

Abstract
Biologically active natural products often contain particularly challenging structural features and functionalities. Perhaps foremost among these difficulties are issues of stereochemistry. A useful strategy for synthesizing these molecules is to devise novel methods of bond-formation that provide new opportunities for enantioselective catalysis. In using this tactic, target structures define the problems to be solved and ultimately drive development of catalysis forward. New enantioselective methods discovered in the context of these total synthesis efforts then contribute to a greater understanding of fundamental bond construction and lead to valuable synthetic technologies useful for a variety of other applications.
Inasmuch as retrosynthetic analysis1 enables synthetic planning toward valuable chemical substances, one can be easily discouraged by a lack of methods to install particular functionalities or structural motifs present in the target molecule. Still more difficult is adapting a strategic approach to incorporate the relatively limited number of catalytic enantioselective transformations to allow preparation of enantioenriched materials for further studies.2,3,4,5,6 However, when encountering the structural challenges presented by important natural products and pharmaceutical compounds a fruitful strategy is to design a synthesis that hinges upon a particular bond disconnection that is beyond the present lexicon of enantioselective transformations. Employing this strategy, the structures of target molecules provide the impetus for the development of novel transformations and lead to a greater fundamental understanding of methods of bond construction and catalysis. In this Review several recent examples of novel catalytic enantioselective transformations demonstrate the effectiveness of this useful strategy for the preparation of important structural motifs found in biologically active molecules. Each example has contributed not only an effective means of accessing a particular target structure, but also a useful new tool for a variety of further applications in synthetic chemistry.
To provide an overview of established catalytic enantioselective methods developed in the context of total synthesis, several notable examples are highlighted in Figure 1. In each of these cases, the target molecules posed particular challenges that had not yet been solved by enantioselective catalysis. Although in some instances (e.g., Diels–Alder reaction, Figure 1a) the methods were developed before their first applications in total synthesis, but the demonstrated value of the transformation served to further highlight the need for enantioselective variants. Subsequent to the development of the [4 + 2] cycloaddition by Diels and Alder in the 1920s,7 many studies of this transformation elucidated several key facets of the stereochemical outcome of the reaction (e.g., the “endo rule”, regioselectivity, and diastereoselectivity). These intrinsic stereochemical control elements proved useful when the Diels–Alder reaction was first featured in a total synthesis with Stork's stereocontrolled synthesis of cantharidin in 1951.8 Subsequently, the thermal Diels–Alder reaction found application in several total syntheses, perhaps most famously in Woodward's landmark synthesis of reserpine.9 Enantioselectivity in this transformation remained elusive, however, and perhaps was considered unattainable. One key practical improvement of the method was Yates' discovery that Lewis acids remarkably increased the reaction rate.10 Many laboratories sought to exploit Yates' discovery to develop asymmetric versions of the Diels–Alder reaction catalyzed by chiral Lewis acids, culminating with Koga's report of the first highly enantioselective catalytic Diels–Alder reaction in 1979.11 The interface between reaction development, mechanistic study, and synthesis is readily apparent from the multitude of chiral Diels–Alder catalysts and accompanying enantioselective total syntheses reported to date.12,13,14 These successes validate the extensive efforts directed toward the realization of this important goal.
Figure 1. Selected examples of enantioselective catalysis in total synthesis.

Natural products and pharmaceuticals often possess challenging structural features and functionalities, particularly stereochemical elements. (a) Corey's synthesis of prostaglandin E2 utilizing an asymmetric Diels–Alder reaction13 and carbonyl reduction.16 Various industrial applications, including (b) the asymmetric hydrogenation toward α-amino acid building blocks,19 (c) the asymmetric epoxidation toward Merck's HIV protease inhibitor Crixivan®,20 and (d) the asymmetric isomerization of allylic amines en route to commodity chemicals,21 have explored the limits of catalysis and efficiency to minimize cost. The asymmetric construction of C–C bonds, apparent in (e) the amino acid-catalyzed intramolecular aldol condensation toward important steroid building blocks,27 and (f) various enantioselecitive intermolecular aldol reactions toward fragments of the cytostatic agent phorboxazole B,28 maximize the use of stereochemical information of precious intermediates through convergency. (Reduced to 45%)
Other methods were developed to address more general synthetic problems (e.g., enantioselective ketone reduction, Figure 1a), however, these key structures are themselves embedded in a variety of important natural products and pharmaceutical compounds. In the case of Corey's approach to the prostaglandins, first reported in the 1960s,15 control of the configuration of the sidechain allylic alcohol at C(15) required stoichiometric chiral reducing agents until a solution to this long-standing problem was disclosed in the 1980s.16 Interestingly, the oxazaborolidine catalyst discovered in these explorations has found other varied applications in synthesis and catalysis13,17 demonstrating the versatility of priveledged molecular frameworks18 in enantioselective catalysis.
The practical impact of enantioselective catalysis is apparent in myriad industrial applications (e.g., Figure 1b–d) where the limits of catalysis must be examined in order to minimize costs. Important industrial applications include synthesis of chiral building blocks (e.g., amino acids, Figure 1b19), novel biologically active pharmaceuticals (e.g., Crixivan®, Figure 1c20), and commodity chemicals with various important uses (e.g., menthol, Figure 1d21). Only the most efficient methods are feasible for large-scale industrial synthesis and in many ways these protocols represent the zenith of modern enantioselective catalysis.22,23,24 A viable commercial operation must account for more than effective asymmetric induction; factors including turnover frequency, catalyst availability, catalyst recovery, catalyst toxicity, and feasible large-scale handling procedures must all be considered for industrial applications. These daunting challenges serve to further underscore the persisting demand for evermore efficient catalyst systems.
To maximize the utility of the stereochemistry imparted by these key asymmetric transformations, subsequent diastereoselective reactions may be used in order to control the formation of many stereocenters based on a single enantioselective transformation (e.g., Figure 1e). The Hajos–Parrish ketone, first prepared in the context of steroid synthesis, has been used extensively in other synthetic efforts, proving to be a versatile chiral pool starting material based on a variety of diastereoselective transformations developed around this versatile bicyclic platform.25,26,27 Interestingly, the amino acid catalyst system developed for this intramolecular aldol condensation provided a sound basis for the recent use of organic molecules as catalysts for a variety of enantioselective transformations (vide infra).
The use of several different enantioselective reactions to prepare enantioenriched fragments of complex molecules enhances efficiency through convergency. The importance of this strategy is shown in the variety of extraordinarily complex polyketide natural products that have been prepared through asymmetric intermolecular aldol reactions (e.g. phorboxazole B, Figure 1f28). The challenging structure of these molecules has required the development of several related protocols to address the subtle differences in substitution patterns and functionality present in substrates, and despite many successes, studies are ongoing.29 Indeed, polyketide natural products exemplify the continuing need for reaction development in order to address stereochemically complex substructures.
The remaining examples in this Review detail recent representative developments of this paradigm for the construction of biologically important target molecules. Most of these involve the formation of C–C bonds, which compose the fundamental structure of organic molecules. These cases were selected to illustrate some of the latest developments in enantioselective catalysis for complex molecule synthesis. Special attention has been given to reactions that address some of the most significant challenges to synthetic chemistry today: increasing functional group tolerance, generating new carbocylic- and heterocyclic-rings, and forming all-carbon quaternary stereocenters. The examples are intended to demonstrate the important symbiosis between total synthesis and method development and to show that improvements in one branch of synthetic chemistry can have a great impact in another.
β-Enamino Amide Hydrogenations – Januvia®
Throughout more than forty years of development, catalytic enantioselective hydrogenation has become one of the most effective and powerful methods for the synthesis of chiral α-amino acids for numerous applications.19 Over the past decade, the utility of their homologous building blocks, β-amino acids, in pharmaceutical, agrochemical, β-peptide, and natural substances has highlighted the need for a general and effective means of their preparation.30,31 Undoubtedly, the implementation of a catalytic, asymmetric hydrogenation N-acyl-β-enamino esters seemed to be the most efficient pathway toward their synthesis, although initial investigations realized poor selectivities.32 Additional syntheses utilizing the chiral pool, auxiliaries, and more recently the catalytic, asymmetric generation of C–C and C–N bonds, have been successful in satisfying the increased demand for β-amino acids.31 These valuable methods enable flexible strategies for the synthesis of a variety of analogues; however, most examples are limited by the requirement that further chemical manipulation is often necessary to reveal the functionality of the desired β-amino acids.
Despite initial difficulties, the asymmetric hydrogenation of N-acyl-β-enamino esters has been developed into a useful method over the past 15 years.33,34 This fruitful endeavour has demonstrated that a number of transition metal and ligand combinations are competent for the preparation of N-acyl-β-amino acids with good to excellent enantioselectivities. A notable drawback to this strategy, however, is the requirement of the seemingly indispensable N-acyl group on the β-enamino esters, which enables metal chelation to improve reactivity and selectivity. The introduction of this moiety often produces enamine olefin isomers that can be difficult to separate, and importantly, the individual isomers are typically hydrogenated with differing rates and selectivities. Moreover, these difficulties were magnified by the necessary removal of this group, a seemingly cumbersome artifact of an otherwise powerful strategy. Nonetheless, this has enabled the preparation of a variety of β-amino acids.31
An innovative solution to this problem was demonstrated by a Merck group en route to Januvia® (sitagliptin phosphate, 8, Figure 2), a recent FDA-approved treatment for type II diabetes mellitus.35 The optimal target contains an unfunctionalized β-amino amide, and thus a strategy was sought to install this moiety directly by means of an asymmetric hydrogenation of unsubstituted β-enamino ester and amide derivatives (e.g., 4).36 A traditional hydrogenation route to amino acids is a proven, cost-effective method for the synthesis of chiral building blocks, and the industrial infrastructure is already in place to realize this goal, however, reduction of unprotected β-enamino acids was not effective with existing chiral catalysts. A critical element to address such limitations is Merck's high-throughput screening facility, a platform technology to rapidly screen catalyst structures and reaction conditions, an essential component for the success of any catalytic, asymmetric process.37 One potential complication for this hydrogenation strategy was avoided when it was observed that the preparation of the β-enamino ester and amide (e.g., 3 → 4) substrates proceeded with complete selectivity for the (Z)-isomer, presumably due to hydrogen bonding in the products.
Figure 2. Asymmetric hydrogenation of β-enamino amides toward Januvia® (8).

The rapid and selective synthesis of β-enamino amide 3 enabled a key rhodium/5-catalyzed enantioselective hydrogenation under mildly acidic conditions to directly reveal the β-amino amide in Merck's synthesis of Januvia®, an FDA-approved treatment for type II diabetes.36,39 DMAP, 4-(N,N-dimethylamino)pyridine; DMA, N,N-dimethylacetamide; PivCl, pivaloyl chloride; COD, cyclooctadiene. (Reduced to 45%)
A survey of transition metals and ligands revealed that rhodium complexes of the Josiphos (e.g., 5) family of ligands efficiently catalyze the hydrogenation of a variety of substrates in high yields with excellent enantioselectivities. The remarkable functional group tolerance of this catalyst enabled the strategic implementation of this asymmetric transformation as the penultimate step of the synthesis, thereby maximizing the utility of the process and materials. Thus, phenylacetic acid derivative 1 was converted into β-ketoamide 3 in a one pot procedure via acylation of Meldrum's acid, followed by treatment with triazole salt 2 (Figure 2).38 Exposure to ammonium acetate converted this into β-enamino amide 4 as a single enamine isomer. Hydrogenation of 4 in the presence of 0.30 mol% of rhodium(I) and ligand 5 provided β-amino amide 7 in >95% conversion and 95% enantiomeric excess (ee). Subsequent recrystallization and salt formation with phosphoric acid gave Januvia® (sitagliptin phosphate, 8). Efforts to optimize efficiency and examine the mechanism of the asymmetric process revealed a reactivity and selectivity dependence on the pH of the reaction solution.39 It was found that ∼1 mol% of a mild acid (i.e., ammonium chloride) was essential for the reaction to proceed reproducibly on large scale. Additionally, it was observed that hydrogenation of a related substrate under identical conditions with a D2 atmosphere resulted in deuterium incorporation at the β-position only, suggesting the intermediacy of an imine (i.e., 7) and that an enamine/imine tautomerization process plays an important role in the mechanism.36 Interestingly, intermediates such as 7 share a striking similarity to asymmetric β-carbonyl hydrogenations pioneered by Noyori.40
This example demonstrates the evolution of asymmetric catalysis to a state of the art science through efficiency by minimization of unnecessary functionality, atom economy, and extremely active catalysts. Moreover, the development of the catalyst system for the synthesis of Januvia® exemplifies the continued need for subtly different catalysts to meet new synthetic demands. Building on the experience attained during the development of highly efficient enamide reduction toward α-amino acids, large-scale industrial synthesis of increasingly important β-amino acids has been a relatively rapid process.
C(sp3)–C(sp3) Cross-Couplings – Fluvirucinine A1
Transition metal-catalyzed cross-coupling reactions have been used extensively for the construction of C–C bonds, and consequently have had a substantial impact on the field of complex molecule synthesis.41,42 The predominance of palladium and nickel catalysts in cross-coupling technologies and their extraordinary functional group tolerance increases the efficiency of this process through convergency by allowing a large degree of functionalization prior to coupling. Moreover, the efficacy of this cross-coupling strategy for streamlining synthesis has enabled retrosynthetic analyses previously thought impossible with standard, non-metal reactions. Until recently, however, the majority of cross-coupling methods involved C(sp2)–C(sp2) or C(sp2)–C(sp) centers, limiting the application potential. Two critical issues associated with expanding the substrate scope to include C(sp3)–C(sp3) couplings are the relative low reactivity of alkyl halides toward oxidative addition, and the propensity of σ-alkyl organometallic complexes to undergo rapid β-H elimination reactions.43,44 Practical solutions to this problem were first presented by Suzuki and Knochel, followed more recently by Fu and others.44,45 In general, the reaction scope now encompasses a variety of primary and secondary halides and pseudohalides as the electrophilic component with organoboranes, boronic acids, alkylmagnesium halides, and alkylzinc halides as the nucleophilic component.44 Although perhaps not developed in the context of a particular target molecule, progress in these cross-coupling methods has allowed retrosynthetic disconnections that were not practical previously. Asymmetric cross-coupling protocols could in turn enable the direct formation of remote stereocenters in relatively unfunctionalized molecules.
Early examples of catalytic, asymmetric cross-coupling reactions involving C(sp3)–C(sp2) centers explored by Kumada and coworkers in the late 1970's were met with modest enantioselectivities.46,47 Despite these initial reports and the subsequent evolution of cross-coupling methods and asymmetric catalysis, a deficiency in the development of catalytic, asymmetric methods for C(sp3)–C(sp3) couplings existed until Fu and coworkers reported an asymmetric Negishi coupling in 2005.48,49 Prior to this report, the Fu lab observed the proficiency of tridentate pybox ligands (e.g., 16, Figure 3) to enable the room-temperature, nickel-catalyzed Negishi coupling of symmetrical secondary alkyl bromides and iodides.50 It was postulated that the tridentate nature of pybox ligands prevented the undesired β-H elimination pathway, which would require a vacant coordination site. Reaction optimization facilitated the development of several asymmetric variations that generate challenging stereocenters applicable to complex molecule synthesis, as demonstrated in Fu's formal total synthesis of fluvirucinine A1 (17), the aglycon of macrolactam antibiotic fluvirucin A1 (18).51 A key nickel(II)-catalyzed asymmetric cross-coupling of racemic allylic chloride 9 and alkylzinc reagent 10 in the presence of (S,S)-16 generated γ-disubstituted enone 11 in excellent yield and 96% ee. Elaboration over two steps to bromide 12, followed by conversion to the alkylzinc and a second nickel(II)-catalyzed asymmetric Negishi cross-coupling with racemic allylic chloride 13, provided ester 14 in good yield and >98% ee with a 15:1 diastereomer ratio (d.r.) A subsequent two-step conversion to aldehyde 15 intersects Suh's52 synthesis of fluvirucinine A1 (17). This method exemplifies the efficiency of the C(sp3)–C(sp3) cross-coupling and presents a creative solution to the particularly difficult challenge of remote stereochemical control.
Figure 3. Enantioselective C(sp3)–C(sp3) cross-coupling toward fluvirucinine A1 (17).
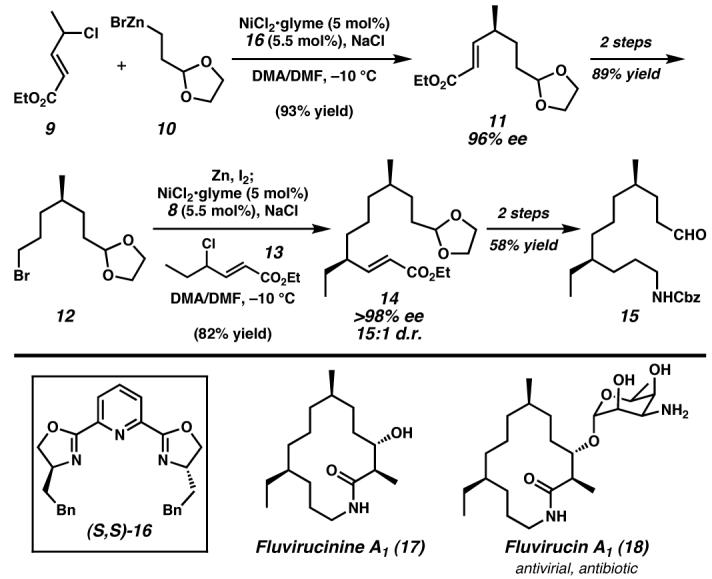
Sequential asymmetric C(sp3)–C(sp3) Negishi cross-couplings of racemic allylic chlorides and alkylzinc reagents catalyzed by nickel/(S,S)-16 enabled the rapid formal synthesis of Fluvirucinine A1 with excellent enantio- and diastereoselectivity, highlighting a creative solution to remote stereochemical control in unfunctionalized systems.51 Glyme, 1,2-dimethoxyethane; DMF, N,N-dimethylformamide. (Reduced to 45%)
At present, the majority of examples of this technology require a stabilizing group adjacent to the site of the putative carbon-centered radical. Eliminating this condition would further improve the utility of this asymmetric cross-coupling method. Additionally, stereogenic organometallic coupling partners (e.g., secondary alkylzinc reagents) have not yet been reported in this asymmetric transformation. A potential goal for this synthetic method would be combination of a racemic secondary alkyl halide and a racemic secondary alkylmetal reagent to form vicinal stereocenters along an alkyl chain with high levels of enantio- and diastereoselectivity.
Intramolecular Heck Cyclizations – Minfiensene
The asymmetric generation of all-carbon quaternary stereocenters represents a significant challenge in synthesis.53 Since quaternary stereocenters are found in many natural product structures, convenient enantioselective methods for their formation would be useful. One such method is the Heck reaction,54 wherein a palladium(0) catalyst promotes the vinylation of an aryl or vinyl halide or triflate. The large body of literature on palladium catalysis and mechanism,41 as well as an ever-growing collection of chiral ligands for transition-metal catalysis, greatly increased the potential of this method toward asymmetric catalysis. Additionally, an abundance of synthetic endeavors employing diastereoselective or non-stereoselective intramolecular Heck reactions have appeared,55 increasing the significance of an enantioselective process. In 1989, the labs of Shibasaki56 and Overman57 independently reported the first variants of an intramolecular catalytic, asymmetric Heck reaction. Initial levels of enantioselectivity were moderate; however, subsequent optimizations realized good to excellent selectivities in the generation of tertiary and all-carbon quaternary stereocenters.58
Indole alkaloids comprise a large number of natural and pharmaceutical substances that possess a wide range of biological activities.59 The plant alkaloid minfiensine (22, Figure 4) represents a compelling example of the all-carbon quaternary stereocenter motif in bioactive natural products. Minfiensine and related alkaloids have been used in traditional medicines and possess promising anticancer activity.60 The intriguing polycyclic structure and biological relevance of minfiensine (22) prompted the Overman laboratory to explore a catalytic, enantioselective Heck reaction to generate the sole quaternary stereocenter at C(9a).61 It was discovered that the palladium-catalyzed intramolecular Heck reaction of dienyl aryl triflate 19 in the presence of phosphinooxazoline ligand (S)-23 under microwave conditions produced indoline 20 in good yield 99% ee. Subsequent acid promoted carbamate cyclization produced the tricyclic core of minfiensine (i.e., 21) that was then elaborated to the natural product. The efficiency and selectivity of the catalytic, asymmetric Heck reaction facilitated completion of the target, where the remaining stereocenters are derived from this initial transformation.
Figure 4. Asymmetric Heck cyclization toward minfiensine (22).

The palladium/23-catalyzed enantioselective intramolecular Heck cyclization of 19 forged the C(9a) all-carbon quaternary stereocenter, and upon acid-mediated cyclization, the polycyclic core of minfiensine. The application of this method allowed the diastereoselective preparation of the remaining stereocenter and completion of the alkaloid.61 PMP, 1,2,2,6,6-pentamethylpiperidine. (Reduced to 45%)
Despite numerous examples of the asymmetric Heck reaction in total synthesis,58 there are several features that could be improved in the coming years. Reactions typically require high temperatures and somewhat high catalyst loadings, and the development of chiral ligands that greatly increase the reactivity of the transition metal while maintaining an adequate asymmetric environment would be greatly beneficial. Since most enantioselective Heck reactions employ an sp2-hybridized organohalide component, another frontier lies in the application of unactivated alkyl carbon electrophiles that possess β-hydrogens in both intramolecular and intermolecular cases, and area currently in its infancy.62
Indole Friedel–Crafts Alkylations – Flustramine B
Numerous methods have been developed for the generation of substituted indoles,63,64 however, enantioselective indole functionalization is far less explored. To address the deficiencies in the indole functionalization literature, Jørgensen65 and MacMillan66 independently developed strategies for asymmetric Friedel–Crafts alkylation of conjugate acceptors with electron-rich heteroaromatics in 2001. The method of MacMillan employs secondary amine catalyst 28 (Figure 5) that facilitates the LUMO-lowering activation of α,β-unsaturated aldehydes for a variety of transformations.67,68 Although imidazolidinone 28 was a sufficient catalyst for the Friedel–Crafts alkylation of pyrroles with good yields and excellent enantioselectivities,66 application of less-activated indole substrates resulted in sluggish reactivity with considerably diminished selectivities.69 Kinetic investigations of iminium-catalyzed reactions revealed that the overall reaction rate was influenced by the efficiency of formation for both the iminium ion and C–C bond, prompting the development of a modified imidazolidinone catalyst (29). This refinement minimized the steric bulk around one face of the catalyst, thereby exposing the nitrogen lone-pair. This structural change translated into increased reactivity that enabled the asymmetric Friedel–Crafts alkylation of a variety of indoles in good to excellent yields with very high enantioselectivities.69
Figure 5. Amine-catalyzed indole alkylation toward flustramine B (27).
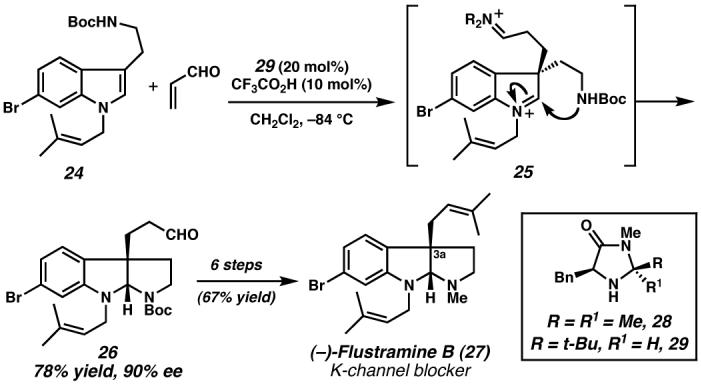
The enantioselective C(3a) alkylation of tryptamine derivative 24 catalyzed by amine 29, upon subsequent intramolecular cyclization, provided the pyrroloindoline core of (−)-flustramine B. This selective cascade process prepared the all-carbon quaternary stereocenter rapidly and efficiently enabling completion of this K-channel blocker.74 (Reduced to 45%)
Pyrroloindoline alkaloids represent a family of polyindole alkaloids of diverse structural complexity and biological relevance.70 Diastereoselective syntheses of this core have focused on the control of the C(3a) all-carbon quaternary stereocenter as a key design element.71,72,73 With a powerful and mild indole alkylation method in hand, MacMillan and coworkers devised a cascade strategy for the catalytic, asymmetric preparation of the C(3a) stereocenter and the pyrroloindoline core of potassium-channel blocker (−)-flustramine B (27) in one step.74 In this key transformation, tryptamine derivative 24 and acrolein, in the presence of catalyst 29, underwent the asymmetric Friedel–Crafts alkylation to provide iminium intermediate 25. Subsequent carbamate cyclization and hydrolysis to regenerate the catalyst provided the core (26) in good yield and 90% ee. Importantly, this enabled completion of (−)-flustramine B (27) in just 6 steps and good overall yield and highlights the efficiency of this cascade approach. It is noteworthy that this strategy is not reserved for the pyrroloindoline architecture, but also has potential for application to various polycyclic indolines such as the diazonamide family of cytotoxic alkaloids.74 It is interesting to note the capacity of both the intramolecular Heck reaction (vide supra) and indole Friedel–Crafts alkylation to generate similar indoline structural motifs despite the drastically different bond-connecting strategies. The success of these dissimilar strategies allows for a great deal of flexibility in synthetic planning.
Although iminium activation methods with chiral amine catalysts have been successful for numerous transformations, catalyst loading, turnover frequency, and excesses of certain reagents limit the large-scale industrial application of these methods. Additionally, in some cases the organic catalyst may be more difficult to remove from the reaction products than a metal catalyst. However, the typically air- and moisture-stable reaction conditions, low cost of some catalysts, and often metal-free conditions are attractive. The variety of asymmetric transformations (some proceeding through substantially different reaction pathways) realized with chiral amine catalysts to date indicates a burgeoning field of useful enantioselective catalysts.
Pictet–Spengler Cyclizations – Harmicine
Since the intramolecular cyclization of an aromatic ring onto an iminium species was reported by Pictet and Spengler in 1911,75 the transformation has been of great use in the synthesis of many important alkaloid natural products.76 Indeed, the need for asymmetric variants of this reaction was recognized and several diastereoselective protocols have been devised.76 A common approach to diastereoselective Pictet–Spengler cyclization has been to employ tryptophan derivatives to control the stereochemistry of the cyclization. However, such a technique in the synthesis of a natural product such as the anti-leishmania compound harmicine (35, Figure 6) necessitates the removal of the stereocontrol element at C(5) following the diastereoselective cyclization. Nonetheless, Allin and coworkers proved this a method viable method in 2007.77 This particular structure, however, highlighted a challenge for enantioselective catalysis and an opportunity to improve synthetic efficiency.
Figure 6. Enantioselective Pictet–Spengler cyclization toward harmicine (35).

Facile prepartion of hydroxylactam 31 facilitated the asymmetric Pictet–Spengler cyclization catalyzed by thiourea 32. The enantioselective N-acyl iminium ion cyclization enabled rapid construction of the alkaloid (+)-harmicine following this key transformation.81 TMSCl, chlorotrimethylsilane; THF, tetrahydrofuran. (Reduced to 45%)
When considering prospects for asymmetric induction, Jacobsen and Taylor considered activated N-acyliminium ions as a template and reasoned that a chiral thiourea derivative might be effective in promoting cyclization. In practice these Brønsted acids,78 as well as other Brønsted acids investigated later by other groups,79,80 proved to be excellent catalysts for enantioselective indole annulations with in situ generated N-acyliminium species (e.g., 33). In later studies by Jacobsen and coworkers it was found that hydroxylactams (e.g., 31) are convenient precursors to N-acyliminium ions and in turn enabled access to various polycyclic structures.81,82 With this effective protocol in hand, an efficient catalytic asymmetric synthesis of harmicine (35) was realized in four steps from tryptamine (30). Several mechanistic experiments have suggested that asymmetric induction is controlled by a complex of the Brønsted acid catalyst (32) and a chloride counterion closely associated with the iminium ion (e.g., 33) that effectively blocks approach to one face of the electrophile. This insight into the remarkable mechanism of this transformation has led to a related C–C bond forming process employing oxocarbenium ions.83 Further exploitation of this unusual proposed catalyst/anion interaction could lead to a variety of other asymmetric addition reactions, for example, intermolecular alkylation N-acyl iminium ions. Paralleling the history of the Diels–Alder reaction (vide supra), the exploration of the Pictet–Spengler cyclization has provided a useful method to access many heterocyclic structures embedded in alkaloid natural products using a classical reaction with well-established synthetic applications.
Phase Transfer Alkylations – Indacrinone
Enolate alkylations exemplify the fundamental utility of the carbonyl group for C–C bond formation. Strategies to induce asymmetry in these reactions have included chiral auxiliaries and chiral ligands, although few examples are catalytic. A particularly challenging class of product targets are all-carbon quaternary stereocenters adjacent to carbonyl groups. One example of an important target bearing this motif is the diuretic drug candidate indacrinone (40, Figure 7).84 Given the lack of efficient methods for synthesizing this structure, researchers at Merck envisioned an enantioselective phase transfer alkylation method based on a quaternary ammonium salt derived from a naturally occurring cinchona alkaloid (e.g., 37). In the event, readily prepared indanone 36 was methylated in 95% yield and 92% ee and then converted to indacrinone (40) in three additional steps. Although successful in achieving enantioselective enolate alkylation, the mechanism of this process appears to be very complicated;85 however, enantiofacial selectivity in the alkylation event may be rationalized through hypothetical transition state 38. Three key interactions are thought to control selectivity: a hydrogen bond between the enolate oxygen and the catalyst hydroxyl group, and two π-stacking interactions between the four aromatic rings. Perhaps a consequence of the complex mechanism, the substrate scope for enolate alkylation is unfortunately limited and other solutions to this problem are still necessary. However, these initial results have led to several related catalytic enantioselective reactions employing cinchoninium salts or related organic ammonium complexes as catalysts.86 The discovery of these useful catalysts has provided not only an alternative to related transformations using metal catalysts, but also a means of accessing chiral environments that are simply not possible with metal-based catalysts. Moreover, eliminating metal waste materials is attractive from an industrial and environmental standpoint. Ultimately, the studies directed toward an enantioselective synthesis of indacrinone (40) demonstrate the versatility of privileged catalysts developed for the synthesis of target molecules for a range of other applications.
Figure 7. Asymmetric phase-transfer alkylation toward indacrinone (40).
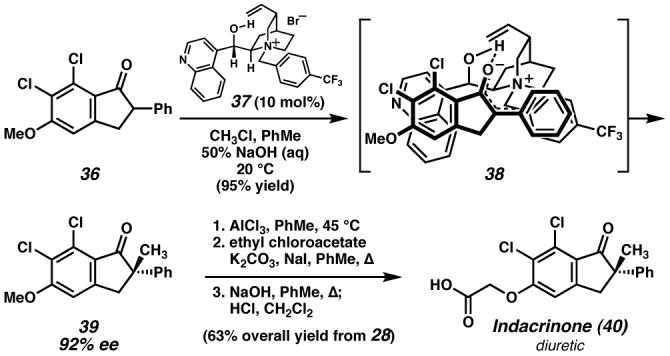
Merck scientists developed an enantioselective alkaloid salt-catalyzed phase-transfer enolate alkylation to gain access to indanone 39 with excellent stereoselectivity en route to the diuretic drug candidate indacrinone.84 (Reduced to 45%)
Pd-Catalyzed Enolate Alkylation – Cyanthiwigin F
A recent case of enantioselective enolate alkylation is the synthesis of the cytotoxic marine natural product cyanthiwigin F (48, Figure 8). The cyanthiwigin family is composed of over thirty diterpenoids, most of which bear two quaternary stereocenters, at C(6) and C(9), and a syn-relationship of the methyl groups in the central ring. These central stereochemical elements are a complicating factor for a convergent strategy that might seek to couple the five- and seven-membered ring portions and subsequently form the six-membered ring. To avoid this difficulty, Enquist and Stoltz chose instead to address these two central stereocenters at an early stage and append the five- and seven-membered rings to the assembled cyclohexane. Accordingly, a synthetic strategy was devised that involved a one-pot, double-enantioselective enolate alkylation reaction to simultaneously form both quaternary stereocenters. Although such enantioselective alkylations have proven difficult, recent research in the Stoltz laboratories and others had identified palladium-catalysts that may provide a solution to this problem and enable the synthesis of a variety of targets containing quaternary carbon stereocenters, including the cyanthiwigins.87,88
Figure 8. Double enantioselective enolate alkylation and the synthesis of cyanthiwigin F (48).
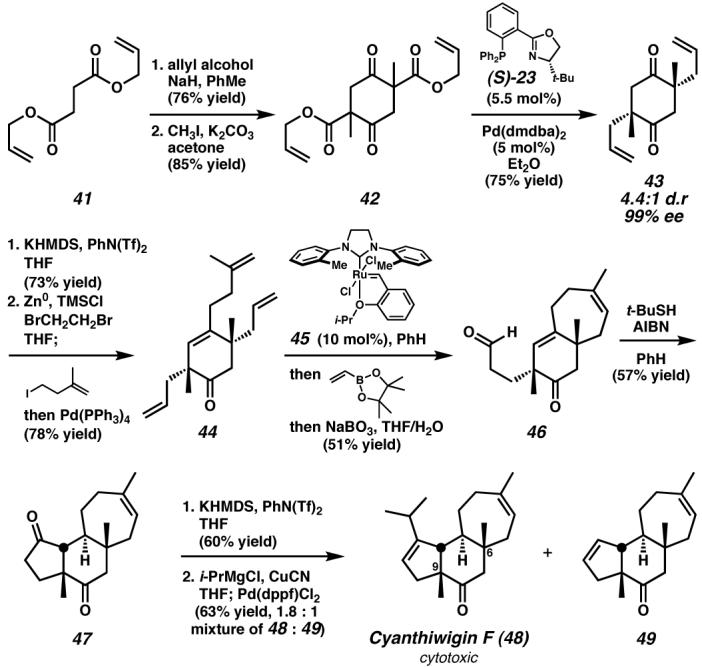
Preparation of a diastereomeric mixture of racemic and meso β-ketoester 42 allowed the strategic application of the palladium/(S)-23-catalyzed double enantioselective enolate alkylation to generate both all-carbon quaternary stereocenters of 43 with excellent stereoselectivity. A subsequent Negishi coupling, ring-closing metathesis, and radical-mediated aldehyde-olefin cyclization rapidly constructed the tricyclic core of cyanthiwigin F, completing a synthesis highlighted by several steps that involve the generation of multiple C–C bonds.87 dmdba, bis(3,5-dimethyoxybenzylidene)acetone; KHMDS, potassium bis(trimethylsilyl)amide; PhN(Tf)2, phenyl bis(trifluoromethane)sulfonimide; AIBN, 2,2′-azobis(isobutyronitrile); dppf, 1,1′-bis(diphenylphosphino)ferrocene. (Reduced to 45%)
The implementation of this plan began with a Claisen–Dieckmann sequence that converted diallyl succinate (41) to bis(β-ketoester) 42 as a 1:1 mixture of racemic and meso diastereomers. Upon exposure to the catalyst derived from Pd(dmdba)2 and phosphinooxazoline ligand (S)-23,89,90 were transformed to bis(allylated) ketone 43 in 75% yield and 99% ee as a 4.4:1 mixture of diastereomers. With both quaternary centers in place, elaboration of this stereochemically rich core structure to the natural product was achieved in six further steps. Enol triflate formation and Negishi coupling preceded a tandem ring-closing metathesis/cross-metathesis sequence with Grubbs' ruthenium catalyst 45. Aldehyde-olefin radical cyclization generated the final ring of the cyanthiwigin core (i.e., 47), and enol triflate formation and palladium-catalyzed cross-coupling formed (−)-cyanthiwigin F (48). Choosing to confront the difficult stereochemical elements of the cyanthiwigin structure at an early stage led to a direct synthetic route proceeding in nine steps from diallyl succinate (41). The strategy herein was enabled by the intriguing reaction mechanism of the enantioselective decarboxylative allylation, in which all three stereoisomers of bis(β-ketoester) 42 are selectively converted to a particular stereoisomer of product (43) with high selectivity through a stereoablative process.91 Additionally, of the nine steps required for the synthesis, seven form C–C bonds, and four of these steps form multiple C–C bonds. Directly addressing the carbon-framework of the target molecule and the stereochemical challenges embedded within ultimately lead to an efficient synthetic sequence to this important molecule.
At this time, the proposed chiral palladium enolate has been intercepted by allyl or proton electrophiles.89 Although the synthesis of cyanthiwigin F (48) demonstrates the versatility of allyl moieties for further derivatization, the direct use of alternative electrophiles would provide a more general and direct method for transition metal-mediated enolate functionalization.
Trimethylenemethane Cyclizations – Marcfortine B
Of the many fundamental approaches to the formation of five-membered rings from acyclic precursors, the [3 + 2] cycloaddition is among the most convergent strategies. A useful method of achieving such a cyclization is via a trimethylenemethane (TMM) intermediate.92 This interesting non-Kekulé molecule was first prepared and studied by Dowd and coworkers through photolytic decomposition of a cyclic diazene precursor. However, the free diyl is prone to several undesired reaction pathways and does not lend itself to asymmetric catalysis. Despite this, intramolecular diyl trapping reactions are valuable methods of cyclopentane formation.92 Recognizing the synthetic utility of TMM, Trost and coworkers developed an array of 2-(trimethylsilyl)-2-propenyl acetate reagents that generate a metal•TMM complex when exposed to a palladium catalyst.93,94 A recent application of this transformation in total synthesis is the approach to marcfortine B (55, Figure 9a), a member of a family of anti-parasitic agents, by Trost and coworkers.95 The strategy employed sought to forge the [2.2.2]bicycle via an intramolecular radical cyclization, and install the spiro all-carbon quaternary stereocenter by the cycloaddition of oxindole 50 with TMM precursor 51. In the event, an excellent yield was observed for the annulation reaction yielding spirooxindole 52 as a 1:1 mixture of diastereomers. Over the course of nine additional steps, spirocycle 52 was transformed into amide 53. Preparation of the xanthate derivative of alcohol 53 enabled radical cyclization generating the challenging [2.2.2]bicycle 54. Seven further steps produced (±)-marcfortine B (55).
Figure 9. Enantioselective trimethylenemethane [3 + 2] cyclization toward marcfortine B (55).
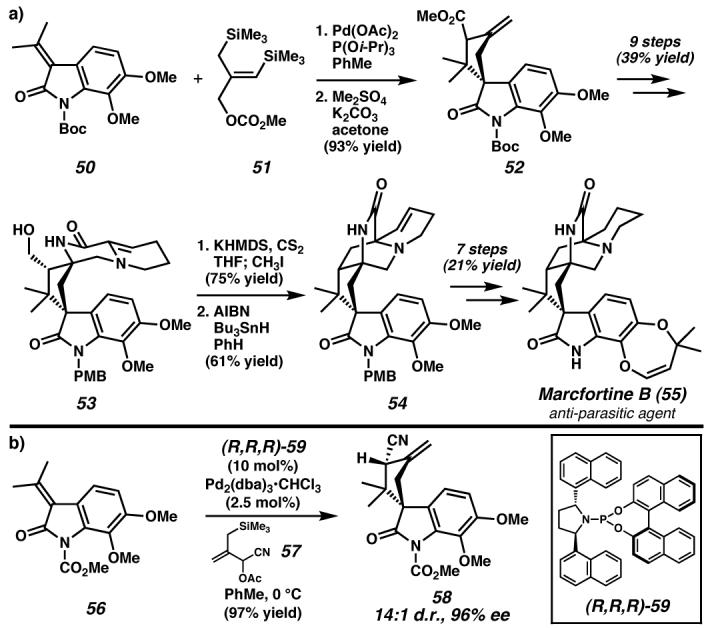
(a) The palladium-catalyzed trimethylenemetane [3 + 2] cyclization of oxindole 50 prepared cyclopentane 52 in excellent yield. Elaboration to tetracycle 53 facilitated an intramolecular radical cyclization to synthesize the core of marcfortine B, which was completed in seven additional steps.95 (b), The recent development of enantioselective palladium-catalyzed TMM cyclizations with (R,R,R)-59 allows access to spiro-annulated products such as 58 with high diastereo- and enantioselecitivity.99 The potential application to marcfortine B has not yet been realized. Boc, tert-butoxycarbonyl; dba, bis(benzylidene)acetone. (Reduced to 45%)
Although this strategy demonstrated several intriguing ring-forming reactions, an asymmetric synthesis of 55 would require an enantioselective variant of the key TMM-[3 + 2] cycloaddition, a goal that has remained elusive.96 Ito and coworkers reported the first asymmetric [3 + 2] cycloaddition with various bis(phosphine) ligands, but observed only moderate enantiomeric excess (up to 78%) and diastereomeric ratio (up to 4:1 trans:cis).97 Thereafter, Trost and coworkers explored bulky monodentate phosphoramidite ligands (e.g., (R,R,R)-59) for the transformation and observed very high enantioselectivity for the first time (Figure 9b).98,99,100 Of particular interest is the enantioselective addition of substituted TMM reagents to functionalized oxindole derivatives.99 The use of oxindole 56 and TMM-precursor 57 in the palladium-catalyzed cyclization with ligand (R,R,R)-59 yielded spirooxindole 58 with 14:1 d.r. and 96% enantiomeric excess for the major diastereomer. Although a completed asymmetric synthesis of marcfortine B (55) from intermediate 58 has not been reported, many of the key functional groups are in place and the challenging spiroquaternary stereocenter has been installed (cf. 52 and 58). The development of this valuable asymmetric transformation highlights the ongoing efforts to devise new and useful techniques toward construction of important molecules.
Conclusion
The representative synthetic efforts presented herein demonstrate the critical interplay between target-directed synthesis and the development of novel reaction methodology. Although many useful asymmetric technologies are currently available, the specific challenges posed by important natural product and pharmaceutical compounds highlight deficiencies in the current technology. Envisioning strategies to construct these relevant molecules through means beyond the current arsenal of enantioselective transformations will facilitate the evolution of both synthetic planning and reaction development. The symbiotic relationship between total synthesis and method development can continue to expand the understanding of synthetic strategy and catalysis on both fundamental and practical levels.
Despite the substantial advances realized to date, significant challenges remain for both multi-step synthesis and catalysis. In addition to enhancements to efficiency and selectivity, better reactivity and handling stability are continually required to implement and improve industrial processes for existing methods. Exceptionally reliable methods will aid in the discovery of new bioactive compounds through high-throughput combinatorial screening techniques that are well established in the pharmaceutical industry, but relatively limited by the number of readily accessible chiral building blocks. Existing methods may be improved by identifying systems with better functional group tolerances that might obviate the need for protecting and masking groups. Similarly, known privileged chiral frameworks may be modified to more effectively control chiral space for especially challenging transformations, a technique conspicuously successful for Trost's TMM cyclizations.
Creative solutions are required to address specific organic transformations that remain significant impediments to efficient syntheses, namely: formation of multiple stereocenters and rings, formation of multiple C–C bonds, generation of vicinal quaternary stereocenters, and C–H and C–C functionalization reactions. Cyclic structures often present particular challenges due to the unique strain and steric elements imparted by their connectivity. As a result, many highly strained or complex polycyclic structures are daunting targets for synthesis. Finally, the disclosure of new natural product possessing novel structures will serve to identify new and different challenges for synthetic chemistry and catalysis.
Acknowledgments
The authors thank NIH-NIGMS (R01GM080269-01), Eli Lilly (predoctoral fellowships to J.T.M. and M.R.K.), Amgen, Abbott, Boeringer Ingelheim, Merck, Bristol–Myers Squibb, and Caltech for financial support.
Footnotes
The authors declare no competing financial interests.
References
- 1.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley; New York: 1995. pp. 1–91. [Google Scholar]
- 2.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis, Vol. I–III. Springer-Verlag; Berlin: 2000. [Google Scholar]
- 3.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis, Supplement 1 & 2. Springer-Verlag; Berlin: 2004. [Google Scholar]
- 4.Hoveyda AH. Asymmetric Catalysis in Target-Oriented Synthesis. In: Vögtle F, Stoddart JF, Shibasaki M, editors. Stimulating Concepts in Chemistry. Wiley-VCH; Weinheim: 2000. pp. 145–160. [Google Scholar]
- 5.Trost BM. Asymmetric Catalysis: An Enabling Science. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5348–5355. doi: 10.1073/pnas.0306715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor MS, Jacobsen EN. Asymmetric Catalysis in Complex Target Synthesis. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5368–5373. doi: 10.1073/pnas.0307893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diels O, Alder K. Synthesen in der Hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928;460:98–122. [Google Scholar]
- 8.Stork G, van Tamalen EE, Friedman LJ, Burgstahler AW. Cantharidin. A Stereospecific Total Synthesis. J. Am. Chem. Soc. 1951;73:4501. [Google Scholar]
- 9.Woodward RB, Bader FE, Bickel H, Frey AJ, Kierstead RW. The Total Synthesis of Reserpine. J. Am. Chem. Soc. 1956;78:2023–2025. [Google Scholar]
- 10.Yates P, Eaton P. Acceleration of the Diels–Alder Reaction by Aluminum Chloride. J. Am. Chem. Soc. 1960;82:4436–4437. [Google Scholar]
- 11.Hashimoto S.-i., Komeshima N, Koga K. Asymmetric Diels–Alder Reaction Catalysed by Chiral Alkoxyaluminium Dichloride. J. Chem. Soc., Chem. Commun. 1979:437–438. [Google Scholar]
- 12.Hayashi Y. Catalytic Asymmetric Diels–Alder Reactions. In: Kobayashi S, Jørgensen KA, editors. Cycloaddition Reactions in Organic Synthesis. Wiley-VCH; Weinheim: 2002. pp. 5–55. [Google Scholar]
- 13.Corey EJ. Catalytic Enantioselective Diels–Alder Reactions: Methods, Mechanistic Fundamentals, Pathways, and Applications. Angew. Chem., Int. Ed. 2002;41:1650–1667. doi: 10.1002/1521-3773(20020517)41:10<1650::aid-anie1650>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 14.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. The Diels–Alder Reaction in Total Synthesis. Angew. Chem., Int. Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 15.Corey EJ, Andersen NH, Carlson RM, Paust J, Vedejs E, Vlattas I, Winter REK. Total Synthesis of Prostaglandins. Synthesis of the Pure dl-E1, -F1α, -F1β, -A1, and -B1 Hormones. J. Am. Chem. Soc. 1968;90:3245–3247. doi: 10.1021/ja01014a053. [DOI] [PubMed] [Google Scholar]
- 16.Corey EJ, Bakshi RK, Shibata S, Chen C-P, Singh VK. A Stable and Easily Prepared Catalyst for the Enantioselective Reduction of Ketones. Applications to Multistep Syntheses. J. Am. Chem. Soc. 1987;109:7925–7926. [Google Scholar]
- 17.Corey EJ, Helal CJ. Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem., Int. Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 18.Yoon TP, Jacobsen EN. Privileged Chiral Catalysts. Science. 2003;229:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 19.Nájera C, Sansano JM. Catalytic Asymmetric Synthesis of α-Amino Acids. Chem. Rev. 2007;107:4584–4671. doi: 10.1021/cr050580o. [DOI] [PubMed] [Google Scholar]
- 20.Senanayake CH, Jacobsen EN. Chiral (Salen)Mn(III) Complexes in Asymmetric Epoxidations: Practical Synthesis of cis-Aminoindanol and Its Application to Enantiopure Drug Synthesis. In: Gadamasetti KG, editor. Process Chemistry in the Pharmaceutical Industry. Marcel Dekker; New York: 1999. pp. 347–368. [Google Scholar]
- 21.Akutagawa S, Tani K. Asymmetric Isomerization of Allylamines. In: Ojima I, editor. Catalytic Asymmetric Synthesis. Wiley-VCH; New York: 2002. pp. 145–161. [Google Scholar]
- 22.Nugent WA, RajanBabu TV, Burk MJ. Beyond Nature's Chiral Pool: Enantioselective Catalysis in Industry. Science. 1993;259:479–483. doi: 10.1126/science.259.5094.479. [DOI] [PubMed] [Google Scholar]
- 23.Farina V, Reeves JT, Senanayake CH, Song JJ. Asymmetric Synthesis of Active Pharmaceutical Ingredients. Chem. Rev. 2006;106:2734–2793. doi: 10.1021/cr040700c. [DOI] [PubMed] [Google Scholar]
- 24.Blaser H-U, Schmidt E, editors. Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 25.Eder U, Sauer G, Wiechert R. New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Angew. Chem., Int. Ed. Engl. 1971;10:496–497. [Google Scholar]
- 26.Hajos ZG, Parrish DR. Stereocontrolled Total Synthesis of 19-Nor Steroids. J. Org. Chem. 1973;38:3244–3249. doi: 10.1021/jo00959a003. [DOI] [PubMed] [Google Scholar]
- 27.Hajos ZG, Parrish DR. Asymmetric Synthesis of Bicyclic Intermediates of Natural Product Chemistry. J. Org. Chem. 1974;39:1615–1621. [Google Scholar]
- 28.Evans DA, Fitch DM, Smith TE, Cee VJ. Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B. J. Am. Chem. Soc. 2000;122:10033–10046. [Google Scholar]
- 29.Palomo C, Oiarbide M, Garcáa JM. The Aldol Addition Reaction: An Old Transformation at Constant Rebirth. Chem.-Eur. J. 2002;8:37–44. doi: 10.1002/1521-3765(20020104)8:1<36::aid-chem36>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 30.Ma J-A. Recent Developments in the Catalytic Asymmetric Synthesis of α- and β-Amino Acids. Angew. Chem., Int. Ed. 2003;42:4290–4299. doi: 10.1002/anie.200301600. [DOI] [PubMed] [Google Scholar]
- 31.Juaristi E, Soloshonok VA, editors. Enantioselective Synthesis of β-Amino Acids. Wiley & Sons; Hoboken: 2005. [Google Scholar]
- 32.Vineyard BD, Knowles WS, Sabacky MJ, Bachman GL, Weinkauff DJ. Asymmetric Hydrogenation. Rhodium Chiral Bisphosphine Catalyst. J. Am. Chem. Soc. 1977;99:5946–5952. [Google Scholar]
- 33.Lubell WD, Kitamura M, Noyori R. Enantioselective Synthesis of β-Amino Acids Based on BINAP–Ruthenium(II) Catalyzed Hydrogenation. Tetrahedron: Asymmetry. 1991;2:543–554. [Google Scholar]
- 34.Drexler H-J, You J, Zhang S, Fischer C, Baumann W, Spannenberg A, Heller D. Chiral β-Amino Acid Derivatives via Asymmetric Hydrogenation. Org. Process Res. Dev. 2003;7:355–361. [Google Scholar]
- 35.Drucker D, Easley C, Kirkpatrick P. Sitagliptin. Nat. Rev. Drug Discovery. 2007;6:109–110. doi: 10.1038/nrd2245. [DOI] [PubMed] [Google Scholar]
- 36.Hsiao Y, Rivera NR, Rosner T, Krska SW, Njolito E, Wang F, Sun Y, Armstrong JD, III, Grabowski EJJ, Tillyer RD, Spindler F, Malan C. Highly Efficient Synthesis of β-Amino Acid Derivatives via Asymmetric Hydrogenation of Unprotected Enamines. J. Am. Chem. Soc. 2004;126:9918–9919. doi: 10.1021/ja047901i. This paper describes the discovery of the rhodium-catalyzed enantioselective hydrogenation of β-enamino amide and ester substrates. [DOI] [PubMed] [Google Scholar]
- 37.Shultz CS, Krska SW. Unlocking the Potential of Asymmetric Hydrogenation at Merck. Acc. Chem Res. 2007;40:1320–1326. doi: 10.1021/ar700141v. [DOI] [PubMed] [Google Scholar]
- 38.Xu F, Armstrong JD, III, Zhou GX, Simmons B, Hughes D, Ge Z, Grabowski EJJ. Mechanistic Evidence for an α-Oxoketene Pathway in the Formation of β-Ketoamides/Esters via Meldrum's Acid Adducts. J. Am. Chem. Soc. 2004;126:13002–13009. doi: 10.1021/ja046488b. [DOI] [PubMed] [Google Scholar]
- 39.Clausen AM, Dziadul B, Cappuccio KL, Kaba M, Starbuck C, Hsiao Y, Dowling TM. Identification of Ammonium Chloride as an Effective Promoter of the Asymmetric Hydrogenation of β-Enamine Amide. Org. Process Res. Dev. 2006;10:723–726. This paper details the scale-up efforts of the enantioselective hydrogenation of a β-enamino amide toward the industrial scale synthesis of diabetes treatment Januvia®, as well as the impact of ammonium chloride as an additive to maintain efficiency and selectivity in this process. [Google Scholar]
- 40.Noyori R, Kitamura M, Ohkuma T. Toward Efficient Asymmetric Hydrogenation: Architectural and Functional Engineering of Chiral Molecular Catalysts. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5356–5362. doi: 10.1073/pnas.0307928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Negishi E.-i., de Meijere A., editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 1. Wiley & Sons; New York: 2002. [Google Scholar]
- 42.de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 1 & 2. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 43.Luh T-Y, Leung M.-k., Wong K-T. Transition Metal-Catalyzed Activation of Aliphatic C–X Bonds in Carbon–Carbon Bond Formation. Chem. Rev. 2000;100:3187–3204. doi: 10.1021/cr990272o. [DOI] [PubMed] [Google Scholar]
- 44.Frisch AC, Beller M. Catalysts for Cross-Coupling Reactions with Non-activated Alkyl Halides. Angew. Chem., Int. Ed. 2005;44:674–688. doi: 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]
- 45.Netherton MR, Fu GC. Palladium-Catalyzed Cross-Coupling Reactions of Unactivated Alkyl Electrophiles with Organometallic Compounds. In: Tsuji J, editor. Topics in Organometallic Chemistry: Palladium in Organic Synthesis. Springer; New York: 2005. pp. 85–108. [Google Scholar]
- 46.Kiso Y, Tamao K, Miyake N, Yamamoto K, Kumada M. Asymmetric Cross-Coupling Reactions of sec-Alkyl Grignard Reagents with Organic Halides in the Presence of a Chiral Phosphine-Nickel Complex as a Catalyst. Tetrahedron Lett. 1974;15:3–6. [Google Scholar]
- 47.Hayashi T, Konishi M, Fukushima M, Mise T, Kagotani M, Tajika M, Kumada M. Asymmetric Synthesis Catalyzed by Chiral Ferrocenylphosphine-Transition Metal Complexes. 2. Nickel- and Palladium-Catalyzed Asymmetric Grignard Cross-Coupling. J. Am. Chem. Soc. 1982;104:180–186. [Google Scholar]
- 48.Fischer C, Fu GC. Asymmetric Nickel-Catalyzed Negishi Cross-Couplings of Secondary α-Bromo Amides with Organozinc Reagents. J. Am. Chem. Soc. 2005;127:4594–4595. doi: 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]
- 49.Arp FO, Fu GC. Catalytic Enantioselective Negishi Reactions of Racemic Secondary Benzylic Halides. J. Am. Chem. Soc. 2005;127:10482–10483. doi: 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Fu GC. Cross-Couplings of Unactivated Secondary Alkyl Halides: Room-Temperature Nickel-Catalyzed Negishi Reactions of Alkyl Bromides and Iodides. J. Am. Chem. Soc. 2003;125:14726–14727. doi: 10.1021/ja0389366. [DOI] [PubMed] [Google Scholar]
- 51.Son S, Fu GC. Nickel-Catalyzed Asymmetric Negishi Cross-Couplings of Secondary Allylic Chlorides with Alkylzincs. J. Am. Chem. Soc. 2008;130:2756–2757. doi: 10.1021/ja800103z. This paper describes the use of two sequential nickel-catalyzed asymmetric Negishi cross-coupling reactions in the formal synthesis of fluvirucinine A1. [DOI] [PubMed] [Google Scholar]
- 52.Suh Y-G, Kim S-A, Jung J-K, Shin D-Y, Min K-H, Koo B-A, Kim H-S. Asymmetric Total Synthesis of Fluvirucinine A1. Angew. Chem., Int. Ed. 1999;38:3545–3547. doi: 10.1002/(sici)1521-3773(19991203)38:23<3545::aid-anie3545>3.3.co;2-s. [DOI] [PubMed] [Google Scholar]
- 53.Douglas CJ, Overman LE. Catalytic Asymmetric Synthesis of All-Carbon Quaternary Stereocenters. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heck RF. Palladium-Catalyzed Reactions of Organic Halides with Olefins. Acc. Chem. Res. 1979;12:146–151. [Google Scholar]
- 55.Overman LE. Application of Intramolecular Heck Reactions for Forming Congested Quaternary Carbon Centers in Complex Molecule Total Synthesis. Pure Appl. Chem. 1994;66:1423–11430. [Google Scholar]
- 56.Sato Y, Sodeoka M, Shibasaki M. Catalytic Asymmetric Carbon–Carbon Bond Formation: Asymmetric Synthesis of cis-Decalin Derivatives by Palladium-Catalyzed Cyclization of Prochiral Alkenyl Iodides. J. Org. Chem. 1989;54:4738–4739. [Google Scholar]
- 57.Carpenter NE, Kucera DJ, Overman LE. Palladium-Catalyzed Polyene Cyclizations of Trienyl Triflates. J. Org. Chem. 1989;54:5846–5848. [Google Scholar]
- 58.Dounay AB, Overman LE. The Asymmetric Intramolecular Heck Reaction in Natural Product Total Synthesis. Chem. Rev. 2003;103:2945–2963. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]
- 59.Saxton JE, editor. Indoles. Part Four. The Monoterpenoid Indole Alkaloids. Wiley & Sons; New York: 1983. [Google Scholar]
- 60.Dounay AB, Overman LE, Wrobleski AD. Sequential Catalytic Asymmetric Heck–Iminium Ion Cyclization: Enantioselective Total Synthesis of the Strychnos Alkaloid Minfiensine. J. Am. Chem. Soc. 2005;127:10186–10187. doi: 10.1021/ja0533895. [DOI] [PubMed] [Google Scholar]
- 61.Dounay AB, Humphreys PG, Overman LE, Wrobleski AD. Total Synthesis of the Strychnos Alkaloid (+)-Minfiensine: Tandem Enantioselective Intramolecular Heck-Iminium Ion Cyclization. J. Am. Chem. Soc. 2008;130:5368–5377. doi: 10.1021/ja800163v. This paper provides a full account of the use of the palladium-catalyzed enantioselective Heck reaction in to total synthesis of alkaloid minfiensine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirmansjah L, Fu GC. Intramolecular Heck Reactions of Unactivated Alkyl Halides. J. Am. Chem. Soc. 2008;129:11340–11341. doi: 10.1021/ja075245r. [DOI] [PubMed] [Google Scholar]
- 63.Sundberg RJ. Indoles. Academic Press; San Diego: 1996. [Google Scholar]
- 64.Humphrey GR, Kuethe JT. Practical Methodologies for the Synthesis of Indoles. Chem. Rev. 2006;106:2875–2911. doi: 10.1021/cr0505270. [DOI] [PubMed] [Google Scholar]
- 65.Jensen KB, Thorhauge J, Hazell RG, Jørgensen KA. Catalytic Asymmetric Friedel–Crafts Alkylation of β,γ-Unsaturated α-Ketoesters: Enantioselective Addition of Aromatic C–H Bonds to Alkenes. Angew. Chem., Int. Ed. 2001;40:160–163. doi: 10.1002/1521-3773(20010105)40:1<160::AID-ANIE160>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 66.Paras NA, MacMillan DWC. New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel–Crafts Alkylation. J. Am. Chem. Soc. 2001;123:4370–4371. doi: 10.1021/ja015717g. [DOI] [PubMed] [Google Scholar]
- 67.Lelais G, MacMillan DWC. Modern Strategies in Organic Catalysis: The Advent and Development of Iminium Activation. Aldrichimica Acta. 2006;39:79–87. [Google Scholar]
- 68.Erkkilä A, Majander I, Pihko PM. Iminium Catalysis. Chem. Rev. 2007;107:5416–5470. doi: 10.1021/cr068388p. [DOI] [PubMed] [Google Scholar]
- 69.Austin JF, MacMillan DWC. Enantioselective Organocatalytic Indole Alkylations. Design of a New and Highly Effective Chiral Amine for Iminium Catalysis. J. Am. Chem. Soc. 2002;124:1172–1173. doi: 10.1021/ja017255c. [DOI] [PubMed] [Google Scholar]
- 70.Hino T, Nakagawa M. Chemistry and Reactions of Cyclic Tautomers of Tryptamines and Tryptophans. In: Brossi A, editor. The Alkaloids. Vol. 34. Academic Press; San Diego: 1988. pp. 1–75. [Google Scholar]
- 71.Overman LE, Paone DV, Stearns BA. Direct Stereo- and Enantiocontrolled Synthesis of Vicinal Stereogenic Quaternary Carbon Centers. Total Syntheses of meso- and (−)-Chimonanthine and (+)-Calycanthine. J. Am. Chem. Soc. 1999;121:7702–7703. [Google Scholar]
- 72.Overman LE, Larrow JF, Stearns BA, Vance JM. Enantioselective Construction of Vicinal Stereogenic Quaternary Stereocenters by Dialkylation: Practical Total Syntheses of (+)- and meso-Chimonanthine. Angew. Chem., Int. Ed. 2000;39:213–215. doi: 10.1002/(sici)1521-3773(20000103)39:1<213::aid-anie213>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 73.Depew KM, Marsden SP, Zatorska D, Zatorski A, Bornmann WG, Danishefsky SJ. Total Synthesis of 5-N-Acetylardeemin and Amauromine: Practical Routes to Potential MDR Reversal Agents. J. Am. Chem. Soc. 1999;121:11953–11963. [Google Scholar]
- 74.Austin JF, Kim S-G, Sinz CJ, Xiao W-J, MacMillan DWC. Enantioselective Organocatalytic Construction Construction of Pyrroloindolines by a Cascade Addition-Cyclization Strategy: Synthesis of (−)-Flustramine B. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5482–5487. doi: 10.1073/pnas.0308177101. This paper recounts the use of imidazolidinone catalysts for enantioselective Friedel–Crafts reactions with indole nucleophiles for the synthesis of the potassium-channel blocker flustramine B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pictet A, Spengler T. Über die Bildung von Isochinolin-derivaten durch Einwirkung von Methylal auf Phenyl-äthylamin, Phenyl-alanin und Tyrosin. Ber. Dtsch. Chem. Ges. 1911;44:2030–2036. [Google Scholar]
- 76.Cox ED, Cook JM. The Pictet–Spengler Condensation: A New Direction for an Old Reaction. Chem. Rev. 1995;95:1797–1842. [Google Scholar]
- 77.Allin SM, Gaskell SN, Elsegood MRJ, Martin WP. A New Asymmetric Synthesis of the Natural Enantiomer of the Indolizidino[8,7-b]indole Alkaloid (+)-Harmicine. Tetrahedron Lett. 2007;48:5669–5671. [Google Scholar]
- 78.Taylor MS, Jacobsen EN. Highly Enantioselective Catalytic Acyl-Pictet–Spengler Reactions. J. Am. Chem. Soc. 2004;126:10558–10559. doi: 10.1021/ja046259p. [DOI] [PubMed] [Google Scholar]
- 79.Seayad J, Seayad AM, List B. Catalytic Asymmetric Pictet–Spengler Reaction. J. Am. Chem. Soc. 2006;128:1086–1087. doi: 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]
- 80.Wanner MJ, van der Haas RNS, de Cuba KR, van Maarseveen JH, Hiemstra H. Catalytic Asymmetric Pictet–Spengler Reactions via Sulfenyliminium Ions. Angew. Chem., Int. Ed. 2007;46:7485–7487. doi: 10.1002/anie.200701808. [DOI] [PubMed] [Google Scholar]
- 81.Raheem IT, Thiara PS, Peterson EA, Jacobsen EN. Enantioselective Pictet–Spengler-Type Cyclizations of Hydroxylactams: H-Bond Donor Catalysis by Anion Binding. J. Am. Chem. Soc. 2007;129:13404–13405. doi: 10.1021/ja076179w. This paper describes development of the thiourea catalyzed enantioselective Pictet–Spengler-type cyclization reaction used in the synthesis of the anti-leishmania compound harmicine. [DOI] [PubMed] [Google Scholar]
- 82.Raheem IT, Thiara PS, Jacobsen EN. Regio- and Enantioselective Catalytic Cyclization of Pyrroles onto N-Acyliminium Ions. Org. Lett. 2008;10:1577–1580. doi: 10.1021/ol800256j. [DOI] [PubMed] [Google Scholar]
- 83.Reisman SE, Doyle AG, Jacobsen EN. Enantioselective Thiourea-Catalyzed Additions to Oxocarbenium Ions. J. Am. Chem. Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dolling U-H, Davis P, Grabowski EJJ. Efficient Catalytic Asymmetric Alkylations. 1. Enantioselective Synthesis of (+)-Indacrinone via Chiral Phase-Transfer Catalysis. J. Am. Chem. Soc. 1984;106:446–447. This paper describes the use of cinchoninium catalysts for enantioselective enolate alkylation toward the diuretic indacrinone. [Google Scholar]
- 85.Hughes DL, Dolling U-H, Ryan KM, Schoenewaldt EF, Grabowski EJJ. Efficient Catalytic Asymmetric Alkylations. 3. A Kinetic and Mechanistic Study of the Enantioselective Phase-Transfer Methylation of 6,7-Dichloro-5-methoxy-2-phenyl-1-indanone. J. Org. Chem. 1987;52:4745–4752. [Google Scholar]
- 86.Maruoka K, editor. Asymmetric Phase Transfer Catalysis. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 87.Enquist JA, Jr., Stoltz BM. The Total Synthesis of (–)-Cyanthiwigin F by means of Double Catalytic Enantioselective Alkylation. Nature. 2008;453:1228–1231. doi: 10.1038/nature07046. This paper details the use of palladium-catalyzed enantioselective enolate alkylation for the total synthesis of the marine natural product cyanthiwigin F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mohr JT, Stoltz BM. Enantioselective Tsuji Allylations. Chem.–Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Behenna DC, Stoltz BM. The Enantioselective Tsuji Allylation. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 90.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Deracemization of Quaternary Stereocenters by Pd-Catalyzed Enantioconvergenent Decarboxylative Allylation of Racemic β-Ketoesters. Angew. Chem., Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 91.Mohr JT, Ebner DC, Stoltz BM. Catalytic Enantioselective Stereoablative Reactions: An Unexploited Approach to Enantioselective Catalysis. Org. Biomol. Chem. 2007;5:3571–3576. doi: 10.1039/b711159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamago S, Nakamura E. [3 + 2] Cycloaddition of Trimethylenemethane and its Synthetic Equivalents. Org. React. 2002;61:1–217. [Google Scholar]
- 93.Trost BM, Chan DMT. New Conjunctive Reagents. 2-Acetoxymethyl-3-allyltrimethylsilane for Methylenecyclopentane Annulations Catalyzed by Palladium(0) J. Am. Chem. Soc. 1979;101:6429–6432. [Google Scholar]
- 94.Trost BM, Chan DMT. 2-Acetoxymethyl-3-allyltrimethylsilane and Palladium(0): A Source of Trimethylenemethane-Palladium Complex? J. Am. Chem. Soc. 1979;101:6432–6433. [Google Scholar]
- 95.Trost BM, Cramer N, Bernsmann H. Concise Total Synthesis of (±)-Marcfortine B. J. Am. Chem. Soc. 2007;129:3086–3087. doi: 10.1021/ja070142u. This paper describes the synthetic route to (±)-marcfortine B. [DOI] [PubMed] [Google Scholar]
- 96.Le Marquand P, Tam W. Enantioselective Palladium-Catalyzed Trimethylenemethane [3 + 2] Cycloadditions. Angew. Chem., Int. Ed. 2008;47:2926–2928. doi: 10.1002/anie.200705481. [DOI] [PubMed] [Google Scholar]
- 97.Yamamoto A, Ito Y, Hayashi T. Asymmetric [3 + 2] Cycloaddition of 2-(Sulfonylmethyl)-2-propenyl Carbonate Catalyzed by Chiral Ferrocenylphosphine-Palladium Complexes. Tetrahedron Lett. 1989;30:375–378. [Google Scholar]
- 98.Trost BM, Stambuli JP, Silverman SM, Schwörer W. Palladium-Catalyzed Asymmetric [3 + 2] Trimethylenemethane Cycloaddition Reactions. J. Am. Chem. Soc. 2006;128:13328–13329. doi: 10.1021/ja0640750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trost BM, Cramer N, Silverman SM. Enantioselective Construction of Spirocyclic Oxindolic Cyclopentanes by Palladium-Catalyzed Trimethylenemethane [3 + 2]-Cycloaddition. J. Am. Chem. Soc. 2007;129:12396–12397. doi: 10.1021/ja075335w. This paper describes the palladium-catalyzed enantioselective trimethylenemethane [3 + 2]-cycloaddition with oxindoles which may provide an enantioselective route to marcfortine B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trost BM, Silverman SM, Stambuli JP. Palladium-Catalyzed Asymmetric [3 + 2] Cycloaddition of Trimethylenemethane with Imines. J. Am. Chem. Soc. 2007;129:12398–12399. doi: 10.1021/ja0753389. [DOI] [PMC free article] [PubMed] [Google Scholar]