

Abstract
Concentrations of circulating 24S-hydroxycholesterol are of interest as a practical measure of cholesterol efflux from the human brain. The current method of choice for 24S-hydroxycholesterol quantification is with GC-MS. LC-MS methods to detect 24S-hydroxycholesterol have been described but they lack rigorous HPLC separation of a closely eluting isomeric oxysterol, 25-hydroxycholesterol. This is important as 25-hydroxycholesterol can be present in significant amounts and MS/MS cannot completely differentiate 24S-hydroxycholesterol. We describe an LC-MS method with rapid chromatographic separation of the oxysterols to permit accurate determination of plasma 24S-hydroxycholesterol. The availability of an LC-MS method offers advantages including simplified sample work-up and analysis.
About one quarter of total body cholesterol can be found as a distinct pool in the human brain [1;2]. Exchange with circulating cholesterol is prevented by the blood-brain barrier. Input of cholesterol into the CNS is almost entirely from de novo synthesis. Synthesis in the brain is normally balanced by elimination of the cholesterol derivative 24S-hydroxycholesterol (24SOHChol) across the blood-brain barrier [3]. Most circulating 24SOHChol has been demonstrated to be of cerebral origin and can therefore be used as a measure of cholesterol efflux from the brain [4]. This is of interest for the study of brain cholesterol homeostasis. Disruption in cholesterol homeostasis is thought to be involved in the development of many neurodegenerative conditions, for example Alzheimer’s Disease [1;5]. To date quantification of plasma or serum 24SOHChol has been mostly accomplished with GC-MS methods [3;4;6]. GC-MS generally requires laborious clean up of biological samples or frequent column maintenance along with derivatization of samples for increased volatility. Derivatization was also a requirement for detection of 24OHChol with an ESI-LC-MS method described recently [7]. In contrast, detection of 24SOHChol can be accomplished without derivatization using LC-MS methods [8;9]. A limitation is that tandem MS (MS/MS) cannot selectively differentiate the oxysterols 24SOHChol and 25-hydroxycholesterol, which demonstrate similar precursor to product ion fragmentations. LC-MS methods have tried to overcome the lack of MS selectivity by utilizing front-end oxysterol separation with C18-HPLC [9]. Careful identification and elimination of 25-hydroxycholesterol is essential for accurate quantification of 24SOHChol, as 25-hydroxycholesterol signal may otherwise contribute as a method interferent. Separation of 24SOHChol and 25-hydroxycholesterol is difficult even when 24R- and 24S-OHChol isomers are separated [9], leading McDonald et al. to conclude in a recent overview of sterol analysis that LC-MS is not viable for 24SOHChol determination compared to GC-MS [9]. The use of isocratic HPLC elution enables oxysterol separation, but methods can suffer from broader peaks, lower signal intensity and lack of rapidity (Figure 1 Panel A). In this article we describe and validate a viable ternary isocratic approach for determination of 24SOHChol with LC-MS. The method allows separation of symmetric peaks for 24SOHChol and 25-hydroxycholesterol within 6.5 min (Figure 1 Panel B) to enable faster quantification of plasma 24SOHChol.
Figure 1.

Both 24OHChol and 25-hydroxycholesterol are detected with MS/MS. To ensure HPLC separation of the oxysterols we initially used an isocratic methanol:acetonitrile:water mobile phase (45:40:35) modified from Burkard et al. [8] with an HPLC column temperature of 55°C. Oxysterol retention times were around 25 min with the 24R- and 24S-OHChol isomers separated from one another (Panel A authentic standards). An alternative eluent of methanol:acetonitrile:water (14:0.6:1) with a column temperature of 10°C enabled separation of 24OHChol from 25-hydroxycholesterol within 6.5 min (Panel B). Also shown are extracted ion chromatograms from plasma samples where significant 25-hydroxycholesterol was detected (Panels C and D).
Racemic 24OHChol 25,26,26,26,27,27,27-d7 internal standard was obtained from Medical Isotopes, Inc (Pelham, NH). All other oxysterols were purchased from Steraloids (Newport, RI). Plasma calibrator concentrations ranged from 38–312ng/mL 24SOHChol (7.6–62ng on-column injection). Calibrators were made using K3EDTA female charcoal-stripped plasma from Bioreclamation, Inc (Hicksville, NY). For development we used NaEDTA male/female pooled plasma from Biological Specialty Corp (Colmar, PA), as well as plasma collected from volunteers consented according to OHSU Institutional Review Board approved polices and procedures. Plasma aliquots of 0.5mL were thawed and spiked to 130ng/mL with 24OHChol-d7 in 50µL of methanol in a manner previously determined as practical for quantification of plasma 24SOHChol with isotope dilution MS [6;8]. Saponification was at 37°C for 1 hour after addition of 2mL of freshly prepared 0.9M sodium hydroxide in 9:1 ethanol:water. Saturated aqueous sodium chloride (1mL) was added and the aqueous phase was extracted twice with 3.5mL of hexane. The dried organic extract residue was reconstituted in 125µL of HPLC mobile phase. The method was validated using a Thermo TSQ Quantum Discovery triple-quadrupole mass spectrometer (San Jose, CA) equipped with an atmospheric pressure chemical ionization (APCI) source. The ionization interface was operated in the positive mode using the following settings: vaporizer temperature, 450°C; corona discharge current, 3 kV; sheath and aux gas flow rates, 30 and 5 respectively; tube lens voltage, 150 V; capillary voltage, 35 V; and capillary temperature, 300°C. An instrument method was created to monitor for the 24SOHChol transition from m/z 385 precursor to m/z 367 product ion and 24OHChol-d7 transition from m/z 392 to m/z 374 ion. The collision energy was 15 and collision gas pressure at 0.8mTorr. The LC-MS system was composed of an in-line Thermo Surveyor auto-sampler and HPLC pump. Oxysterols were separated using a 250×2.1 (i.d.) mm, 5m BetaBasic C18-HPLC column (ThermoHypersil, Waltham, MA) with guard and a pre-column 0.5µm filtering frit. The HPLC column temperature was 10°C. The isocratic mobile phase consisted of methanol:acetonitrile:water (14:0.6:1 by volume, modified from a 14:1:1 system used by Pulfer et al. [10] and 14:1:1.1 system by Raith et al. [11]) delivered at a flow rate of 0.4mL/min. A 15 min column wash was included. The injection volume was 50µL in mobile phase.
Ammonium acetate [M+NH4]+ adduct ions have been utilized for oxysterol detection with Applied BioSystems instruments equipped with a TurboV™ ESI source [9;10] (Foster City, CA). Adduct ions were not generated via electrospray ionization with the Thermo TSQ ionization source. The phenomenon of instrument dependent in-source adduct formation was previously noted [9]. Maximum 24OHChol ion intensity with the TSQ was achieved using APCI in the positive mode [8;11]. Full scan mass spectra exhibited prominent decay ions corresponding to the loss of one and two water molecules. No [M+H]+ ion was observed for 24SOHChol, as previously described [8]. When 24OHChol-d7 was detected with LC-MS experiments monitoring for [M+H-2H2O]+ m/z 374 ion [8], an extracted ion chromatogram from un-spiked plasma demonstrated endogenous interference at the retention time of 24SOHChol-d7 (supplemental data). When monitoring for [M+H-H2O]+ m/z 392 ion unsatisfactory signal-to-noise was obtained due to bis(2-ethylhexyl) or di-isooctyl phthalate contamination commonly encountered at m/z 391 [12]. Monitoring for the transition from m/z 392 precursor to a strong m/z 374 product ion gave an adequate signal-to-noise ratio and eliminated endogenous interference. When monitoring for the 24SOHChol transition from m/z 385 precursor to m/z 367 product ion, 25-hydroxycholesterol was also detected. Use of an isocratic methanol:acetonitrile:water mobile phase at a column temperature of 10°C enabled resolution of the oxysterols. The 24SOHChol signal-to-noise ratio for the lowest charcoal-stripped plasma calibrator was 6:1.
The within- and between-run precision (%RSD) for calculated 24SOHChol was <8.5% for six calibrators across the range 38–312ng/mL. Within-run RSDs ranged from 0.4–8%. Between-run RSDs for calibrators analyzed over a month ranged from 2.6–8.5%. Calibrators were from charcoal-stripped plasma certified as low sterol content. The endogenous 24SOHChol level in stripped plasma was calculated to be 38ng/mL from the y-intercept concentrations for multiple calibration curves. The lower limit of quantification from plasma was 38ng/mL with the between-run RSD determined as 8.5%. A weighted (1/X) linear regression of peak area ratio (24SOHChol/24OHChol-d7) versus concentration of 24SOHChol (ng/mL) was used for calibration. Plasma calibrators were included with each sample set and monitored over one month. Calibration curves were reproducible with a typical linear regression equation of y = 0.007+0.075x. Acceptable linearity was observed up to 1120ng/mL with characteristic correlation coefficients (r2) >0.998. An initial oxysterol isolation step from plasma was performed using liquid-liquid extraction with hexane or with C18-SPE [6]. To determine liquid-liquid extraction efficiency plasma was thawed and spiked with additional amounts of 24SOHChol, along with 65ng 24SOHChol-d7 internal standard, in 50µL methanol prior to saponification. After the plasma baseline signal for 24SOHChol was subtracted the calculated 24SOHChol recovery at 33ng spiked per mL was 85%, 66ng was 100%, 132ng was 101%, 264ng was 100%, 528ng was 101%, and 1055ng was 100%. From the same experiment mean absolute 24OHChol-d7 recovery averaged 65% across the calibrator range. Plasma 24SOHChol was relatively stable after three freeze (−80°C)/thaw cycles with peak area ratios differing from a baseline ratio by an average of +5.3% (range 3.7–6.8%). Plasma 24SOHChol was stable for at least one year when samples were stored at −80°C. Storage stability of reconstituted extracts in the auto-sampler at 4°C was evaluated by analysis at time zero and after 24 hours. 24SOHChol and 24OHChol-d7 exhibited a 30% loss in absolute signal but demonstrated <5% variation in isotope dilution ratios for up to 24 hours. Extracts in mobile phase were not stable after storage at −80°C for one week. 24SOHChol and 24OHChol-d7 standards diluted in methanol were stable at −80°C for at least one year.
Concentrations of 24SOHChol determined using the LC-MS method were compared with those previously obtained by GC-MS. Quantification of 24SOHChol with GC-MS was from 0.2mL of plasma and was accomplished by saponification, extraction and oxysterol derivatization to trimethylsilyl ether as described previously [13]. A Bland-Altman difference plot [14] (Figure 2 Panel A) suggests a systematic inter-method bias estimated by the mean difference of 3.8ng/mL. Random variation around the mean difference is constant across the range of measurement. The results were also evaluated using Passing-Bablock regression analysis with no special assumptions regarding the distribution of samples and measurement errors [15] (Figure 2 Panel B). A close correlation existed between LC-MS and GC-MS (R = 0.9963, p < 0.001). The Passing-Bablock regression line was y=0.991x-2.267. The LC-MS method demonstrated satisfactory accuracy with a regression analysis that closely followed the line of identity. Although the sample size was small (n=10) the method comparison was acceptable.
Figure 2.
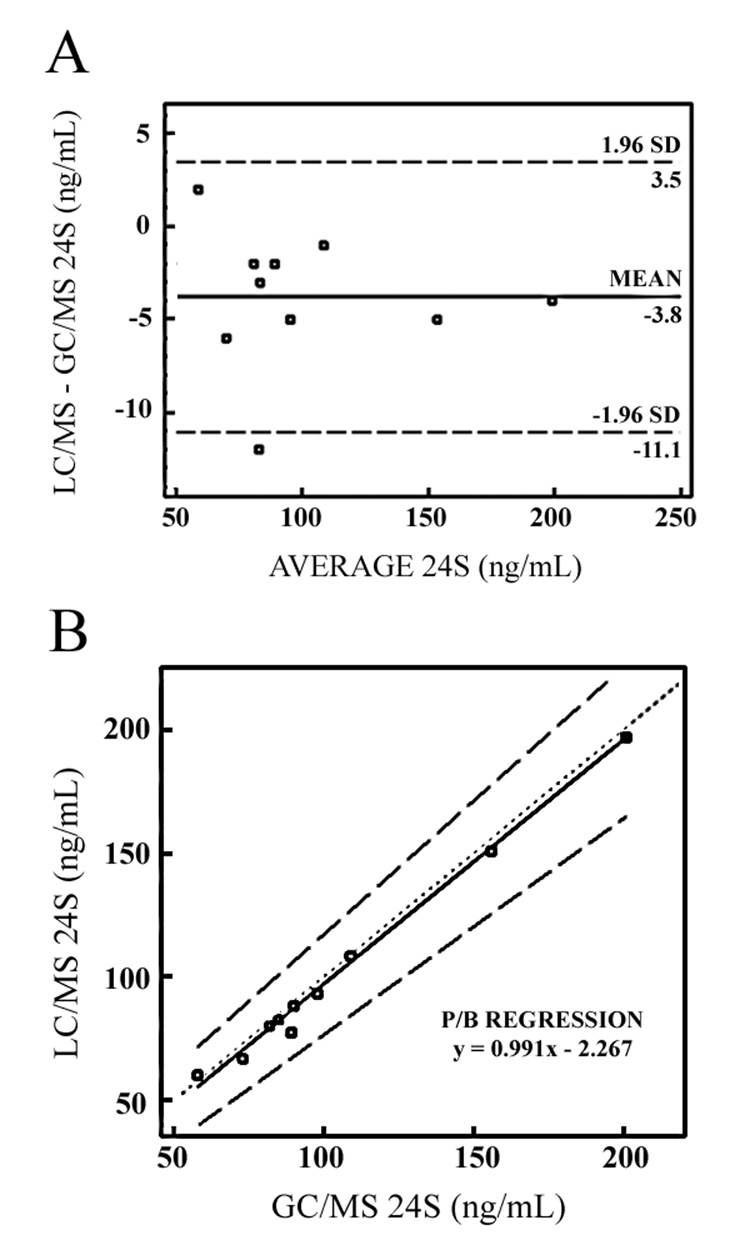
Panel A is a Bland-Altman plot for the comparison of 24SOHChol concentration measured by GC/MS and LC/MS. The mean difference of −3.8ng/mL is indicated by the solid line, the 95% confidence intervals by dashed lines. Panel B is a Passing-Bablok regression analysis.
Detailed in this report is an LC-MS method for determining plasma 24SOHChol. The method demonstrates satisfactory precision and eliminates any endogenous 25-hydroxycholesterol interference to ensure accurate 24SOHChol determination, as evidenced by comparison of the method with a GC-MS reference method [13]. In summary we have developed and validated an APCI-LC-MS method suitable for quantification of 24SOHChol from 0.5mL plasma across the physiological 24SOHChol range with a 30ng/mL lower limit of quantification.
Supplementary Material
MS profiles for 24OHChol and 24OHChol-d7 exhibit abundant [M+H-H2O]+ and [M+H-2H2O]+ ions (Panels A and B respectively). Racemic 24OHChol-d7 can by detected by monitoring for m/z 374 ion. This can result in extracted ion chromatograms from internal standard-spiked and un-spiked plasma (Panel C) that demonstrate an endogenous interferent peak at the retention time of 24SOHChol-d7. By monitoring for the transition from m/z 392 precursor to m/z 374 product ion the interference is eliminated (Panel D).
ACKNOWLEDGMENTS
This work was supported by a NIH grant (R01-HL073980 to RS) and was accomplished using instrumentation housed in the Department of Physiology and Pharmacology Bioanalytical Shared Resource.
ABBREVIATIONS
- 24SOHChol
24S-hydroxycholesterol
- MS/MS
tandem mass spectrometry
- APCI
atmospheric pressure chemical ionization
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004;24:806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- 2.Lütjohann D. Cholesterol metabolism in the brain: importance of 24S-hydroxylation. Acta. Neurol. Scand. Suppl. 2006;185:33–42. doi: 10.1111/j.1600-0404.2006.00683.x. [DOI] [PubMed] [Google Scholar]
- 3.Lütjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U, Björkhem I. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Björkhem I, Lütjohann D, Diczfalusy U, Stahle L, Ahlborg G, Wahren J. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 1998;39:1594–1600. [PubMed] [Google Scholar]
- 5.Björkhem I. Rediscovery of cerebrosterol. Lipids. 2007;42:5–14. doi: 10.1007/s11745-006-1003-2. [DOI] [PubMed] [Google Scholar]
- 6.Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal. Biochem. 1995;225:73–80. doi: 10.1006/abio.1995.1110. [DOI] [PubMed] [Google Scholar]
- 7.Griffiths WJ, Wang Y, Alvelius G, Liu S, Bodin K, Sjövall J. Analysis of oxysterols by electrospray tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2006;17:341–362. doi: 10.1016/j.jasms.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Burkard I, Rentsch KM, von Eckardstein E. Determination of 24S- and 27-hydroxycholesterol in plasma by high-performance liquid chromatography-mass spectrometry. J. Lipid Res. 2004;45:776–781. doi: 10.1194/jlr.D300036-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.McDonald JG, Thompson BM, McCrum EC, Russell DW. Extraction and analysis of sterols in biological matrices by high performance liquid chromatography electrospray ionization mass spectrometry. Methods Enzymol. 2007;432:145–170. doi: 10.1016/S0076-6879(07)32006-5. [DOI] [PubMed] [Google Scholar]
- 10.Pulfer MK, Taube C, Gelfand E, Murphy RC. Ozone exposure in vivo and formation of biologically active oxysterols in the lung. J Pharmacol. Exp. Ther. 2005;312:256–264. doi: 10.1124/jpet.104.073437. [DOI] [PubMed] [Google Scholar]
- 11.Raith K, Brenner C, Farwanah H, Muller G, Eder K, Neubert RH. A new LC/APCI-MS method for the determination of cholesterol oxidation products in food. J. Chromatogr. A. 2005;1067:207–211. doi: 10.1016/j.chroma.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 12.Verge KM, Agnes GR. Plasticizer contamination from vacuum system O-rings in a quadrupole ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 2002;13:901–905. doi: 10.1016/S1044-0305(02)00386-0. [DOI] [PubMed] [Google Scholar]
- 13.Thelen KM, Lütjohann D, Vesalainen R, Janatuinen T, Knuuti J, von Bergmann K, Lehtimaki T, Laaksonen R. Effect of pravastatin on plasma sterols and oxysterols in men. Eur. J. Clin. Pharmacol. 2006;62:9–14. doi: 10.1007/s00228-005-0068-9. [DOI] [PubMed] [Google Scholar]
- 14.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1:307–310. [PubMed] [Google Scholar]
- 15.Bablock Passing H. A new biometrical procedure for testing the equality of measurements from two different analytical methods. Application of linear regression procedures for method comparison in clinical chemistry, Part 1. J. Clin. Chem. Biochem. 1983;21:709–720. doi: 10.1515/cclm.1983.21.11.709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MS profiles for 24OHChol and 24OHChol-d7 exhibit abundant [M+H-H2O]+ and [M+H-2H2O]+ ions (Panels A and B respectively). Racemic 24OHChol-d7 can by detected by monitoring for m/z 374 ion. This can result in extracted ion chromatograms from internal standard-spiked and un-spiked plasma (Panel C) that demonstrate an endogenous interferent peak at the retention time of 24SOHChol-d7. By monitoring for the transition from m/z 392 precursor to m/z 374 product ion the interference is eliminated (Panel D).