Lin et al., 2004; Shefer et al., 2007; Theimer et al., 2005). Although one of the two stems involved in such a triple helix is actually formed by two base pairing regions that are arranged in the same direction (5′ to 3′), our approach will allow the stem to be modeled with an individual CM the same way as modeling a regular stem, without the need of additional, new techniques.
Second, the current implementation of program does not allow the search for an instance of ncRNA in the target genome that differs in structure significantly from those in the training set; nor can the current program consider alternative or optional substructures in RNAs. One solution to this will be to develop probabilistic profiling of variable substructures that may occur in the structure model. In particular, our modeling method makes it possible to characterize and implement the structure of an RNA family with a graph model that contains probabilistic edges to specify variable substructures. This will bear similarity to earlier methods by Holmes (2004) and Rivas (2005) but with the ability to include pseudoknots.
Funding
National Institutes of Health (BISTI grant No: R01GM072080-01A1).
Conflict of Interest: none declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the anonymous referees for their constructive comments that have greatly improved this manuscript. We also thank the authors of Infernal, RSEARCH and FastR who have made their software packages publicly available to use.
REFERENCES
- Bafna V, Zhang S. Proceedings of the 3rd IEEE Computational Systems Bioinformatics Conference. London: Imperial College Press; 2004. FastR: fast database search tool for non-coding RNA; pp. 52–61. [DOI] [PubMed] [Google Scholar]
- Brown JW. The Ribonuclease P database. Nucleic Acids Res. 1999;27:314. doi: 10.1093/nar/27.1.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Wilson C. RNA pseudoknot modeling using intersections of stochastic context free grammars with applications to database search. In: Hunter L, Klein T, editors. Proceedings of Pacific Symposium on Biocomputing. Singapore: World Scientific Publishing Co; 1996. [PubMed] [Google Scholar]
- Cai L, et al. Stochastic modeling of RNA pseudoknotted structures: a grammatical approach. Bioinformatics. 2003;19(Suppl. 1):i66–i73. doi: 10.1093/bioinformatics/btg1007. [DOI] [PubMed] [Google Scholar]
- Chen L, Greider CW. An emerging consensus for telomerase RNA structure. Proc. Natl Acad. Sci. USA. 2004;101:14683–14684. doi: 10.1073/pnas.0406204101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35:D169–D172. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandjinou AT, et al. A phylogenetically based secondary structure for the yeast telomerase RNA. Curr. Biol. 2004;14:1148–1158. doi: 10.1016/j.cub.2004.05.054. [DOI] [PubMed] [Google Scholar]
- Durbin R, et al. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press; 1998. [Google Scholar]
- Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, Pace NR. Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Annu. Rev. Biochem. 1998;67:153–180. doi: 10.1146/annurev.biochem.67.1.153. [DOI] [PubMed] [Google Scholar]
- Gautheret D, Lambert A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol. 2001;313:1003–1011. doi: 10.1006/jmbi.2001.5102. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S. Annotating noncoding RNA genes. Annu. Rev. Genomics Hum. Genet. 2007;8:279–298. doi: 10.1146/annurev.genom.8.080706.092419. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, et al. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JK, et al. New insight into RNase P RNA structure from comparative analysis of the archaeal RNA. RNA. 2001;7:220–232. doi: 10.1017/s1355838201001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL. RNAs everywhere: geonom-wide annotation of structured RNAs. Genome Inform. 2006;17:281–282. [PubMed] [Google Scholar]
- Holmes I. A probabilistic model for the evolution of RNA structure. BMC Bioinformatics. 2004;5:166. doi: 10.1186/1471-2105-5-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infernal: inference of RNA alignments. (2008)http://infernal.janelia.org/(last accessed date June 30, 2008). [Google Scholar]
- Klein RJ, Eddy SR. RSEARCH: finding homologs of single structured RNA sequences. BMC Bioinformatic. 2003;4:44. doi: 10.1186/1471-2105-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, et al. A universal telomerase RNA core structure including structured motifs required for binding the telomerase reverse transcriptase protein. Proc. Natl Acad. Sci. USA. 2004;101:14713–14718. doi: 10.1073/pnas.0405879101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, et al. Proceedings of Life Science Society Computational Systems Biology Conference (CSB 2006) London: Imperial College Press; 2006. Efficient annotation of non-coding RNA structures including pseudoknots via automated filters; pp. 99–110. [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SD, Sauer RT. The tmRNA system for translational surveillance and ribosome rescue. Annu. Rev. Biochem. 2007;76:101–124. doi: 10.1146/annurev.biochem.75.103004.142733. [DOI] [PubMed] [Google Scholar]
- Nameki N, et al. Functional and structural analysis of a pseudoknot upstream of the tag-encoded sequence in E. coli tmRNA. J. Mol. Biol. 1999;286:733–744. doi: 10.1006/jmbi.1998.2487. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, et al. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- Rivas E. Evolutionary models for insertions and deletions in a probabilistic modeling framework. BMC Bioinformatic. 2005;6:63. doi: 10.1186/1471-2105-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- Rivas E, Eddy SR. The language of RNA: a formal grammar that includes pseudoknots. Bioinformatics. 2000;16:334–340. doi: 10.1093/bioinformatics/16.4.334. [DOI] [PubMed] [Google Scholar]
- Rivas E, Eddy SR. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatic. 2001;2:8. doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E, et al. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 2001;11:1369–1373. doi: 10.1016/s0960-9822(01)00401-8. [DOI] [PubMed] [Google Scholar]
- Robertson N, Seymour PD. Graph minors II. Algorithmic aspects of tree-width. J. Algorithms. 1986;7:309–322. [Google Scholar]
- Shefer K, et al. A triple helix within a pseudoknot is a conserved and essential element of telomerase RN. Mol. Cell Biol. 2007;27:2130–2143. doi: 10.1128/MCB.01826-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, et al. Proc. IEEE Comput. Syst. Bioinform. Conf. IEEE Computer Society Press; 2005. Tree decomposition based fast searching for RNA structures with and without pseudoknots; pp. 223–234. [DOI] [PubMed] [Google Scholar]
- Song Y, et al. Efficient parameterized algorithms for biopolymer structure-sequence alignment. IEEE/ACM Trans. Comput. Biol. Bioinform. 2006;3:423–431. doi: 10.1109/tcbb.2006.52. [DOI] [PubMed] [Google Scholar]
- Theimer CA, et al. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol. Cell. 2005;17:671–682. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Uemura Y, et al. Tree adjoining grammars for RNA structure prediction. Theor. Comput. Sci. 1999;210:277–303. [Google Scholar]
- Washietl S, et al. Fast and reliable prediction of noncoding RNAs. Proc. Natl Acad. Sci. USA. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Ruzzo WL. Exploiting conserved structure for faster annotation of non-coding RNAs without loss of accuracy. Bioinformatics. 2004;20(Suppl. 1):I334–I341. doi: 10.1093/bioinformatics/bth925. [DOI] [PubMed] [Google Scholar]
- Weinberg Z, Ruzzo WL. Sequence-based heuristics for faster annotation of non-coding RNA families. Bioinformatics. 2006;22:35–39. doi: 10.1093/bioinformatics/bti743. [DOI] [PubMed] [Google Scholar]
- Yang Z, et al. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- Zhang S, et al. Searching genomes for noncoding RNA using FastR. IEEE/ACM Trans. Comput. Biol. Bioinform. 2005;2:366–379. doi: 10.1109/TCBB.2005.57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}