

Abstract
Paclitaxel (PTX) is an important cancer chemotherapeutic agent that binds to β-tubulin and prevents mitosis through microtubule overstabilization. Recent evidence also implicates PTX in the induction of apoptosis of cancer cells via the TLR4 innate immune pathway. The TLR4 accessory protein, MD-2, is an essential component for the species-specific proinflammatory activity of PTX on murine cells. However, whether PTX binds to human MD-2 and how MD-2 and TLR4 interact with PTX are not well defined. Recombinant human MD-2 (rhMD-2) was produced in a Pichia pastoris expression system, and the interaction between rhMD-2 and PTX was assessed by an enzyme-linked immunosorbent assay to show that PTX binds rhMD-2. Formation of the latter complex was found to be dose-dependent and inhibited by anti-MD-2 antibody but not by an isotype control antibody. As measured by human tumor necrosis factor α production, human THP-1 monocytes expressing TLR4 and MD-2 were poorly responsive to the addition of PTX, but murine macrophages expressing TLR4 and MD-2 responded in a dose-dependent manner. Human embryonic kidney (HEK293) cells transfected with both human TLR4 and human MD-2 or human MD-2 and murine TLR4 were also poorly responsive to PTX (10 μm). However, HEK293 cells transfected with murine MD-2 and human TLR4 or murine MD-2 and murine TLR4 were highly responsive to PTX (10 μm), indicating that the murine MD-2/PTX interaction is required for TLR4 activation. To further define the structural differences for MD-2/TLR4 activation, crystal structures of both murine and human MD-2 were subjected to PTX docking by computational methods. These models indicate that PTX binds in the pocket of both human and mouse MD-2 structures. The species-specific difference between human and murine MD-2 activation of TLR4 by PTX can be explained by alterations of surface charge distribution (i.e. electrostatic potential), binding pocket size, and the locus of PTX binding within the MD-2 pocket, which results in reorganization of the 123-130 amino acid loop. In particular, Phe126 appears to operate as a bridge for TLR4·MD-2 dimerization in the mouse but not the human protein.
Paclitaxel (PTX)3 (Fig. 1) is a potent anti-cancer agent derived from the Pacific yew tree, which acts through overstabilization of cellular microtubules. The natural product leads to disruption of mitotic machinery and inhibition of cell growth (1-3). Structurally, the PTX molecule and its interaction with β-tubulin have been well characterized (4-6). PTX also exhibits antiangiogenic properties, and these have expanded the application of the family of drugs known as taxanes to a variety of tumor types (breast, prostate, ovarian, lung) (7) and to treatment of coronary atherosclerosis via paclitaxel-coated coronary stents (8). The contribution of innate immunity pathways to the mechanism of action of PTX has not been well studied. However, the wide ranging applications of this family of drugs and the known interaction of PTX with TLR4 (Toll-like receptor 4) (9, 10) suggest a potential anti-inflammatory mechanism for some of the observed biological effects. TLR4 is a critical component of the innate immune response to bacterial endotoxins (11, 12). Activation of TLR4 by endotoxin requires association with the accessory protein MD-2, an N-glycosylated (13) 19-27-kDa protein that is expressed in both a soluble and a membrane-bound form (14). Binding of endotoxin (lipopolysaccharide (LPS) or lipooligosaccharide (LOS)) to MD-2 in association with TLR4 can lead to dimerization or oligomerization of two or more TLR4 receptors and subsequent cellular activation (15, 16). Although much is known about the MD-2 protein, its interaction with TLR4, and the hypothesized binding site of MD-2 for endotoxin (17-21), the specific structural requirements of endotoxin and other TLR4 ligands for MD-2 association and TLR4 activation are not fully understood.
FIGURE 1.

Paclitaxel. a, topological structure of PTX. b, three-dimensional conformation of PTX as observed in β-tubulin (i.e. T-Taxol (6)). This conformer is one of the many possible options for PTX binding to MD-2 as revealed by Glide docking.
MD-2 belongs to a family of proteins that express an ML (MD-2-related lipid-recognition) domain (22). The protein contains 160 amino acids, which form a “clamshell” binding site (Fig. 2) for hydrophobic ligands inserted between two β-pleated sheets (22, 23). These proteins include MD-1 associated with RP105 on B cells; the dust mite antigens, Der p2 (24) and Der f2 (25); NPC2 (Niemann-Pick disease C2) (26); and the GM2-activating protein important in Tay-Sachs gangliosidosis (27, 28). Der p2 exhibits the most homology to MD-2, and its NMR-determined solution structure includes a β-folded binding pocket for an unidentified lipid ligand (23, 25). MD-2 is thus predicted to directly interact with the lipid A of endotoxin. Several important crystal structures have been published recently (human MD-2 bound to the TLR4 antagonist, lipid IVa (29); the human TLR4·MD-2 complex associated with the endotoxin antagonist eritoran (30); and mouse MD-2 complexed to mouse TLR4 (30)). These enhance our understanding of the structure of MD-2 and offer an opportunity to explore the interaction between MD-2 and various TLR4 agonists and antagonists.
FIGURE 2.

MD-2 structures. a, hMD-2 crystal structure, ligand removed (Protein Data Bank code 2Z65). b, mMD-2 crystal structure (Protein Data Bank code 2Z64).
MD-2 Confers Species-specific Ligand Recognition—The species-specific discrimination of TLR4 ligands by MD-2 is exemplified by PTX, a proinflammatory murine TLR4·MD-2 ligand. Murine MD-2 binding to paclitaxel is an essential step for species-specific activation of the mouse TLR4 with subsequent inflammatory cytokine response (9, 31, 32). As demonstrated by chimeric experiments in which human TLR4 was expressed with mouse MD-2, activation of TLR4 by PTX requires the mouse MD-2 protein and is independent of TLR4 species (9, 32). Species-specific activation of the mouse TLR4 receptor complex by Salmonella enterica lipid A has also been described by Muroi et al. (33) and is dependent upon MD-2 rather than TLR4.
Recombinant human MD-2 (rhMD-2) produced in a Pichia pastoris expression system is able to confer responsiveness on TLR4-expressing cells and bind to meningococcal endotoxin (34). We show that rhMD-2 also binds PTX in a dose-dependent fashion. To further understand the species-specific difference in PTX activation of TLR4, we have employed the recently reported crystal structures of hMD-2 and mMD-2 to construct models of the interactions between PTX and these proteins. Differences in MD-2 electrostatic potential surfaces, hydrophobicity, binding pocket size, and mouse versus human conformational gating of the 123-130 amino acid loop are predicted to be responsible for the species-specific activation of TLR4 by PTX.
EXPERIMENTAL PROCEDURES
RPMI 1640 medium, Dulbecco's modified Eagle's medium, fetal bovine serum, penicillin/streptomycin, sodium pyruvate, and nonessential amino acids were obtained from Cellgro Mediatech (Herndon, VA). Phorbol 12-myristate 13-acetate was purchased from Invitrogen. Interleukin-8 and tumor necrosis factor α (TNFα) ELISA kits were obtained from R&D Systems (Minneapolis, MN). THP-1 and HEK293 cell lines were obtained from ATCC (Manassas, VA). P. pastoris strain GS115 and yeast expression kit (EasySelect Pichia expression kit), including pPICZB expression vector, were obtained from Invitrogen. Media used for the culture of yeast were as follows: yeast extract peptone dextrose medium (1% yeast extract, 2% peptone, and 2% dextrose), buffered glycerol complex medium (1% yeast extract, 2% peptone, 100 mm potassium phosphate buffer, pH 6.0, 1.34% yeast nitrogen base without amino acids, 0.4 mg/liter biotin, and 1% glycerol). Buffered methanol complex medium was made with 0.5% methanol instead of glycerol. Human MD-2 and TLR4 plasmids (pEFBOS MD-2 and pEFBOS TLR4) were kind gifts from K. Miyake (University of Tokyo). Murine TLR4 and murine MD-2 plasmids, pUNOMD-2 and pUNOTLR4, were purchased from Invivogen. Nickel-nitrilotriacetic acid slurry and protein purification columns were obtained from Qiagen (Valencia, CA). Wash buffers and elution buffers were made following the manufacturers' instructions (Qiagen). Mouse anti-human MD-2 antibody was obtained from eBioscience (San Diego, CA). Biotinylated anti-His6 antibody was purchased from Sigma. Endotoxin from the serogroup B N. meningitidis (NMB) (encapsulated, L2/L4 immunotype) (35) was initially extracted by the phenol/water method (35). These preparations were further purified and quantified based on lipid A content, as previously described (36). Stock concentrations of 10 μm or 100 nm were diluted in pyrogen-free water. Semi-synthetic paclitaxel (<0.5% PTX degradation products) was purchased from Sigma and dissolved in 1% DMSO and pyrogen-free water.
Expression and Purification of Recombinant Human MD-2 from P. pastoris
MD-2 cDNA was amplified by PCR from pEFBOS (37) MD-2, and the correct nucleotide sequence PCR product was confirmed by DNA sequencing and inserted into the pPICZB expression vector. The cloned product (pPICZB-MD-2) in the expression vector flanked by the yeast AOXI gene and a His6 selection marker was confirmed by sequencing using the AOXI forward and reverse primers (Invitrogen). As previously described (34), cell pellets were washed with breaking buffer (50 mm sodium phosphate, pH 7.4, 1 mm EDTA, 5% glycerol, 1 mm phenylmethylsulfonyl fluoride) in the presence of a protease inhibitor mixture with broad specificity for the inhibition of serine, cysteine, aspartic, and aminopeptidases and thermolysin-like activities (Sigma). P. pastoris were vortexed with one-fifth volume 0.5-mm glass beads seven times and incubated on ice. Lysed cells were then centrifuged at 12,000 × g for 10 min, and the supernatant was harvested for purification. Five ml of cleared supernatant was mixed with 1 ml of nickel-nitrilotriacetic acid slurry (Qiagen) and gently shaken at 200 rpm on a rotary shaker at 4 °C for 2 h. Washing and elution of rhMD-2 was done following the manufacturer's instructions.
SDS-PAGE and Western Blotting
The eluted MD-2 proteins were analyzed by 10% SDS-PAGE, according to the method of Laemmli (38), and Coomassie Blue staining, and protein concentration was estimated using the Bradford assay (39) comparing elutions of rhMD-2 with known concentrations of BSA. For Western blotting, resolved proteins in the gel were transferred to a polyvinylidene difluoride membrane using a semidry electroblot apparatus (Bio-Rad) and recognized by mouse anti-hMD-2 antibody or anti-His6 antibody diluted at 1:1000. The identity of rhMD-2 was confirmed by nano-liquid chromatography-tandem mass spectrometry (Emory University microchemical facility). The samples were digested in-gel overnight with trypsin at 37 °C. The samples were analyzed by nano-liquid chromatography-tandem mass spectrometry on the quadruple time offlight (QTOF) mass spectrometer instrument. The data from peptide sequences were used to search the Sprot data base (human taxa).
Cell Cultures
THP-1 human macrophage-like cell lines were grown in RPMI 1640 with l-glutamate supplemented with 10% fetal bovine serum, 50 IU/ml penicillin, 50 μg/ml streptomycin, 1% sodium pyruvate, and 1% nonessential amino acids. Murine macrophages (RAW264.7) and human embryonic kidney (HEK293) cells were grown in Dulbecco's modified Eagle's medium supplemented as noted above. Culture flasks were incubated at 37 °C with humidity under 5% CO2.
Cytokine Induction
THP-1 human monocytes were differentiated into macrophage-like cells using phorbol myristate acetate at a final concentration of 10 ng/106 cells and incubated at 37 °C for 24 h. Differentiated macrophages were washed with PBS, counted, adjusted to 106 cells/ml, and transferred into a 24-well tissue culture plate (1 ml/well). The cells were stimulated with 0.56 pmol of LOS or PTX in the indicated concentrations (0.125-100 μm) overnight at 37 °C with 5% CO2. Cell culture supernatants were harvested and saved at -20 °C. RAW 246.7 macrophages adherent to the flask were washed with phosphate-buffered saline. Harvested cells were resuspended in Dulbecco's complete medium. Macrophages (106 cells/ml) were transferred to 24-well tissue culture plates and stimulated with serial dilutions of PTX (0.19-100 μm). Induced RAW 246.7 macrophages were incubated overnight at 37 °C with 5% CO2. Cell culture supernatants were harvested and saved at -20 °C.
HEK293 Transfection with TLR4 and MD-2
HEK293 cells seeded in 12-well plates (5 × 105 cells/well) were transiently transfected with 0.5 μg/well of the pEFBOS-human TLR4, the pEFBOS-human MD-2, pUNO-mouse MD-2, pUNO-mouse TLR4, or a combination of two plasmids together and SuperFect transfection reagent (Qiagen, Inc., Valencia, CA) for 3 h. An empty vector was used as a control. Fresh medium was applied, and the cells were allowed to recover for 12-18 h before stimulation with NMB (0.56 pmol/ml) or PTX (10 μm) overnight at 37 °C with 5% CO2. Cell culture supernatants were harvested and saved at -20 °C.
Cytokine and Chemokine Quantification by ELISA
Human or murine TNF-α (THP-1 or RAW 246.7 supernatants, respectively) and human interleukin-8 (HEK293 supernatants) Duoset kits (R&D Systems) were used for cytokine quantification according to the manufacturer's instructions. Maxisorp ELISA plates were obtained from Nalge Nunc International (Rochester, NY).
PTX Binding to Immobilized rhMD-2 (See Fig. 5a)
FIGURE 5.

PTX binds to recombinant human MD-2. a, human rMD-2 was coated on Maxisorp plates. PTX (1 or 10 μm) was added, and unbound rhMD-2 was recognized by anti-MD-2 antibody. NMB (0.3 μg/ml) and BSA were used as controls. Error bars, ±S.D. from the mean. b, meningococcal LOS, PTX, or BSA were coated on Maxisorp plates. The binding of human rMD-2 to wild-type meningococcal LOS (0.17 nmol of NMB; closed diamonds) and PTX (10 μm; closed squares) was inhibited by increasing concentrations of anti-MD-2 antibody (0-2 μg/ml) (x axis). Isotype control antibody (0-2 μg/ml) did not inhibit binding (open symbols). Human rMD-2 did not bind to BSA-coated wells. Error bars, ±S.D. from the mean. c, PTX was coated on Maxisorp plates. Increasing concentrations of PTX (0, 10, and 100 μm) were recognized by human rMD-2 (0-100 μg/ml). Binding of rhMD-2 to PTX was recognized by anti-His6 antibody. Error bars, ±S.D. from the mean.
96-well Maxisorp microtiter plates were coated overnight with 100 μl of increasing concentrations of MD-2 (0-100 μg/ml) or BSA (10 mg/ml) in 0.2 m sodium acetate buffer, pH 5.0. Plates were then washed three times with washing buffer (1 mg/ml BSA, 50 mm HEPES, 0.15 m NaCl, pH 7.4) and blocked for 1 h at 37°C with blocking buffer (10 mg/ml BSA, 50 mm HEPES, 0.15 m NaCl, pH 7.4). Plates were washed three times in wash buffer and then incubated with 100 μl of NMB (0.3 μg/well) or PTX (1 μm or 10 μm) for 3 h at 37 °C. Plates were again washed three times and incubated with 100 μl of anti-His6 antibody (Sigma) diluted 1:500 in wash buffer for 2 h at room temperature. After three washings, 100 μl of a secondary antibody, alkaline phosphatase-conjugated goat anti-mouse IgG (1:10,000 in wash buffer) was added for 1 h at room temperature. The ELISA was developed with p-nitrophenyl phosphate in 0.5 m diethanolamine buffer containing 0.5 mm MgCl2, pH 9.5, and was read at A405 nm.
Human rMD-2 Binding to PTX and NMB (See Fig. 5b)
Maxisorp microtiter plates were coated for 3 h with PTX (10 μm), NMB (0.3 μg/ml), or BSA (10 mg/ml) in 0.1 m Na2CO3, 20 mm EDTA, pH 9.6, at 37 °C. Plates were washed with water, allowed to air-dry, and then blocked for 3 h with blocking buffer. Plates were then washed three times with washing buffer and incubated for 3 h at 37°C with 100 μl/well rhMD-2 (10 μg/ml). rhMD-2 was preincubated on ice without or with increasing concentrations of anti-human MD-2 antibody or an isotype control antibody (0-2 μg/well). Plates were then washed three times with wash buffer, incubated with biotinylated anti-His6 antibody (Sigma) (1:200) for 2 h at room temperature, again washed three times with wash buffer, and incubated with 100 μl/well 1:200 streptavidin-conjugated horseradish peroxidase (R&D Systems) for 30 min at room temperature. The ELISA was developed with substrate solution (R&D Systems), stopped with 50 μl/well of 2 n H2SO4, and read at A450 nm.
Dose-dependent rhMD-2 Binding to PTX
Maxisorp microtiter plates were coated for 3 h with increasing concentrations of PTX (1, 10, and 100 μm) or BSA (10 mg/ml) in 0.1 m Na2CO3, 20 mm EDTA, pH 9.6, at 37 °C. Plates were washed with water, allowed to air-dry, and then blocked for 3 h with blocking buffer. Plates were then washed three times with washing buffer and incubated for 3 h at 37°C with 100 μl/well of rhMD-2 ranging from 0 to 100 μg/ml. Plates were then washed three times with wash buffer, incubated with biotinylated anti-His6 antibody (Sigma) (1:200) for 2 h at room temperature, again washed three times with wash buffer, and incubated with 100 μl/well 1:200 streptavidin-conjugated horseradish peroxidase (R&D Systems) for 30 min at room temperature. The ELISA was developed with substrate solution (R&D Systems), stopped with 50 μl/well of 2 n H2SO4, and read at OD 450 nm.
Homology Modeling
A homology model of mouse MD-2 was constructed by application of Prime version 1.6 with the Maestro interface (Schrödinger, LLC) and by using the crystal structure of hMD-2 complexed with lipid IVa (PDB code 2E59; 79% sequence similarity) (29) as a template. The resulting homology model was refined by backbone and side chain energy optimizations using the OPLS force field within Prime. A WHATIF (40) analysis demonstrated very similar Z-scores between the mMD-2 homology model and the hMD-2 crystal structure in terms of first and second generation packing quality, χ-1/χ-2 rotamer normality, and backbone conformation.
Computational Ligand-Protein Docking
Glide Docking—Previously established NAMFIS (NMR analysis of molecular flexibility in solution) conformations of PTX (41) were docked separately into the cavity regions of the human MD-2 crystal structure (Protein Data Bank code 2E59) (29) devoid of lipid IVa and the mMD-2 homology model described above using Glide with SP precision (Schrödinger, LLC). This methodology regards the structure of the protein as a rigid body but treats the ligand as a conformationally flexible molecule. The same PTX conformers were similarly docked into the ligand-free MD-2 x-ray structures derived from the hybrid TLR4·hMD-2·eritoran complex (Protein Data Bank code 2Z65) and the mTLR4·mMD-2 complex (Protein Data Bank code 2Z64) (30). Neither MD-2 docking exercise led to NAMFIS conformations as the favored binding pose. Glide was also used to examine the relative energies of PTX docked to the external surfaces of the MD-2 proteins.
Induced Fit Docking—To improve the fit between protein interior and PTX and to achieve deeper residence of the ligand within the cavity, induced fit docking with Prime version 1.6 (Schrödinger) was performed. This approach allows both protein side chain movement and ligand flexibility. Duplicate poses were removed with MacroModel (Schrödinger), and the resulting MD-2·PTX complexes were sorted energetically with the MMGBSA scoring algorithm (42).
Molecular Volumes
The ligand volumes were calculated by extracting the structures from their respective protein x-ray or electron crystallo-graphic (e.g. PTX T-Taxol conformation) complexes as Protein Data Bank files, followed by importing into the Spartan software (Wavefunction, Inc., Irvine, CA). A single point energy calculation provides the volume of the corresponding CPK model. The volumes of the MD-2 cavities were obtained by the World Wide Web-based CASTp package. (44).
Statistical Analysis
Mean values ± S.D. and p values (Student's t test) of at least three independent determinations were calculated with Microsoft Excel software.
RESULTS
Activation of TLR4·MD-2 by PTX Is Species-specific—Ba/F3 cells transfected with murine TLR4·MD-2 demonstrate proinflammatory response to PTX via the TLR4·MD-2 pathway, whereas human TLR4·MD-2 transformants do not (9). To confirm this observation in murine and human macrophage cell lines, RAW264.7 cells were stimulated with serial dilutions of PTX (0.19-100 μm). These cells produced mouse TNFα measured by ELISA in a dose-response manner (Fig. 3a). In contrast, human THP-1 monocytes are poorly responsive to 100 μm concentrations of PTX (Fig. 3b). As expected, a robust TNFα response to wild-type meningococcal endotoxin (NMB) was observed. Wild-type meningococcal LOS is a potent activator of the TLR4 and MyD88-dependent and-independent pathways through MD-2 binding (45).
FIGURE 3.

Proinflammatory response to PTX is species-specific. a, RAW264.7 cells (106 cells/ml) were stimulated with serial dilutions of PTX (100-0.19 μm). Murine TNFα production was measured by ELISA. PTX activated murine macrophages cells in a dose-dependent manner. Unstimulated cells were used as a control. Error bars, ±S.D. from the mean. b, differentiated THP-1 monocytes (106 cells/ml) were stimulated with 0.56 pmol of wild-type meningococcal LOS (NMB) or dilutions of PTX (12.5-100 μm). Human TNFα production was measured by ELISA. Human monocytes were poorly responsive to PTX, as measured by TNFα release. Unstimulated cells were used as a control. Error bars, ±S.D. from the mean.
HEK293 Cells Transfected with Human TLR4 and MD-2 Do Not Respond to PTX—HEK293 cells (106 cell/ml) were transfected with plasmids for human TLR4 and human MD-2 or mouse TLR4 and mouse MD-2. Transfectants were stimulated with wild-type meningococcal LOS (NMB) (0.56 nm) or PTX (10 μm). Interleukin-8 production (pg/ml) was measured by ELISA. Both human and murine transfectants responded to LOS; however, only murine transfectants demonstrated a significant interleukin-8 response to PTX (Fig. 4). Untransfected cells were unresponsive.
FIGURE 4.

HEK293 cells transfected with mouse but not human TLR4 and MD-2 respond to PTX. HEK293 cells (106 cell/ml) were transfected with plasmids for human TLR4 and human MD-2 or mouse TLR4 and mouse MD-2. Transfectants were stimulated with wild-type meningococcal LOS (NMB) (0.56 nm) or PTX (10 μm). Interleukin-8 production (pg/ml) was measured by ELISA. Both human and murine transfectants responded to NMB; however, only murine transfectants responded to PTX. Untransfected cells were unresponsive. Error bars, ±S.D. from the mean.
PTX Binds to rhMD-2 and Binding Is Dose-dependent—The activation of TLR4 by endotoxin requires the binding of endotoxin to MD-2 (46), presumably as the result of MD-2 interacting with lipid A (23). The species-specific activation of TLR4-expressing cells requires murine MD-2; thus, PTX is presumed to bind to murine MD-2 (47). The interaction between PTX and human MD-2 has not been studied. Wild-type N. meningitidis LOS (0.3 μg/well ∼0.17 nmol based on lipid A content) interfered with recognition of increasing concentrations of human rMD-2 (0-100 μg/well) coated on ELISA plates by an anti-His6 antibody (Fig. 5a, closed squares). Compared with BSA (crosses), wild-type meningococcal LOS significantly inhibited rhMD-2 recognition by the antibody at all concentrations of the MD-2 protein. PTX at a concentration of 10 μm (closed circles) similarly blocked recognition of immobilized rhMD-2 by the anti-His6 antibody, whereas a lower concentration of PTX (1 μm)(open squares) was similar to the BSA control. In further support of these observations, increasing concentrations of anti-MD-2 antibody (0-2 μg/well) (closed symbols) inhibited the interaction of rhMD-2 with PTX and meningococcal LOS in a dose-dependent manner (Fig. 5b). An isotype control antibody (open symbols) had no effect on rhMD-2 recognition of PTX or NMB, and rhMD-2 did not bind to BSA-coated wells. Binding between rhMD-2 and PTX was dose-dependent. Increasing concentrations of rhMD-2 (0-100 μg/ml) bound to increasing concentrations of immobilized PTX (0, 10, and 100 μm) (Fig. 5c). Neither rhMD-2 nor PTX bound to BSA (No PTX).
MD-2 Models and Glide Docking—Paclitaxel (molecular weight of 854) is a globular hydrophobic molecule with a molecular volume of 831 Å3 in its microtubule binding conformation (4) (Spartan; Wavefunction). By comparison, lipid IVa, with a molecular weight of 1405 (48) and a molecular volume of 1451 Å3, is a highly flexible structure that can adopt a flat and laminate-like shape for its four lipid appendages. In principle, both ligands can occupy the MD-2 pockets, since the cavity volumes (44) for human (x-ray) (29) and mouse (homology model) MD-2 are 1622 and 1683 Å3, respectively. Attempts to dock PTX into either of the two proteins with Glide leads to complexes in which the ligand rests at the mouth of the cavities but does not penetrate within. The entrance passages into both mouse and human MD-2 are clearly too small to accommodate the globular ligand.
During the course of this work, several additional MD-2 crystal structures were released: hybrid TLR4·hMD-2·eritoran complex (Protein Data Bank code 2Z65) and mTLR4·mMD-2 complex (PDB code 2Z64). (30) The MD-2 proteins revealed CASTp (44) pocket volume prediction estimates of 1906 and 1922 Å3, respectively. The cavity volumes have increased by almost 300 Å3 relative to that for the hMD-2·lipid IVa complex, suggesting a significant change in protein conformation. Interestingly, superposition of hMD-2·lipid IVa (Protein Data Bank code 2E59) (29) and hMD-2·eritoran (Protein Data Bank code 2Z65) (30) illustrates that the backbones overlay quite well, but several side chains near the cavity entrance adopt different orientations. Particularly critical for LPS docking are the Arg90 and Glu92 residues located in this vicinity. The residues are substantially altered in their spatial orientations in the two human MD-2 crystal structures.
The x-ray structures of both lipid IVa and eritoran bound to hMD-2 show that, whereas the lipid moieties of the molecules extend deep into the protein cavities, the diglucosamine backbone and phosphate groups remain in solution outside the mouth of the cavities (Fig. 6a). As previously noted (29), residues around the pocket entrance assist in anchoring the ligand to MD-2. In the case of lipid IVa (2E59), interactions on both edges of the pocket mouth clamp the ligand in place. Fig. 6a illustrates that Lys122 interacts electrostatically with both phosphates at NH-O distances of 4.2 and 5.6 Å. On the opposite lip, Arg90 sustains similar NH-O separations of 4.7 and 5.4 Å while simultaneously associating with Glu92 (4.5 Å). Eritoran, on the other hand, makes use of the charged residues on only one lip of the cavity opening, as depicted by Fig. 6b. Both Lys122 and Lys125 participate in local contact with the sugar phosphates (4.4-6.3 Å), but Arg90 and Glu92 do not associate with the ligand. Instead, again as electrostatic partners, the two residues are directed away from the mouth and reside in aqueous solvent. In all of these associations, the somewhat stretched salt bridges may be complemented by water molecules bridging the charged centers by means of classic hydrogen bonds. The different arrangement of acyl side chains within the MD-2 pocket can be ascribed to variations in acyl chain length for the lipid IVa and eritoran structures. Lipid IVa possesses four C14 extensions, whereas eritoran displays two C10 and single C14 and C18 hydrocarbon chains. The folding organization of these acyl chains in the MD-2 pocket alters the positions of the phosphate head groups just outside the pocket with the concomitant interactions shown in Fig. 6.
FIGURE 6.

Lipid molecules in hMD-2 bind differently to the cavity mouth. a, lipid IVa in hMD-2 (Protein Data Bank code 2E59) clamped by Lys122 at the upper lip of the cavity mouth and by Arg90 and Glu92 at the lower lip. b, eritoran in hMD-2 (Protein Data Bank code 2Z65), illustrating columbic interactions with Lys122 and Lys125 on the upper lip but no interactions with Arg90 and Glu92.
The implication of conformational mobility around the MD-2 entrance channel is significant. Glide docking of PTX into hMD-2-eritoran (Protein Data Bank code 2Z65) illustrates that the molecule docks almost completely within the binding cavity. The basis for the docking differences between MD-2/2E59 (lipid IVa) and MD-2/2Z65 (eritoran) can be traced largely to the Arg90 and Glu92 partners. Adjustment of the side chain torsion angles of the single Arg90 residue in hMD-2-lipid IVa to the values in hMD-2-eritoran increases the cavity volume by 100 Å3. Reorientation of nearby Glu92 similarly leads to a total volume increase of 200 Å3, indicating that these two residues alone appear to control access to the cavity. As a result, the entrance of human MD-2 opens up sufficiently to fit PTX in the pocket very similar to that observed for the MD-2 protein in the x-ray structure of 2Z65 (eritoran).
PTX was subjected to docking into mMD-2 from the mTLR4·mMD-2 complex (Protein Data Bank code 2Z64) with a similar but not identical result relative to the human protein. Although the overall backbones of the human and mouse MD-2 structures (Protein Data Bank codes 2Z65 and 2Z64, respectively) are essentially superimposable, the properties of the individual surfaces display important differences. Comparison of the electrostatic surfaces shows that the cavities for both structures are close to electroneutral, but the outer edge of mMD-2 is more electronegative than hMD-2, especially in the Cys95-Cys105 loop, which is critical for the MD-2/TLR4 interaction. (30) Fig. 7a depicts the loop in mMD-2 as being able to present a larger negatively charged surface than hMD-2. In addition, the x-ray structures of the MD-2·TLR4 complexes display close association of the two proteins in the case of the mouse structure but a significant gap for the human complex (Fig. 7b). If the latter represents the cellular situation, both the relative ES and contact surfaces would favor the mMD-2/TLR4 association and promotion of activation. Furthermore, the electrostatic surface of human MD-2 displays three electropositive patches corresponding to Lys58, Lys122, and Lys125, which are absent on the mouse MD-2 surface (three small red patches on the protein surface at the right in Fig. 7a). The electrostatic discrepancy in this peptide segment is also predicted to be partially responsible for the differences in the mouse and human docking surfaces. Finally, although both cavities appear to have similar hydrophobic properties (Fig. 8), certain regions on the external surface of mMD-2, particularly Phe126, contribute a hydrophobic character, which may be responsible for species specificity.
FIGURE 7.

Electrostatic potential energy surfaces of MD-2 and MD-2·TLR4 models. Left, mouse; right, human. a, electrostatic surfaces (blue is negative) of the MD-2 proteins in the MD-2·TLR4 x-ray complexes (m2Z64 and h2Z65) (30) were produced with SYBYL software. The surfaces are shown with the binding cavities circled in white and the Cys95-Cys105 loops in yellow; b, complexes of MD-2·TLR4 in which the MD-2 is pictured to the right of each graphic as a solid surface, whereas the TLR4 protein is shown as translucent. The corresponding Cys95-Cys105 loops are circled in yellow. In the human MD-2·TLR4 complex, the latter loop is not in direct van der Waals contact with TLR4 residues.
FIGURE 8.

Lipophilic and hydrophobic surfaces of the MD-2 models. Lipophilic (LP; blue) and hydrophobic (brown) surfaces of MD-2 (2Z64 and 2Z65) were obtained with SYBYL software. The surfaces are shown from the front (top; cavities are circled for clarity) and from the back (bottom).
Importantly, none of the top 50 poses arising from Glide docking into h2Z65 and m2Z64 were found outside the pockets of the MD-2 proteins. Certain PTX conformers not in the top 50 poses were docked on the outer surfaces, but the energies derived from the Glide scoring function were considerably higher than poses within the pocket. Thus, for MD-2 proteins in the absence of contact with TLR4, we regard such ligand-protein interactions as low probability.
Induced Fit Docking of PTX into MD-2—Results from induced
fit docking of PTX to h2Z65 (green) and m2Z64 (blue) are
shown in Fig. 9a. Side
chain movements around the cavity opening, resulting from the induced fitting
procedure, cause a slight expansion of the entry channel relative to the x-ray
structure (induced fit cavity volumes for hMD-2 and mMD-2 are 1945 and 1933
Å3, respectively, corresponding to modest protein backbone
root mean square deviations from the original structures of 0.35 and 0.45
Å). One of these, involving Phe126, as shown in
Fig. 9b, portrays a
PTX docking pose for h2Z65. From the most favorable docking poses in
Fig. 9a, comparison of
human and mouse MD-2 illustrates that although the bulk of the PTX baccatin
core occupies a similar position in the cavity, the two C-13 phenyl groups
extend in different directions. This may contribute to the species-specific
differential activation of TLR4 by PTX. In the hMD-2 top pose, the benzamido
phenyl group (NHC(O)Ph) is very close to Phe126, suggesting that a
hydrophobic interaction may exist between them. In addition, the
(CH2)4 moiety of the Lys125 side chain
appears to associate with this phenyl ring by hydrophobic contact. The C-13
phenyl group in PTX exhibits a π-cation interaction with Lys122,
since the terminal 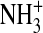 group is
directed toward the plane of the phenyl group at an NH-C distance of only 2.6
Å. In addition, three intermolecular hydrogen bonds serve to anchor the
ligand into the protein, as depicted in
Fig. 10. The apparent
electrostatic association of PTX with MD-2 contrasts with the largely
hydrophobic binding demonstrated by PTX in β-tubulin.
(4). In MD-2, the multiple
interactions attract the Gly123-Lys130 loop so as to
form a concave surface facing the docked PTX.
group is
directed toward the plane of the phenyl group at an NH-C distance of only 2.6
Å. In addition, three intermolecular hydrogen bonds serve to anchor the
ligand into the protein, as depicted in
Fig. 10. The apparent
electrostatic association of PTX with MD-2 contrasts with the largely
hydrophobic binding demonstrated by PTX in β-tubulin.
(4). In MD-2, the multiple
interactions attract the Gly123-Lys130 loop so as to
form a concave surface facing the docked PTX.
FIGURE 9.

MD-2·PTX docking poses. a, the top docking poses for PTX in mouse MD-2 (green) and human MD-2 (blue) (Protein Data Bank codes 2Z64 and 2Z65, respectively); b, detailed view of the top PTX docking pose in human MD-2 (Protein Data Bank code 2Z65) illustrating the Phe126-benzamido phenyl hydrophobic interaction and the Lys122-C13 phenyl π-cation interaction.
FIGURE 10.

Detailed view of the top PTX docking pose in human MD-2 (Protein Data Bank code 2Z65) illustrating hydrogen bonds for Ser120-C1 hydroxyl, Glu92-C7 hydroxyl, and the Arg90-C13 hydroxyl.
The same loop in the mouse protein is oriented in the reverse direction. Comparison of the most favorable mMD-2 docking pose with the mMD-2 crystal structure reveals that loop 123-130 experiences movement outward as a consequence of PTX binding, as depicted in Fig. 11.
FIGURE 11.

Comparison between mMD-2 top docking pose (blue) and mMD-2 crystal structure (orange). PTX docking shifts the 123-130 loop (circled in yellow), including Phe126 outward (blue) relative to the crystal structure.
Importantly, the residues in the critical LPS recognition sequence Phe119-Gly123 are conserved in all species of MD-2 except Lys122. In our exploration of the PTX occupation of the MD-2 cavity, this residue plays a crucial role for both hMD-2 and mMD-2, as reflected by Fig. 6. However, in the mouse protein, position 122 is occupied by Glu instead of Lys. Thus, the absence of a π-cation interaction (Fig. 9b) between PTX and Glu122 in the mouse-PTX complex causes an entirely different binding pose for PTX, resulting in a qualitatively dissimilar interaction between the ligand and the Gly123-Lys130 loop. The loop dislocation suggested by Fig. 11 is accommodated by this observation. As we discuss below, movement of the loop is most likely necessary for TLR4 dimerization and activation.
DISCUSSION
Although the anti-cancer mechanism of action of PTX has long been attributed to effects at the level of β-tubulin, growing evidence supports innate immune activation and possibly antitumor effects of PTX mediated through TLR4·MD-2 (10, 49). The LPS mimetic activity of PTX in murine systems has been extensively documented (9, 31, 32). Activation of the TLR4·MD-2 pathway by PTX has been demonstrated using murine macrophages, (50-52) transfected cell lines (9, 31), and mouse cancer cell lines (53). Further, C3H/HeJ cells, which are known to be unresponsive to LPS due to a mutation in the tlr4 receptor gene, fail to respond to PTX (54). In contrast, cells transfected with human TLR4 and human MD-2 fail to mount a proinflammatory response to PTX (9, 31). The species-specific difference is attributed to the MD-2 protein (32, 55).
This study demonstrates that although PTX does not induce a proinflammatory cytokine response through human MD-2, binding to human MD-2 does occur. Recombinant human MD-2, which confers responsiveness to endotoxin with human TLR4-expressing cells (data not shown), bound PTX in a dose-dependent fashion, and binding was inhibited by an anti-human MD-2 antibody. Further support for PTX binding to human MD-2 was furnished by molecular modeling comparing the docking probabilities of PTX in the human MD-2 molecule with those in the mouse MD-2 protein. These two proteins share 56% identity at the amino acid level, and both are necessary for activation of TLR4 by bacterial endotoxin, yet they discriminate PTX as a TLR4 ligand. As elaborated below, discrimination may be traced, in part, to amino acid sequence variations that result in conformational differences between the proteins accompanied by alterations of surface charge distribution (i.e. electrostatic potential), binding pocket size, and location of PTX binding within the MD-2 pocket, which results in reorganization of the 123-130 amino acid loop.
The observation that PTX associates with human MD-2 in vitro without promoting TLR4 activation is in contrast with the proinflammatory action of PTX on mouse MD-2/TLR4. With the help of molecular modeling, an explanation for the species-specific differences in the proinflammatory TLR4-mediated response by this drug is suggested. The recent model for TLR4·MD-2 aggregation by Kim et al. (30) highlights the function of Phe126 as a bridge to connect MD-2 and TLR4, leading to formation of a dimer. In the present study, PTX binding to human MD-2 is characterized by a π-cation interaction with Lys122 and hydrophobic interaction with Phe126, causing the latter to reside inside the pocket. The electrostatic and lipophilic properties of hMD-2 likewise illustrate the positive character of Lys122 (Figs. 7 and 8) while emphasizing burial of Phe126 within the hydrophobic cavity (Figs. 6b, 9b, and 10). In murine MD-2, the positively charged patch on the outer surface is absent; the hydrophobic orientation for Phe126 is directed away from the protein, and loop 123-130, the bridge for TLR4 dimerization, forms a convex surface to facilitate oligomerization (Fig. 11). A commonly accepted sequence of events believed to activate TLR4 includes the binding of a ligand (i.e. PTX) to soluble MD-2, followed by binding of the PTX·MD-2 complex to TLR4, subsequent homodimerization of TLR4, and signal transduction (56, 57). TLR4 dimerization is a critical step in signal transduction. PTX lacks interaction with amino acid loop 123-130 in mMD-2. Thus, in this protein, PTX does not draw the 123-130 loop inside the mouth of the pocket, and loop 123-130 is forced outward with a concomitant change in the conformation of Phe126 (Figs. 11 and 12). This dynamic process might serve as the primary step for TLR4 dimer formation upon PTX docking. This suggests that agonist binding to mMD-2 may cause a similar conformational change in mMD-2 to facilitate homodimer formation.
FIGURE 12.

Molecular surface of MD-2 models. a, mouse MD-2 protein (Protein Data Bank code 2Z64) with Phe126 in red; b, human MD-2 protein (Protein Data Bank code 2Z65) with Phe126 in red.
The recently disclosed x-ray crystal structure of hMD-2 bound by lipid IVa (29) supports this hypothesis. Insertion of two additional acyl chains into the hydrophobic cavity (e.g. lipid A) is predicted to lead to enlargement of the protein cavity, most certainly accompanied by reorganization of loop 123-130 and giving rise to subsequent signal propagation (29). Thus, the MD-2 binding of molecules with varying molecular volumes can be expected to promote different local conformational changes in the protein as illustrated for lipid IVa, eritoran, and PTX. In addition, much bulkier molecules, such as the lipid A and other LPS agonists, require a flexible binding pocket in order to fully engage the protein.
A final point concerns Gln-22 shown experimentally to be critical to PTX-mediated mMD-2 activity (32). We have not discussed this residue, since it resides on the external surface of MD-2, distant from both the internally docked PTX ligand and the MD-2/TLR4 interface, as illustrated by the recently disclosed complex x-ray structures (30). However, the same paper discusses possible models for the MD-2·TLR4 receptor dimerization. The favored third model (Fig. 7a) positions glutamine at the interface between MD-2 in one complex and TLR4 in the other. Analysis of this interaction is under way in our laboratory.
Thus, species-specific differences in signaling influenced by the binding geometry of PTX to MD-2 are predicted to affect activation of TLR4 at the level of its ectodomain and the recruitment of downstream effector molecules. Differential TLR effector molecule signaling in response to traditional ligands has been described (45, 58). New TLR4 ligands with anti-cancer and apoptotic effects have been identified, further supporting the potential for a TLR4-mediated anti-cancer mechanism of PTX (43, 59, 60). The binding of PTX to human MD-2 also supports a role for PTX in the human system and may lead to new uses for this important family of anti-cancer and anti-inflammatory agents.
Acknowledgments
We thank Lane Pucko for administrative assistance and Xianhe Bai for technical assistance. We are grateful to Prof. Dennis Liotta (Emory University) for encouragement and support. Additionally, we thank U. Ohto and colleagues for advance release of the Protein Data Bank files of the crystal structures of human MD-2.
This work was supported, in whole or in part, by National Institutes of Health Grants K08 AI060966-03 (to S. M. Z.), R01 AI033517-10 (to D. S. S.), and R01 CA-69571 (to J. P. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PTX, paclitaxel; LPS, lipopolysaccharide; LOS, lipooligosaccharide; TNFα, tumor necrosis factor α; ELISA, enzyme-linked immunosorbent assay; hMD-2, human MD-2; rMD-2, recombinant MD-2; rhMD-2, recombinant human MD-2; BSA, bovine serum albumin; NMB, endotoxin from serogroup B N. meningitidis; GM2, gangliosidosis type II.
References
- 1.Wani, M. C., Taylor, H. L., Wall, M. E., Coggon, P., and McPhail, A. T. (1971) J. Am. Chem. Soc. 93 2325-2327 [DOI] [PubMed] [Google Scholar]
- 2.Schiff, P. B., Fant, J., and Horwitz, S. B. (1979) Nature 277 665-667 [DOI] [PubMed] [Google Scholar]
- 3.Horwitz, S. B., Lothstein, L., Manfredi, J. J., Mellado, W., Parness, J., Roy, S. N., Schiff, P. B., Sorbara, L., and Zeheb, R. (1986) Ann. N. Y. Acad. Sci. 466 733-744 [DOI] [PubMed] [Google Scholar]
- 4.Snyder, J. P., Nettles, J. H., Cornett, B., Downing, K. H., and Nogales, E. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 5312-5316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganesh, T., Guza, R. C., Bane, S., Ravindra, R., Shanker, N., Lakdawala, A. S., Snyder, J. P., and Kingston, D. G. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 10006-10011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganesh, T., Yang, C., Norris, A., Glass, T., Bane, S., Ravindra, R., Banerjee, A., Metaferia, B., Thomas, S. L., Giannakakou, P., Alcaraz, A. A., Lakdawala, A. S., Snyder, J. P., and Kingston, D. G. I. (2007) J. Med. Chem. 50 713-725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fishman, M. N., Garrett, C. R., Simon, G. R., Chiappori, A. A., Lush, R. M., Dinwoodie, W. R., Mahany, J. J., Dellaportas, A. M., Cantor, A., Gollerki, A., Cohen, M. B., and Sullivan, D. M. (2006) Clin. Cancer Res. 12 523-528 [DOI] [PubMed] [Google Scholar]
- 8.Moses, J. W., Mehran, R., Nikolsky, E., Lasala, J. M., Corey, W., Albin, G., Hirsch, C., Leon, M. B., Russell, M. E., Ellis, S. G., and Stone, G. W. (2005) J. Am. Coll. Cardiol. 45 1165-1171 [DOI] [PubMed] [Google Scholar]
- 9.Kawasaki, K., Akashi, S., Shimazu, R., Yoshida, T., Miyake, K., and Nishijima, M. (2000) J. Biol. Chem. 275 2251-2254 [DOI] [PubMed] [Google Scholar]
- 10.Wang, J., Kobayashi, M., Han, M., Choi, S., Takano, M., Hashino, S., Tanaka, J., Kondoh, T., Kawamura, K., and Hosokawa, M. (2002) Br. J. Haematol. 118 638-645 [DOI] [PubMed] [Google Scholar]
- 11.Medzhitov, R., Preston-Hurlburt, P., and Janeway, C. A., Jr. (1997) Nature 388 394-397 [DOI] [PubMed] [Google Scholar]
- 12.Chow, J. C., Young, D. W., Golenbock, D. T., Christ, W. J., and Gusovsky, F. (1999) J. Biol. Chem. 274 10689-10692 [DOI] [PubMed] [Google Scholar]
- 13.Ohnishi, T., Muroi, M., and Tanamoto, K. (2001) J. Immunol. 167 3354-3359 [DOI] [PubMed] [Google Scholar]
- 14.Shimazu, R., Akashi, S., Ogata, H., Nagai, Y., Fukudome, K., Miyake, K., and Kimoto, M. (1999) J. Exp. Med. 189 1777-1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ozinsky, A., Underhill, D. M., Fontenot, J. D., Hajjar, A. M., Smith, K. D., Wilson, C. B., Schroeder, L., and Aderem, A. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 13766-13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visintin, A., Mazzoni, A., Spitzer, J. A., and Segal, D. M. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 12156-12161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishitani, C., Mitsuzawa, H., Hyakushima, N., Sano, H., Matsushima, N., and Kuroki, Y. (2005) Biochem. Biophys. Res. Commun. 328 586-590 [DOI] [PubMed] [Google Scholar]
- 18.Visintin, A., Latz, E., Monks, B. G., Espevik, T., and Golenbock, D. T. (2003) J. Biol. Chem. 278 48313-48320 [DOI] [PubMed] [Google Scholar]
- 19.Re, F., and Strominger, J. L. (2003) J. Immunol. 171 5272-5276 [DOI] [PubMed] [Google Scholar]
- 20.Viriyakosol, S., Tobias, P. S., Kitchens, R. L., and Kirkland, T. N. (2001) J. Biol. Chem. 276 38044-38051 [DOI] [PubMed] [Google Scholar]
- 21.Viriyakosol, S., Tobias, P. S., and Kirkland, T. N. (2006) J. Biol. Chem. 281 11955-11964 [DOI] [PubMed] [Google Scholar]
- 22.Inohara, N., and Nunez, G. (2002) Trends Biochem. Sci 27 219-221 [DOI] [PubMed] [Google Scholar]
- 23.Gruber, A., Mancek, M., Wagner, H., Kirschning, C. J., and Jerala, R. (2004) J. Biol. Chem. 279 28475-28482 [DOI] [PubMed] [Google Scholar]
- 24.Derewenda, U., Li, J., Derewenda, Z., Dauter, Z., Mueller, G. A., Rule, G. S., and Benjamin, D. C. (2002) J. Mol. Biol. 318 189-197 [DOI] [PubMed] [Google Scholar]
- 25.Ichikawa, S., Takai, T., Inoue, T., Yuuki, T., Okumura, Y., Ogura, K., Ina-gaki, F., and Hatanaka, H. (2005) J. Biochem. (Tokyo) 137 255-263 [DOI] [PubMed] [Google Scholar]
- 26.Friedland, N., Liou, H. L., Lobel, P., and Stock, A. M. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 2512-2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wright, C. S., Zhao, Q., and Rastinejad, F. (2003) J. Mol. Biol. 331 951-964 [DOI] [PubMed] [Google Scholar]
- 28.Bertoni, C., Appolloni, M. G., Stirling, J. L., Li, S. C., Li, Y. T., Orlacchio, A., and Beccari, T. (1997) Mamm. Genome 8 90-93 [DOI] [PubMed] [Google Scholar]
- 29.Ohto, U., Fukase, K., Miyake, K., and Satow, Y. (2007) Science 316 1632-1634 [DOI] [PubMed] [Google Scholar]
- 30.Kim, H. M., Park, B. S., Kim, J. I., Kim, S. E., Lee, J., Oh, S. C., Enkhbayar, P., Matsushima, N., Lee, H., Yoo, O. J., and Lee, J. O. (2007) Cell 130 906-917 [DOI] [PubMed] [Google Scholar]
- 31.Kawasaki, K., Akashi, S., Shimazu, R., Yoshida, T., Miyake, K., and Nishijima, M. (2001) J. Endotoxin Res. 7 232-236 [PubMed] [Google Scholar]
- 32.Kawasaki, K., Gomi, K., and Nishijima, M. (2001) J. Immunol. 166 11-14 [DOI] [PubMed] [Google Scholar]
- 33.Muroi, M., Ohnishi, T., and Tanamoto, K. (2002) Infect. Immun. 70 3546-3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimmer, S., Zughaier, S., Tzeng, Y.-L., and Stephens, D. (2007) Glycobiology 17 847-856 [DOI] [PubMed] [Google Scholar]
- 35.Rahman, M. M., Stephens, D. S., Kahler, C. M., Glushka, J., and Carlson, R. W. (1998) Carbohydr. Res. 307 311-324 [DOI] [PubMed] [Google Scholar]
- 36.Zughaier, S. M., Tzeng, Y. L., Zimmer, S. M., Datta, A., Carlson, R. W., and Stephens, D. S. (2004) Infect. Immun. 72 371-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mizushima, S., and Nagata, S. (1990) Nucleic Acids Res. 18 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cleveland, D. W., Fischer, S. G., Kirschner, M. W., and Laemmli, U. K. (1977) J. Biol. Chem. 252 1102-1106 [PubMed] [Google Scholar]
- 39.Bradford, M. M. (1976) Anal. Biochem. 72 248-254 [DOI] [PubMed] [Google Scholar]
- 40.Vriend, G. (1990) J. Mol. Graph. 8 52-56 [DOI] [PubMed] [Google Scholar]
- 41.Snyder, J. P., Nevins, N., Cicero, D. O., and Jansen, J. (2000) J. Am. Chem. Soc. 122 724-725 [Google Scholar]
- 42.Jayaram, B., Sprous, D., and Beveridge, D. L. (1998) J. Phys. Chem. B 102 9571-9576 [Google Scholar]
- 43.Okamoto, M., and Sato, M. (2003) J. Med. Invest. 50 9-24 [PubMed] [Google Scholar]
- 44.Dundas, J., Ouyang, Z., Tseng, J., Binkowski, A., Turpaz, Y., and Liang, J. (2006) Nucleic Acids Res. 34 W116-W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zughaier, S. M., Zimmer, S. M., Datta, A., Carlson, R. W., and Stephens, D. S. (2005) Infect. Immun. 73 2940-2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyakushima, N., Mitsuzawa, H., Nishitani, C., Sano, H., Kuronuma, K., Konishi, M., Himi, T., Miyake, K., and Kuroki, Y. (2004) J. Immunol. 173 6949-6954 [DOI] [PubMed] [Google Scholar]
- 47.Kawasaki, K., Gomi, K., Kawai, Y., Shiozaki, M., and Nishijima, M. (2003) J. Endotoxin Res. 9 301-307 [DOI] [PubMed] [Google Scholar]
- 48.Raetz, C. R. H., Purcell, S., Meyer, M. V., Qureshi, N., and Takayama, K. (1985) J. Biol. Chem. 260 6080-6088 [PubMed] [Google Scholar]
- 49.Kelly, M. G., Alvero, A. B., Chen, R., Silasi, D. A., Abrahams, V. M., Chan, S., Visintin, I., Rutherford, T., and Mor, G. (2006) Cancer Res. 66 3859-3868 [DOI] [PubMed] [Google Scholar]
- 50.Manthey, C. L., Perera, P. Y., Salkowski, C. A., and Vogel, S. N. (1994) J. Immunol. 152 825-831 [PubMed] [Google Scholar]
- 51.Manthey, C. L., Qureshi, N., Stutz, P. L., and Vogel, S. N. (1993) J. Exp. Med. 178 695-702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vogel, S., Hirschfeld, M. J., and Perera, P. Y. (2001) J. Endotoxin Res. 7 237-241 [PubMed] [Google Scholar]
- 53.Zaks-Zilberman, M., Zaks, T. Z., and Vogel, S. N. (2001) Cytokine 15 156-165 [DOI] [PubMed] [Google Scholar]
- 54.Manthey, C. L., Brandes, M. E., Perera, P. Y., and Vogel, S. N. (1992) J. Immunol. 149 2459-2465 [PubMed] [Google Scholar]
- 55.Kawasaki, K., Nogawa, H., and Nishijima, M. (2003) J. Immunol. 170 413-420 [DOI] [PubMed] [Google Scholar]
- 56.Gioannini, T. L., Teghanemt, A., Zhang, D., Coussens, N. P., Dockstader, W., Ramaswamy, S., and Weiss, J. P. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4186-4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pugin, J., Stern-Voeffray, S., Daubeuf, B., Matthay, M. A., Elson, G., and Dunn-Siegrist, I. (2004) Blood 104 4071-4079 [DOI] [PubMed] [Google Scholar]
- 58.Mata-Haro, V., Cekic, C., Martin, M., Chilton, P. M., Casella, C. R., and Mitchell, T. C. (2007) Science 316 1628-1632 [DOI] [PubMed] [Google Scholar]
- 59.Hung, L. C., Lin, C. C., Hung, S. K., Wu, B. C., Jan, M. D., Liou, S. H., and Fu, S. L. (2007) Biochem. Pharmacol. 73 1957-1970 [DOI] [PubMed] [Google Scholar]
- 60.Okamoto, M., Oshikawa, T., Tano, T., Ohe, G., Furuichi, S., Nishikawa, H., Ahmed, S. U., Akashi, S., Miyake, K., Takeuchi, O., Akira, S., Moriya, Y., Matsubara, S., Ryoma, Y., Saito, M., and Sato, M. (2003) J. Natl. Cancer Inst. 95 316-326 [DOI] [PubMed] [Google Scholar]