

Abstract
γ-Glutamyl cysteine ligase (GCL) is the rate limiting enzyme in glutathione (GSH) synthesis. A GAG repeat polymorphism in the 5′ UTR of the gene coding for the catalytic subunit of GCL (GCLC) has been associated with altered GSH levels in vitro. Thus, we hypothesized that this polymorphism is associated with altered GCL activity and blood GSH levels in vivo. A total of 256 healthy US black and white adults were genotyped for the GAG polymorphism and blood GSH levels were measured. In a subset of 107 individuals, blood GCL activity was determined. Five alleles with 4, 7, 8, 9 and 10 GAG repeats were observed. The most prevalent genotype was 7/9 (40%) followed by 7/7 (32%) and 9/9 (11%). GSH levels were 15% lower in 9/9 individuals than 7/9 individuals (p = 0.05). GCL activity was 21% lower in 9/9 individuals than 7/7 individuals (p= 0.04). A decreasing trend of GCL activity was observed in the order of 7/7 > 7/9 > 9/9 (p=0.04). These findings show that 9/9 individuals have lower blood GSH levels, which is likely due to a decrease in GCL activity. Such individuals might be more susceptible to oxidative stress-related diseases than individuals with other genotypes.
Keywords: γ-glutamylcysteine ligase, GAG trinucleotide repeat polymorphism, Short tandem repeat polymorphism, Glutathione, GCL activity
Introduction
Glutathione (γ-glutamylcysteinylglycine, GSH), is the most abundant non-protein thiol in cells and has an array of functions involved in the maintenance of cellular homeostasis. These functions include detoxifying drugs [1], protecting macromolecules from reactive oxygen species [2] and modulating protein structure and function [3]. More recently, GSH has been found to play important roles in signal transduction [4], cell proliferation [5], apoptosis [6] and gene expression [7]. Based upon these many functions, maintenance of optimal GSH concentration in cells and tissues represents a critical factor in the maintenance of health and susceptibility to diseases. Decreased GSH levels have been implicated in numerous diseases such as heart disease [8], arthritis [8, 9], diabetes [8, 9] and cancers [10-12]. While large interindividual variation in GSH levels has been observed [13, 14], little is known about the intrinsic and extrinsic factors responsible for this variation, particularly as it relates to disease risk or treatment toxicities involving GSH depletion.
Two enzymes are responsible for the biosynthesis of GSH in cells and tissues. The initial and rate-limiting step, formation of γ-glutamylcysteine, is catalyzed by γ-glutamylcysteine ligase (GCL, EC # 6.3.2.2), a heterodimer consisting of a catalytic (GCLC) and a modulatory subunit (GCLM). Glutathione synthetase (EC # 6.3.2.3) catalyzes the addition of glycine to the dipeptide, γ-glutamylcysteine, to form GSH. Polymorphisms have been identified in both the GCLC and GCLM genes, some of which have been associated with decreased GSH levels in vitro and altered susceptibility to certain diseases [15-17]. A trinucleotide (GAG) tandem repeat polymorphism, identified in the 5′ untranslated region (exon 1, 10 base pairs upstream of the ATG start site) of GCLC has been identified with alleles consisting of 4, 7, 8, 9 and 10 consecutive GAG repeats; 7 and 9 being most prevalent [18]. It has been observed that the distribution of the allele with the 4 GAG-repeat is restricted to blacks and the 10 GAG-repeat allele restricted to blacks and Hispanics [18]. This polymorphism has been associated with several disease conditions such as schizophrenia [19], chronic beryllium disease [20] and diabetes [21]. Walsh et al [18] have demonstrated an association between certain GAG alleles and GSH levels in tumor cell lines. However, the in vivo functional significance of the GAG polymorphism on GSH biosynthesis has not been previously examined. This study was conducted to determine if the presence of specific GAG repeat alleles are associated with altered GCL enzyme activity or GSH levels in the blood of healthy adults.
Materials and Methods
Study subjects
A total of 256 healthy US blacks and whites were recruited with their informed consent from the city of Mt. Vernon, NY. as part of our comprehensive study of tobacco smoke biomarkers described previously [22]. All study protocols were approved by our Institutional Human Subjects Committee. Age and smoking characteristics of the study subjects are provided in Table 1. The subjects were 18-57 years old, consisted of 86% smokers and nearly equal numbers of men versus women and blacks versus whites. Due to incomplete data for some individuals, sex and race specific numbers are lower for some analyses.
Table 1. Characteristics of Study Subjects.
| Blacks [Number of subjects (%)] | Whites [Number of subjects (%)] | |||||
|---|---|---|---|---|---|---|
| Men | Women | Total | Men | Women | Total | |
| N | 66 (100) | 67 (100) | 133 (100) | 56 (100) | 59 (100) | 116 (100) |
| Age (yr): | ||||||
| <34 | 28 (42.4) | 23 (34.3) | 51 (38.3) | 31 (55.3) | 29 (49.0) | 61 (52.6) |
| 34 – 44 | 26 (39.4) | 30 (44.7) | 56 (42.1) | 16 (28.5) | 16 (27.0) | 32 (27.6) |
| 45 – 57 | 12 (18.2) | 14 (20.8) | 26 (19.5) | 9 (16.0) | 14 (23.7) | 23 (19.8) |
| Smoking Status: | ||||||
| Non- smokers | 5 (7.5) | 19 (28.3) | 24 (18.0) | 0 (0) | 2 (3.4) | 3 (2.5) |
| Smokers | 61 (92.5) | 48 (71.6) | 109 (82.0) | 56 (100) | 57 (96.6) | 113 (97.4) |
Note: Percentages shown are across age and smoking status.
Biospecimen collection
Exfoliated buccal mucosal cells were obtained by having the subjects rinse their mouth with distilled water, brush their cheeks and gums with a soft tooth brush, and rinse with 20 ml of saline. The collected rinse was stored at 4°C until centrifugation (6000 × g for 10 min) on-site within 1 h after collection. Cells were washed three times with saline and packed cells were stored at -80° C until analysis. Venous blood was collected from the antecubital vein into Vacutainer tubes (Becton Dickinson, Franklin Lakes, NJ) containing K-EDTA as an anticoagulant. The subjects were also asked to fill out a detailed questionnaire on their lifestyle, alcohol consumption, diet (food frequency) and smoking habits.
Determining GCLC genotype
a. DNA amplification
To isolate DNA, exfoliated buccal epithelial cells were lysed in lysis-buffer (10 mM Tris, 10 mM EDTA, 0.1 M NaCl, 2 % SDS) and incubated with proteinase K (0.1 mg/mL) at 58° C for 3 h. DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1) and precipitated with ethanol as previously described [23]. Buccal cell DNA containing the GAG trinucleotide repeat region was amplified by P32-labeled PCR amplification. Each reaction was performed in a 0.2 mL PCR tube by adding 200 μM each of the four dNTPs, 0.8 μCi of deoxycitidine 5′-triphosphate [α32P] (3000 mCi/mmole, Perkin-Elmer Life and Analytical Science, Boston), 0.2 μM each of the forward and reverse primers (5′-GGCTGAGTGTCCGTCTCG- 3′and 5′-GTGGTAGATGTGCAGGAACT-3′, respectively; Integrated DNA Technologies Inc., Coralville, IA), 2.5 U of Taq DNA polymerase, 5 μL of 5X Taq master, 5 μL of 10X Taq buffer with Mg2+ (Eppendorf, Westbury, NY), Q. S. to 50 μL with distilled water. Thermocycling parameters for PCR were as follows: initial denaturation for 3 min at 95° C, 35 cycles of 94° C for 30 sec, 60° C for 30 sec and 72° C for 30 sec, and a final extension for 7 min at 72° C. Successful amplification of the PCR product in each reaction was checked by visualizing ethidium bromide stained-PCR product under UV light.
b. Polyacrylamide gel electrophoresis
Amplified samples were genotyped by electrophoresis of the PCR products, as previously described, in 38×50 cm 10% non-denatured polyacrylamide gels using a Sequi-Gen GT Sequencing Cell apparatus (Biorad, Hercules, CA) [24]. Negative (no DNA template) and positive (β-actin primers) controls were run along with the samples on each gel. Allelic ladders, DNA samples with known genotypes (confirmed through automated sequencing), representing the five possible alleles, were run along with samples on each gel. A DNA marker, (PhiX174 DNA/HinfI, Fermentas, Glen Burnie, MD) consisting of DNA fragments of length ranging from 48 to 726 nuleotides, was 5′-end labeled with 50 mCi adenosine 5′-triphosphate, [γ-32P] (3000 mCi/mmole, Perkin-Elmer Life and Analytical Science, Boston, MA) per 1 pmol of DNA fragments, and was run on each gel along with samples. Polyacrylamide gels were run for 5 h 30 min at a constant voltage of 1500 V. After electrophoresis, the gels were dried in a gel drier (Model 583, Biorad, Hercules, CA) and exposed to a phosphor screen for 2.5 h. Images were obtained by scanning the screens in a Typhoon 9200 phosphorimager (GE Healthcare Bio-Sciences Corp., Piscataway, NJ) and subsequently analyzed for GAG genotypes based upon fragment length of allelic ladders (174, 183, 186, 189 and 192 bp representing 4, 7, 8, 9 and 10 GAG repeats, respectively).
Determination of blood glutathione
Total glutathione (GSH and glutathione disulfide) was assayed enzymatically as previously described [14]. Briefly, total protein in blood samples was precipitated by adding 4 volumes of 5% metaphosphoric acid (MPA) followed by centrifugation (14000 g/3 min). The supernatant obtained was diluted 40-fold with assay buffer (0.1 M sodium phosphate plus 5 mM EDTA, pH 7.5) and mixed with 5,5′-dithiobis-2-nitrobenzoic acid (50 μL of 0.5 mg/mL) and GSH reductase (50 μL of 2.5 units/mL) in assay buffer. NADPH (50 μL of 0.72 mM) was added to each well and the initial reaction rates were measured at 405 nm on a microtiter plate reader (Synergy HT, Biotek, Winooski, VT). A standard curve was generated using different concentrations of pure GSH in the same plate. Protein-bound GSH in the MPA insoluble pellet was reduced by adding 500 μL of 1.3 M potassium borohydride to each sample and incubating at 40° C for 60 min. Reduced pellets were again treated with metaphosphoric acid and analyzed for GSH as described above. Hemoglobin concentration was determined by hemolyzing an aliquot of blood in distilled water and analyzing spectrophotomoetrically with Drabkin's reagent [25]. GSH levels were expressed as μmoles per gram hemoglobin.
Determination of GCL activity
GCL activity was determined by measuring the amount of the GCL product, γ-glutamylcysteine, formed after incubating the RBC protein lysates with cysteine and glutamic acid. Protein concentration in the RBC lysates was measured by the bicinchonic acid procedure (Pierce, Rockford, IL). Protein lysates from RBC were purified by centrifugal ultrafiltration (molecular weight cutoff 3000; 12,000 g/25 min) and were incubated for 30 min with 20-fold of 100 mM Tris buffer containing substrates glutamic acid (20 mM), cysteine (5 mM) and ATP (10 mM). The reaction was stopped by precipitating proteins with 1 volume of 5% MPA. Precipitated proteins were separated by centrifugation and the concentrations of γ-glutamylcysteine in supernatants were determined by HPLC with coulometric detection using a Bio-Sil ODS-5S, 5-μm, 4.0 × 250 mm, C18 column (Bio-Rad, Life Science Research Group, Hercules, CA) eluted with a mobile phase consisting of 50 mM NaH2PO4, 0.05 mM octane sulfonic acid, 1% (v/v) acetonitrile and 0.5% N,N dimethylformamide (v/v) (pH 2.52) at a flow rate of 1 ml/min. The CoulArray detector contained 8-channels set at 250, 400, 450, 500, 550, 600, 650, and 700 mV. The GCL activity was expressed as nmol/min/g.
Statistical Methods
The chi-square test for goodness of fit was used to find genetic equilibrium. The Student's t-test was used to compare GSH levels and GCL activity data among different genotypes, while the chi square test was used to test for trends. Data are expressed as mean ± standard error (S.E.). A p value of ≤ 0.05 was considered as significant.
Results
A typical image obtained by scanning is presented in Figure 1. A total of five alleles consisting of 4, 7, 8, 9 and 10 GAG repeats were found in the study subjects (Table 2). Allele frequencies in this study were comparable to previous reports [18, 26]. Earlier studies have reported that the 4 and 10 repeat alleles were restricted to blacks while 7, 8 and 9 repeat alleles were found both in blacks and whites. However, in our study, one 4-repeat allele was observed in a white female. The most frequent allele contained 7 GAG repeats (55.3%), followed by 9 (34.9%) and 8 GAG repeats (7.2%). A significantly higher frequency of the 9 GAG-repeat allele was observed in blacks compared to whites, while the 7 GAG-repeat allele was more prevalent in whites than in blacks (p = 0.005).
Figure 1. Representative Genotyping Analysis for GCLC GAG-Repeat Polymorphism.

P32–labeled PCR products were electrophorosed in 10% non-denatured polyacrylamide gels. A positive control (β-actin) and a negative control (water), indicated by white triangles, were run on each gel. Samples representing all alleles (indicated by black triangles) and 5′ γ-32P labeled ΦX174 DNA/HinfI (1st lane) were used as allelic and DNA markers, respectively.
Table 2. GAG-Repeat Allele Frequencies in the Study Population.
| Present Study | Walsh et al | Willis et al | ||||
|---|---|---|---|---|---|---|
| Allele Type a | Blacks b | Whites b | Blacks | Whites | Blacks | Whites |
| 4 | 2.7 (7) | 0.4 (1) | N/A | 0 | 2.6 | 0 |
| 7 | 48.0 (122) | 63.3 (138) | N/A | 54 | 44.7 | 61.7 |
| 8 | 4.7 (12) | 10.0 (22) | N/A | 11 | 1.8 | 12.9 |
| 9 | 42.9 (109) | 26.1 (57) | N/A | 35 | 46.1 | 25.4 |
| 10 | 1.5 (4) | 0 (0) | N/A | 0 | 4.8 | 0 |
Number of GAG repeats
Values are percent (number of alleles).
N/A: Study includes only whites.
Based upon the five alleles observed, a total of 15 genotypes are possible. Of these 15 genotypes, 10 were found in blacks and 7 in whites (Figure 2). The most frequent genotype was 7/9 (40%), followed by 7/7 (32%) and 9/9 (11%). The frequency of the 7/9 and 9/9 genotypes were greater in blacks compared to whites, consistent with the greater allelic frequency for the 9 repeat in blacks. Chi-square test for goodness of fit shows that the allele and genotype frequencies in the study population are in Hardy-Weinberg equilibrium (p > 0.99, df = 14, χ2 = 0.1). No significant difference was found between the observed and expected genotype frequencies.
Figure 2. GAG-Repeat Genotype Frequency in Study Subjects.

Genotypes were determined by separating (32P-dCTP labeled) PCR products containing the GAG repeat region in 10% non-denatured polyacrylamide gels. A total of 250 black and white adults were genotyped.
GSH levels ranged from 3.5 to 21.3 μmol/g hemoglobin in the study population with a mean ± S. E. of 9.6 ± 0.25. The distribution of total blood GSH levels (Figure 3) was normal and similar to previous reports [14]. Total blood GSH levels were significantly different between certain genotypes (Table 3). Among the most common genotypes, a decreasing trend in the blood GSH concentration in the order 7/9 > 7/7 > 9/9 (p < 0.05) was observed (Figure 4a). Compared to the 7/7 and 7/9 genotype, individuals with the 9/9 genotype had 11.6% and 14.6% less GSH/Hb, respectively (p = 0.05). Among homozygous individuals, those with the 8/8 genotype had the highest GSH levels. The GSH levels of subjects with the 8/8 genotype was 39.9% and 31.3% greater than those with the 9/9 and 7/7 genotypes, respectively (p = 0.05).
Figure 3. Distribution of GSH Levels and GCL Activity in Study Subjects.
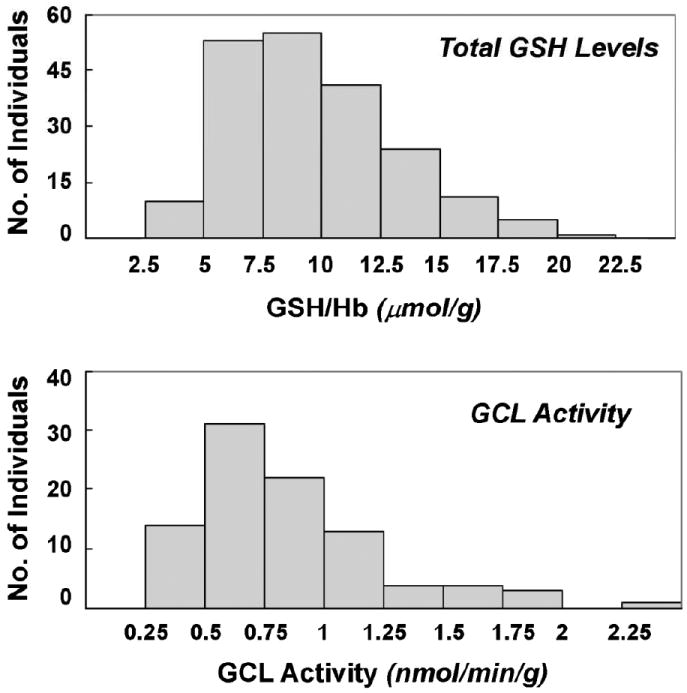
Blood GSH was measured in a total of 256 healthy black and white adults. Blood GCL activity was measured in 107 of the 256 adults.
Table 3. Total Blood GSH Levels in Individuals with Different GAG Genotypes.
| GCLC
Genotype a |
GSH/Hb (μmol/g) b | ||
|---|---|---|---|
| Blacks | Whites | Total | |
| 4/7 | 7.4 ± 1.0 (3) | 15.6 (1) | 9.4 ± 2.1 (4) |
| 4/9 | 7.5 ± 1.0 (3) | N/A | 7.5 ± 1.0 (3) |
| 7/7 | 9.0 ± 0.5 (26) | 9.6 ± 0.7 (32) | 9.3 ± 0.4 (58) |
| 7/8 | 9.6 (1) | 8.6 ±1.1(7) | 8.7 ±0.9 (8) |
| 7/9 | 9.4 ± 0.4 (46) | 10.0 ±1.2 (30) | 9.6 ± 0.3 (76) |
| 7/10 | 11.9, 5.5 (2) | N/A | 11.9, 5.5 (2) |
| 8/8 | 18.4, 11.2 (2) | 17.7, 7 | 13.6 ±2.7 (4) |
| 8/9 | 9.6 ± 1.8 (5) | 10.2 ± 1.4 (6) | 10.0 ± 1.1 (11) |
| 9/9 | 9.3 ± 0.8 (15) | 10.0 ± 1.5 (5) | 8.2 ± 0.5 (20) |
| 9/10 | 10.5 (1) | N/A | 10.5 (1) |
GAG repeat genotype
Values are mean ± SEM (N). Where n ≤ 2, actual values are given.
N/A: No individuals were present in that category.
Figure 4. Association of Total Blood Glutathione Levels and GCL Activities with GAG Repeat Genotypes.
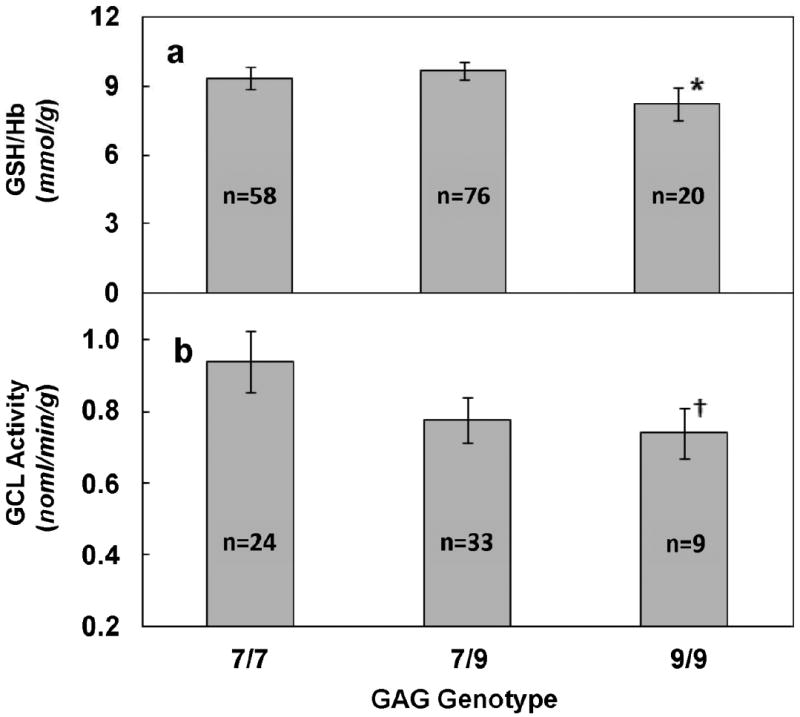
GSH levels (a) and GCL activity (b) were determined as described in the text. * Significantly different from 7/9 genotype (p=0.05). † Significantly different from 7/7 genotype (p < 0.05).
GCL activity levels ranged from 0.27 – 2.4 nmol/min/g with a mean ± S.E. of 0.86 ± 0.04. Associations between GCL activity by GAG genotypes are summarized in Table 4. GCL activity varied significantly among the major genotypes (Figure 4b). Individuals with the 9/9 genotype had significantly lower GCL activity levels than those with the 7/7 genotype (p<0.05). The number of subjects with the 8/8 genotype for which we had GCL activity data were too low to assess for differences among individuals with this homozygous genotype.
Table 4. GCL Activity in Individuals with Different GAG Genotypes.
| GCLC
Genotype a |
GCL Activity b (nmol/min/g) Mean ± S.E.(N) | ||
|---|---|---|---|
| Blacks | Whites | Total | |
| 4/7 | N/A | 0.81 (1) | 0.81 (1) |
| 4/9 | 1.7 (1) | N/A | 1.7 (1) |
| 7/7 | 0.87 ± 0.09 (10) | 0.99 ± 0.14 (14) | 0.94 ± 0.09 (24) |
| 7/8 | N/A | 0.74 ± 0.06 (3) | 0.74 ± 0.06 (3) |
| 7/9 | 0.83 ± 0.07 (19) | 0.70 ± 0.11 (14) | 0.78 ± 0.06 (33) |
| 7/10 | 1.18 (1) | N/A | 1.18 (1) |
| 8/8 | 0.74 (1) | N/A | 0.74(1) |
| 8/9 | 0.95, 1.05 (2) | 0.65 ± 0.11 (4) | 0.77 ± 0.10 (6) |
| 9/9 | 0.82 ± 0.10 (5) | 0.65 ± 0.08 (4) | 0.74 ± 0.07 (9) |
| 9/10 | 1.04 (1) | N/A | 1.04 (1) |
GAG repeat genotype.
Values are mean ± SEM (N). Where n ≤ 2, actual values are given.
N/A: No individuals were present in that category.
Discussion
Several studies have suggested that the GAG repeat polymorphism in GCLC may have a functional significance based on its association with disease risk/severity [21, 27, 28] and intracellular GSH levels [18]. We hypothesized that the GAG polymorphism in GCLC will affect GSH biosynthetic capacity in cells and will be reflected by differences in GCL activity and GSH levels in blood of healthy adults. Results from our study support this hypothesis by demonstrating an association between GAG genotype and both GSH levels and GCL activity. Individuals with the 9/9 genotype exhibited lower levels of GSH and GCL activity compared to other genotypes. Results obtained from this study suggest that, due to low GSH levels and GCL activity, the 9/9 genotype may contribute to higher risk for GSH-related or oxidative stress-induced diseases or toxicities compared to the 7/7 or 7/9 genotypes. Also since previous studies have demonstrated in colon, ovarian and small cell lung cancer cells that GCLC expression is inversely associated with resistance to chemotherapeutic drugs such as carboplatin and cisplatin, cancer patients with the 9/9 genotype might exhibit a greater response to tumoricidal chemotherapeutics and thus have a better prognosis [17].
There are few studies associating GCLC genotype with GSH levels or GCLC activity. Our findings are consistent with previous observations that individuals with schizophrenia, a disease characterized by low tissue levels of GSH [29, 30], had a lower frequency (30%) of the 7/7 GAG genotype than controls (50%) and higher frequency of the GAG 9/9 genotype (6.1%) than controls (0%) [19]. In the same study, individuals with 7/7 and 7/9 genotypes considered together had significantly higher GCLC mRNA, GCLC activity and GSH levels in cultured fibroblasts compared to individuals with all other genotypes together. These results are not consistent with the in vitro study by Walsh et al. [18], which demonstrated that cell lines with the 7/7 genotype had significantly lower intracellular GSH than cell lines with the 9/9 genotype. However, the panel of neoplastic cell lines used in this study may not be relevant to in vivo conditions in normal cells. In fact, GSH is well known to be induced in most cancer cells as a result of multiple changes in the regulation of its biosynthesis and metabolism [31].
We observed different distributions of GAG genotypes in blacks and whites. The higher frequency of the 9/9 genotype found among blacks is suggestive of a greater susceptibility to oxidative stress and its related diseases. Higher incidence rates for numerous such diseases have been observed for blacks compared to whites including cardiovascular diseases [32], diabetes [33] and cancers at several sites [34]. While few studies have examined differences in GSH status between blacks and whites, black diabetic patients were found to have significantly lower GSH levels than white patients [35].
While the mechanism of action is not known, it is unlikely that the GAG polymorphism affects the transcription of GCLC as it exists downstream of its transcriptional start sites [36]. Post-transcriptionally, the GAG repeats may differentially affect the translation efficiency and/or structure or stability of GCLC mRNA. Under physiological conditions, it has been speculated that the GAG repeats will form stable hair pin structures [37]. Such structures might impede scanning by the 40s ribosome of the 5′ UTR during translation as shown for CTG repeats in a reporter gene [38]. Transport of mRNA into the cytoplasm may be affected due to formation of nuclear foci consisting of clustered mRNA resulting from the occurrence of an abnormal number of repeats, thus decreasing the total mRNA available for translation [39]. However, this later affect has only been observed for polymophisms consisting of very high numbers of repeats such as thousands of CTG repeats in the DMPK gene instead of a normal number of 5-35 repeats. In a number of diseases where trinucleotide repeats are involved, a progressive change in the disease severity or phenotype is observed as the number of repeats increase [40, 41]. However, this might not be the case with GAG repeats in GCLC, as the number of repeats in alleles do not differ by more than six repeats as opposed to a difference of more than a hundred repeats in these other genes [42]. Also, the results from our study suggest that the relation between the GAG repeat polymorphism and GSH levels is not linear as individuals with the 8/8 genotype have significantly higher GSH levels than individuals with either the 7/7 and 9/9 genotypes. It is also possible that the mechanism of action might be directed by some other polymorphism such as a SNP, insertion or a deletion that is linked to the GAG repeats. For example, the -129 C/T (upstream transcription start site) polymorphism in the GCLC gene has been linked to GAG repeats [21].
To our knowledge, this study is the first to investigate the functionality of the GAG repeat polymorphism in the GCLC gene in healthy adults. This study establishes that individuals with the 9/9 genotype have significantly lower blood GSH levels and GCL activity compared to individual having the 7/7 or 7/9 genotype. It has yet to be evaluated if individuals with the 9/9 genotype are at higher risk for diseases involving oxidative stress. Also, additional studies are required to reveal the mechanism of action of the GAG repeats or its linkage with any other polymorphisms that affect GCL activity and GSH levels.
Acknowledgments
This study was supported in part by USPHS Grants P01CA68384 and K07CA104231.
We thank Dr. Thomas Spratt for his assistance with 32P scanning.
List of Abbreviations
- ATP
adenosine triphosphate
- DNA
deoxyribo nucleic acid
- EDTA
ethylene diamine tetra acetic acid
- GAG
the repeating unit of trinucleotide repeat polymorphism
- GCL
g-glutamyl cysteine lilgase
- GCLC
catalytic sub unit of g-glutamyl cysteine lilgase
- GCLM
modulatory sub unit of γ-glutamyl cysteine lilgase
- GSH
glutathione
- Hb
hemoglobin
- HPLC
high pressure liquid chromatography
- MPA
meta phosphoric acid
- mRNA
messenger ribonucleic acid
- NADPH
nicotinamide adenine dinucleotide triphosphate
- RBC
red blood cells
- SDS
sodium dodocyl sulphate
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sailendra N. Nichenametla, Cancer Prevention and Control Program, Penn State Cancer Institute, Department of Public Health Sciences, Penn State University College of Medicine, Hershey PA 17033
Irina Ellison, Department of Biology, St. Francis College, Brooklyn Heights, NY 11201.
Ana Calcagnotto, Cancer Prevention and Control Program, Penn State Cancer Institute, Department of Public Health Sciences, Penn State University College of Medicine, Hershey PA 17033.
Philip Lazarus, Department of Pharmacology, Penn State University College of Medicine, Hershey PA 17033.
Josh E. Muscat, Cancer Prevention and Control Program, Penn State Cancer Institute, Department of Public Health Sciences, Penn State University College of Medicine, Hershey PA 17033
John P. Richie, Jr., Cancer Prevention and Control Program, Penn State Cancer Institute, Department of Public Health Sciences, Penn State University College of Medicine, Hershey PA 17033
Literature Cited
- 1.Schroder CP, Godwin AK, O'Dwyer PJ, Tew KD, Hamilton TC, Ozols RF. Glutathione and drug resistance. Cancer Invest. 1996;14:158–168. doi: 10.3109/07357909609018891. [DOI] [PubMed] [Google Scholar]
- 2.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a coordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 3.Hill BG, Bhatnagar A. Role of glutathiolation in preservation, restoration and regulation of protein function. IUBMB Life. 2007;59:21–26. doi: 10.1080/15216540701196944. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn RV, Spitz DR, Liu X, Galoforo SS, Sim JE, Ridnour LA, Chen JC, Davis BH, Corry PM, Lee YJ. Metabolic oxidative stress activates signal transduction and gene expression during glucose deprivation in human tumor cells. Free Radic Biol Med. 1999;26:419–430. doi: 10.1016/s0891-5849(98)00217-2. [DOI] [PubMed] [Google Scholar]
- 5.Cotgreave IA, Gerdes RG. Recent trends in glutathione biochemistry--glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem Biophys Res Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- 6.Bojes HK, Datta K, Xu J, Chin A, Simonian P, Nunez G, Kehrer JP. Bcl-xL overexpression attenuates glutathione depletion in FL5.12 cells following interleukin-3 withdrawal. Biochem J. 1997;325(Pt 2):315–319. doi: 10.1042/bj3250315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arrigo AP. Gene expression and the thiol redox state. Free Radic Biol Med. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 8.Julius M, Lang CA, Gleiberman L, Harburg E, DiFranceisco W, Schork A. Glutathione and morbidity in a community-based sample of elderly. J Clin Epidemiol. 1994;47:1021–1026. doi: 10.1016/0895-4356(94)90117-1. [DOI] [PubMed] [Google Scholar]
- 9.Nuttall SL, Martin U, Sinclair AJ, Kendall MJ. Glutathione: in sickness and in health. Lancet. 1998;351:645–646. doi: 10.1016/s0140-6736(05)78428-2. [DOI] [PubMed] [Google Scholar]
- 10.Beutler E, Gelbart T, Kondo T, Matsunaga AT. The molecular basis of a case of gamma-glutamylcysteine synthetase deficiency. Blood. 1999;94:2890–2894. [PubMed] [Google Scholar]
- 11.Lang CA, Mills BJ, Mastropaolo W, Liu MC. Blood glutathione decreases in chronic diseases. J Lab Clin Med. 2000;135:402–405. doi: 10.1067/mlc.2000.105977. [DOI] [PubMed] [Google Scholar]
- 12.Lang CA, Naryshkin S, Schneider DL, Mills BJ, Lindeman RD. Low blood glutathione levels in healthy aging adults. J Lab Clin Med. 1992;120:720–725. [PubMed] [Google Scholar]
- 13.Coban T, Mabsout A, Eke BC, Bulbul D, Berberoglu U, Iscan M. Glutathione and lipid peroxidation levels in human breast tumors. Neoplasma. 1998;45:151–156. [PubMed] [Google Scholar]
- 14.Richie JP, Jr, Skowronski L, Abraham P, Leutzinger Y. Blood glutathione concentrations in a large-scale human study. Clin Chem. 1996;42:64–70. [PubMed] [Google Scholar]
- 15.Custodio HM, Broberg K, Wennberg M, Jansson JH, Vessby B, Hallmans G, Stegmayr B, Skerfving S. Polymorphisms in glutathione-related genes affect methylmercury retention. Arch Environ Health. 2004;59:588–595. doi: 10.1080/00039890409603438. [DOI] [PubMed] [Google Scholar]
- 16.Koide S, Kugiyama K, Sugiyama S, Nakamura S, Fukushima H, Honda O, Yoshimura M, Ogawa H. Association of polymorphism in glutamate-cysteine ligase catalytic subunit gene with coronary vasomotor dysfunction and myocardial infarction. J Am Coll Cardiol. 2003;41:539–545. doi: 10.1016/s0735-1097(02)02866-8. [DOI] [PubMed] [Google Scholar]
- 17.Yang P, Ebbert JO, Sun Z, Weinshilboum RM. Role of the glutathione metabolic pathway in lung cancer treatment and prognosis: a review. J Clin Oncol. 2006;24:1761–1769. doi: 10.1200/JCO.2005.02.7110. [DOI] [PubMed] [Google Scholar]
- 18.Walsh AC, Feulner JA, Reilly A. Evidence for functionally significant polymorphism of human glutamate cysteine ligase catalytic subunit: association with glutathione levels and drug resistance in the National Cancer Institute tumor cell line panel. Toxicol Sci. 2001;61:218–223. doi: 10.1093/toxsci/61.2.218. [DOI] [PubMed] [Google Scholar]
- 19.Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P, Deppen P, Preisig M, Ruiz V, Steullet P, Tosic M, Werge T, Cuenod M, Do KQ. Impaired glutathione synthesis in schizophrenia: Convergent genetic and functional evidence. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0706778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bekris LM, Viernes HM, Farin FM, Maier LA, Kavanagh TJ, Takaro TK. Chronic beryllium disease and glutathione biosynthesis genes. J Occup Environ Med. 2006;48:599–606. doi: 10.1097/01.jom.0000201845.02369.ba. [DOI] [PubMed] [Google Scholar]
- 21.Bekris LM, Shephard C, Janer M, Graham J, McNeney B, Shin J, Zarghami M, Griffith W, Farin F, Kavanagh TJ, Lernmark A. Glutamate cysteine ligase catalytic subunit promoter polymorphisms and associations with type 1 diabetes age-at-onset and GAD65 autoantibody levels. Exp Clin Endocrinol Diabetes. 2007;115:221–228. doi: 10.1055/s-2007-970574. [DOI] [PubMed] [Google Scholar]
- 22.Muscat JE, Djordjevic MV, Colosimo S, Stellman SD, Richie JP., Jr Racial differences in exposure and glucuronidation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Cancer. 2005;103:1420–1426. doi: 10.1002/cncr.20953. [DOI] [PubMed] [Google Scholar]
- 23.Park LY, Muscat JE, Kaur T, Schantz SP, Stern JC, Richie JP, Jr, Lazarus P. Comparison of GSTM polymorphisms and risk for oral cancer between African-Americans and Caucasians. Pharmacogenetics. 2000;10:123–131. doi: 10.1097/00008571-200003000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Fang JL, Lazarus P. Correlation between the UDP-glucuronosyltransferase (UGT1A1) TATAA box polymorphism and carcinogen detoxification phenotype: significantly decreased glucuronidating activity against benzo(a)pyrene-7,8-dihydrodiol(-) in liver microsomes from subjects with the UGT1A1*28 variant. Cancer Epidemiol Biomarkers Prev. 2004;13:102–109. doi: 10.1158/1055-9965.epi-03-0070. [DOI] [PubMed] [Google Scholar]
- 25.Fairbanks VF, Klee GG. Biochemical aspects of hematology. In: Tietz NW, editor. Fundamentals of clinical chemistry. Philadelphia: WB Saunders; 1987. pp. 789–824. [Google Scholar]
- 26.Willis AS, Freeman ML, Summar SR, Barr FE, Williams SM, Dawson E, Summar ML. Ethnic diversity in a critical gene responsible for glutathione synthesis. Free Radic Biol Med. 2003;34:72–76. doi: 10.1016/s0891-5849(02)01178-4. [DOI] [PubMed] [Google Scholar]
- 27.McKone EF, Shao J, Frangolias DD, Keener CL, Shephard CA, Farin FM, Tonelli MR, Pare PD, Sandford AJ, Aitken ML, Kavanagh TJ. Variants in the glutamate-cysteine-ligase gene are associated with cystic fibrosis lung disease. Am J Respir Crit Care Med. 2006;174:415–419. doi: 10.1164/rccm.200508-1281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang P, Bamlet WR, Ebbert JO, Taylor WR, de Andrade M. Glutathione pathway genes and lung cancer risk in young and old populations. Carcinogenesis. 2004;25:1935–1944. doi: 10.1093/carcin/bgh203. [DOI] [PubMed] [Google Scholar]
- 29.Engelen MP, Schols AM, Does JD, Deutz NE, Wouters EF. Altered glutamate metabolism is associated with reduced muscle glutathione levels in patients with emphysema. Am J Respir Crit Care Med. 2000;161:98–103. doi: 10.1164/ajrccm.161.1.9901031. [DOI] [PubMed] [Google Scholar]
- 30.Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Dis Markers. 2006;22:83–93. doi: 10.1155/2006/248387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. 2006;43:143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- 32.Melikian N, Wheatcroft SB, Ogah OS, Murphy C, Chowienczyk PJ, Wierzbicki AS, Sanders TA, Jiang B, Duncan ER, Shah AM, Kearney MT. Asymmetric dimethylarginine and reduced nitric oxide bioavailability in young Black African men. Hypertension. 2007;49:873–877. doi: 10.1161/01.HYP.0000258405.25330.80. [DOI] [PubMed] [Google Scholar]
- 33.Mehrotra S, Ling KL, Bekele Y, Gerbino E, Earle KA. Lipid hydroperoxide and markers of renal disease susceptibility in African-Caribbean and Caucasian patients with Type 2 diabetes mellitus. Diabet Med. 2001;18:109–115. doi: 10.1046/j.1464-5491.2001.00416.x. [DOI] [PubMed] [Google Scholar]
- 34.Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–280. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]
- 35.Jain SK, McVie R. Effect of glycemic control, race (white versus black), and duration of diabetes on reduced glutathione content in erythrocytes of diabetic patients. Metabolism. 1994;43:306–309. doi: 10.1016/0026-0495(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 36.Gipp JJ, Mulcahy RT. Structure of the human glutamate-L-cysteine ligase catalytic (GLCLC) subunit gene. Cytogenet Cell Genet. 2000;88:130–132. doi: 10.1159/000015505. [DOI] [PubMed] [Google Scholar]
- 37.Mitas M. Trinucleotide repeats associated with human disease. Nucleic Acids Res. 1997;25:2245–2254. doi: 10.1093/nar/25.12.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raca G, Siyanova EY, McMurray CT, Mirkin SM. Expansion of the (CTG)(n) repeat in the 5′-UTR of a reporter gene impedes translation. Nucleic Acids Res. 2000;28:3943–3949. doi: 10.1093/nar/28.20.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harper PS. Trinucleotide repeat disorders. J Inherit Metab Dis. 1997;20:122–124. doi: 10.1023/a:1005388218625. [DOI] [PubMed] [Google Scholar]
- 41.Tohgi H, Utsugisawa K, Kawamorita A, Yamagata M, Saitoh K, Hashimoto K. Effects of CTG trinucleotide repeat expansion in leukocytes on quantitative muscle histopathology in myotonic dystrophy. Muscle Nerve. 1997;20:232–234. doi: 10.1002/(sici)1097-4598(199702)20:2<232::aid-mus16>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 42.Siianova E, Mirkin SM. Expansion of trinucleotide repeats. Mol Biol (Mosk) 2001;35:208–223. [PubMed] [Google Scholar]