

Abstract
Genetic background influences the outcome of L. major infection. C57BL/6 mice mount a Th1 response and resolve infection. In contrast, BALB/c mice mount a Th2 response and develop chronic lesions. This susceptible phenotype is seen even though BALB/c mice generate IFN-γ-producing T cells at proportions similar to C57BL/6 mice in their lymph nodes early after infection. We had previously shown that chemokine receptor CXCR3 mediates immunity against L. major by recruiting IFN-γ-producing T cells to the lesions of C57BL/6 mice. Therefore, we hypothesized that IFN-γ-secreting T cells in BALB/c mice are unable to confer protection because they may be defective in up-regulating CXCR3. To test this hypothesis, we analyzed kinetics of CXCR3-expressing T cells in the lymph nodes and lesions of BALB/c and C57BL/6 mice during L. major infection. Additionally, we compared ability of T cells from BALB/c and C57BL/6 mice to up-regulate CXCR3 upon activation. We found that resolution of L. major infection in C57BL/6 mice was associated with an increase in the proportion of CXCR3+ T cells in regional lymph nodes and lesions whereas disease progression in BALB/c mice was associated with a decrease in these populations. Anti-CD3/CD28-activated T cells from naïve BALB/c but not C57BL/6 mice were defective in up-regulating CXCR3. Impaired induction of CXCR3 on BALB/c T cells was not due to lack of IFN-γ and was mediated partially by IL-10 but not IL-4 or IL-13. We propose that defective CXCR3 up-regulation on T cells in BALB/c mice may contribute to L. major-susceptibility.
Keywords: Chemokines, cell surface markers, T cells, Th1/Th2 cells
Introduction
Leishmania are obligate intracellular parasites that cause a wide range of diseases such as cutaneous, mucocutaneous and visceral leishmaniasis. In humans, cutaneous leishmaniasis caused by L. major usually manifests as a localized, self-resolving lesion associated with development of long-term immunity (1,2). The murine model of cutaneous L. major infection has been well characterized and frequently has been used as a functional model of Th1 and Th2 cell responses (3). Most inbred mice such as C57BL/6, C3H and CBA/J are genetically resistant to L. major and spontaneously resolve infection (4) because they mount a protective IL-12 induced Th1-type response. In contrast, susceptible BALB/c mice develop large non-healing lesions and mount a Th2 response that is associated with the production of the cytokines IL-4 and IL-10. Nevertheless, previous studies have shown that BALB/c mice are capable of mounting a Th1 response and contain significant number of IFN-γ producing T cells in their lymph nodes comparable to C57BL/6 mice during the early phase of L. major infection (5,6). C57BL/6 mice also produce high levels of IL-4 similar to BALB/c mice early after infection. Furthermore, some studies have found that deletion of the IL-4 or IL-4R gene in BALB/c mice has no effect on the outcome of L. major infection (7,8). Taken together, these findings suggest that genetically regulated mechanisms other than Th1/Th2 cytokine production may also control outcome of L. major infection in BALB/c mice.
We had previously found that the CXC chemokine receptor 3 (CXCR3), which binds CXCL9, CXCL10 and CXCL11, plays a critical role in immunity against L. major in C57BL/6 mice by mediating recruitment of IFN-γ producing T cells from the lymph nodes to lesion (9). This was perhaps not surprising since CD4+ Th1 cells have been shown to preferentially express CXCR3 (10). Because CXCR3 is critical for Th1 cell migration and L. major-infected BALB/c mice fail to restrict early parasite growth in the skin despite containing high number of IFN-γ producing T cells in their lymph nodes, we hypothesized that BALB/c mice may have a defect in up-regulating CXCR3 on T cells. Such a defect may prevent recruitment of Th1 cells to the lesion and therefore contribute to L. major-susceptibility. To test this hypothesis we analyzed CXCR3 expressing T cells the lesions and lymph nodes from BALB/c and C57BL/6 mice during L. major infection. In addition, we compared the ability of naïve CD4+ and CD8+ T cells from BALB/c and C57BL/6 mice to up-regulate CXCR3 upon anti-CD3/anti-CD28 activation in vitro. Our results indicate that both CD4+ and CD8+ T cells from susceptible BALB/c, but not resistant C57BL/6 mice have an intrinsic defect in efficiently up-regulating CXCR3 upon activation, which may contribute to susceptibility to L. major
Materials and Methods
Mice and Parasites
Sex, age matched C57BL/6 and BALB/c mice were purchased from Harlan (Indianapolis, IN) and maintained at The Ohio State University according to institutional regulations. IL-4Rα−/− mice where kindly provided by Alison Finnegan of Rheuma, Chicago, Illinois. For intradermal infections, 1 × 104 L. major promastigotes (LV39) were injected into the ears of C57BL/6 and BALB/c mice.
Leukocyte isolation and flow cytometry
At time-points post infection, 3−4 C57BL/6 and BALB/c mice were sacrificed. dLN were excised and single-cell suspensions of LN cells were obtained by teasing through a 70μm mesh. Lesion leukocytes were isolated as described previously (9). Cells were stained with PE-conjugated anti-mouse CXCR3 (R and D systems, MN) and either FITC-labeled anti-CD4 or anti-CD8 antibodies (Biolegend, CA). Flow cytometric analysis of LN and lesion cells was performed with a BD FACScalibur.
In vitro stimulation and treatment of T cells
Cell suspensions were obtained from the excised LNs and spleens of uninfected mice. 90−95% pure CD3+ T cells or CD4+ and CD8+ T cells populations were obtained via nylon wool columns and immunomagnetic isolation (Mitenyi Biotec USA, CA) from either wild type C57BL/6, BALB/c, IL-4R−/− or IL-10−/− BALB/c mice (11). T cells were incubated 48 hours at 0.5−2.5 ×106 cells/well in a 24-well plate pre-coated with 3 and 4 μg/ml anti-CD3 (clone 145−2C11) and anti-CD28 (clone 37.51) antibodies (Biolegend, CA), respectively. Following in vitro activation, cells were transferred to uncoated wells and rested in their conditioned media for 24 hours prior to flow cytometry analysis. Briefly, in vitro activated cells were recovered and washed in cold PBS before staining with PE-labeled anti-mouse CXCR3 antibodies or a rat IgG2a-PE control (R and D Systems, MN). Stained cells were either analyzed immediately or fixed in 1% paraformaldehyde for short-term storage.
Cytokine ELISA
Culture supernatants were sampled after 48 hours of incubation at 37°C, 5% CO2 and then analyzed for the presence of IFN-γ, IL-4, and IL-10 by sandwich ELISA as described previously (9).
Semi-Quantitative Real Time PCR
Total RNA was extracted from T-cells using TRIZOL Reagent (Invitrogen) and cDNA was synthesized and amplified as described previously (12). Primers sequences were found using the Harvard's PRIMER BANK website and oligonucleotides were purchased.
Statistical analysis
Statistically significant differences were determined by an unpaired Student's t test. A P value <0.05 was considered significant.
Results
L. major-infected BALB/c mice contain lower frequencies of CXCR3-expressing T cells in their regional nodes and lesions than C57BL/6 mice
CXCR3 is preferentially expressed on activated Th1-promoting cells (10). Previously, our group found that during L. major infection, CXCR3-deficiency results in more severe non-resolving disease compared to wild type mice. This heightened susceptibility coincided with deficient T cell mobilization and local IFN-γ production (9). Others have reported that resistant C57BL/6 mice produce high levels of CXCR3 ligands in response to L. major infection while susceptible BALB/c mice do not (13). These findings indicate that CXCR3 plays a critical role in immunity against L. major in a resistant mouse and that L. major-susceptible and resistant mice differ in their ability to produce CXCR3 ligands. However, whether CXCR3 is differentially expressed by susceptible and resistant mice remains to be investigated.
Therefore, we infected L. major-susceptible BALB/c and resistant C57BL/6 mice intradermally by inoculating 1×104 L. major promastigotes into ear dermis and frequency of CXCR3 expressing T cells in the lymph nodes (dLN) and lesions temporally using flow cytometry. Throughout the course of infection, L. major-infected C57BL/6 mice displayed higher frequencies of CXCR3 expressing CD4+ and CD8+ lymphocytes in their dLNs compared to BALB/c mice (Figure 1A). Similar trends in CXCR3+ T cell frequencies were also noted between the lesions of BALB/c and C57BL/6 mice (Figure 1B). Furthermore, analysis of the dLN and lesion reveled that CXCR3+ T cells comprised greater percentages of the CD4+ and CD8+ compartments of infected C57BL/6 mice than BALB/c mice (Figure 1C, and data not shown). Also, C57BL/6 mice contained more CXCR3+ CD4+ and CD8+ T cells in their dLN (Figure 2A) and the lesions than BALB/c mice (Figure.2B) although L. major-infected C57BL/6 mice showed a decline in CXCR3-expressing T cells in the dLNs and lesions as they resolved the infection (data not shown). These results demonstrate that resolution of L. major infection in C57BL/6 mice is associated with an increase in CXCR3+ T cells in their dLNs and lesions whereas disease progression in BALB/ c mice is associated with no or only modest increase in these cell populations.
Figure 1.

C57BL/6 derived T cells express CXCR3 more readily than those of BALB/c mice during L. major infection. Cells of the draining LN (A) and (B) lesions of C57BL/6 mice infected with L. major were analyzed by flow cytometry and were found to contain higher percentages of CXCR3+/CD4+ and CXCR3+/CD8+ lymphocytes than those of infected BALB/c mice after 3 weeks of L. major infection. Also CXCR3+ cells comprised a larger percentage of the CD4+ and CD8+ T cell compartments in the C57BL/6 dLN throughout the observed course of infection (C). For panels A and B, shown are representative results from one of five independent experiments. Numbers indicate the percent of lymphocytes. Panel C depicts the mean percentage of either CD4+ or CD8+ T cells that also express CXCR3+ (+/−SEM) from 3−5 independent trails. *= a P value < 0.05.
Figure 2.

The draining lymph nodes and lesions of C57BL/6 mice contain greater numbers of CXCR3+ T cells than those of BALB/c mice during L. major infection. From flow cytometry analysis, the absolute number of CXCR3+/CD4+ and CXCR3+/CD8+ T cells were calculated for C57BL/6 and BALB/c mice infected with L. major. Higher numbers of C57BL/6 dLN (A) and lesion cells (B) express CXCR3 compared to the cells of BALB/c mice. Shown are the mean results (+/−SEM) from at least two independent trials. *= a P value < 0.05.
Naïve T cells from BALB/c but not C57BL/6 mice are less efficient in up-regulating CXCR3 upon activation
To determine whether T cells from C57BL/6 and BALB/c mice inherently differ in their ability to up-regulate CXCR3, naïve T cells were isolated from the spleens of naïve mice and stimulated with plate bound anti-CD3/anti-CD28 antibodies as described previously (11). Expression of CXCR3 on these activated T cells was compared by flow cytometry. Following in vitro stimulation, T cells from C57BL/6 mice efficiently up-regulated CXCR3. In contrast, the increase in CXCR3 was minimal in similarly stimulated T cells from BALB/c mice (Figure 3A). Moreover, low CXCR3 expression on activated BALB/c T cells also correlated with low CXCR3 mRNA levels suggesting that suppression of CXCR3 expression was at the transcript level (Figure 3B). This defect in CXCR3 expression was observed in CD4+ and CD8+ T cells of BALB/c mice purified and activated separately (data not shown). Baseline levels of CXCR3 were low on pre-stimulated T cells from both BALB/c and C57BL/6 mice (data not shown) as previously reported (12, 16).
Figure 3.

Upon in vitro mitogenic stimulation, C57BL/6 derived T cells dramatically up-regulate CXCR3 while those of BALB/c mice do not. Briefly, T cells purified from uninfected BALB/c and C57BL/6 mice were stimulated in vitro by plate-bound anti-CD3/anti-CD28 antibodies for 48 hours prior to removal from stimulation and a 24 hour rest period. Flow cytometric staining of these activated cells revealed that C57BL/6 derived T cells (solid peak) but not BALB/c T cells (hollow gray peak) stained intensely for surface CXCR3 (A). Isotype controls were denoted by dotted hollow peaks. Real Time-PCR measurement of CXCR3 mRNA showed that this trend extended to the transcript level since C57BL/6 and BALB/c derived T cells induced CXCR3 mRNA at comparable levels (B). In panel A, a representative result from one of five trials is shown. Panel B represents the averaged result of three independent trials (+/−SEM). *= a P value < 0.05.
Reduced CXCR3 expression on T cells of BALB/c mice is not due to lack of IFN-γ
It has been shown that IFN γ and the transcription factor T-bet are required for efficient induction of CXCR3 on certain T cell subsets (16-18). To determine whether failure of BALB/c T cells to up-regulate CXCR3 was associated with reduced production of IFN-γ, we analyzed levels of Th1-associated IFN-γ as well as Th2-associated IL-4 and IL-10 in the above culture supernatants by ELISA. Also we quantified mRNA levels of T-bet in T cells by real time PCR.
Both C57BL/6 and BALB/c derived T cells produced high and comparable levels of IFN-γ upon activation (Figure 4A). However, activated T cells from BALB/c mice produced significantly more IL-4 and IL-10 as compared to similarly activated T cells from C57BL/6 mice (Figure 4B, C). Also mRNA levels of T-bet and IFN-γR (Figure 4D and 4E) as well as surface levels of IFN-γR (data not shown) were comparable in T cells from both groups. These results show that even though BALB/c T cells produce higher levels of Th2-associated cytokine, they are not deficient in IFN-γ production, T-bet induction or IFN-γR expression compared to C57BL/6 derived cells. Furthermore, they suggest that neither lack of IFN-γ production or deficient expression of IFN-γR is responsible for the inability of BALB/c derived T cells to up-regulate CXCR3.
Figure 4.

Despite higher IL-4 and IL-10 production by BALB/c derived T cells, both strains are similarly capable of IFN-γ, IFN-γR, and Tbet expression. At various time points after stimulation was ceased, the culture supernatants of activated T cells were analyzed by ELISA and at each sampling, BALB/c derived T cells activated in vitro with anti-CD3/anti-CD28 make levels of IFN-γ similar to C57BL/6 (A) but T cells of BALB/c mice made significantly more IL-10 and IL-4 (B, C). Additionally T-bet and IFN-γR mRNA levels were found to be comparable in activated T cells of both groups by RT-PCR (D and E, respectively). Data are presented as the mean results from 2−3 independent experiments (+/−SEM). *= a P value < 0.05.
Blockade of IL-4 or IL-4Ra deficiency does not restore expression of CXCR3 on activated T cells from BALB/c mice
Between 10 and 48 hours post stimulation C57BL/6 derived T cells rapidly induced CXCR3 surface protein (Figure 5A) while their BALB/c derived counterparts expressed only marginal levels of CXCR3 after 48 hours (Figure 5B). Interestingly, the slight increase in CXCR3 staining of BALB/c derived T cells coincided with reduction in IL-4 concentrations at later time points post- stimulation (Figure 4B, C).
Figure 5.
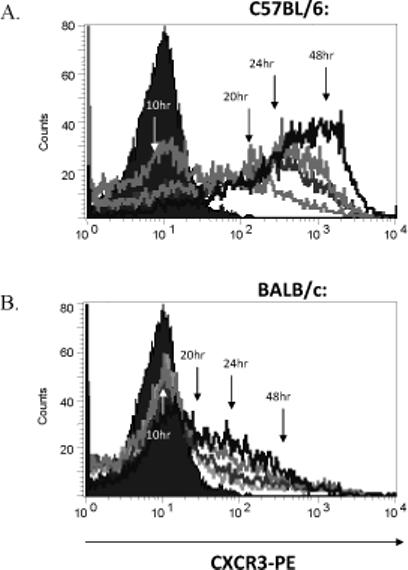
The kinetics of CXCR3 up-regulation by C57BL/6 and BALB/c derived T cells. At the time points specified in Figure 4, T cells were also sampled and CXCR3 expression was evaluated by flow cytometry. While at 10 hours post-stimulation both groups expressed low levels of CXCR3, by 24−48 hours after stimulation, the majority of C57BL/6 (A), but not BALB/c derived T cells (B) expressed high levels of CXCR3. The histograms shown are the representative results of two independent experiments with similar results.
To determine whether IL-4 was involved in preventing up-regulation of CXCR3 on BALB/c T cells, we stimulated BALB/c and C57BL/6 T cells in the presence of 10μg/ml anti-IL-4 or an isotype matched control antibody and examined the expression of CXCR3 by flow cytometry. In addition, we analyzed CXCR3 expression on T cells from WT BALB/c and IL-4Rα−/− BALB/c mice following in vitro activation with anti-CD3/CD28. Blockade of IL-4 (Figure 6A) or lack of IL-4Rα (Figure 6B) failed to increase expression of CXCR3 on BALB/c derived T cells indicating that IL-4 and IL-13 were not responsible for suppression of CXCR3. As expected, expression of CXCR3 on the T cells of C57BL/6 mice were unaffected by IL-4 neutralization (Figure 6C).
Figure 6.
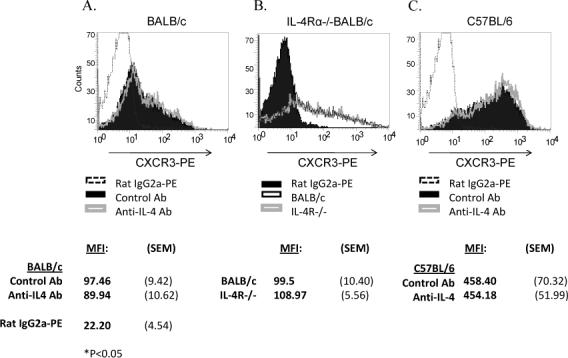
High levels of IL-4 are not responsible for reduced CXCR3 expression in BALB/c derived T cells. Neutralizing IL-4 with monoclonal antibodies (clone 11B.11, 10−40 μg/ml) during and post stimulation did not affect CXCR3 surface expression by BALB/c (A) derived T cells as measured by flow cytometry. Solid black peaks represent control antibody treated cells, while gray hollow peaks and dotted lines represent anti-IL-4 treated cells and straining isotype controls, respectively. Likewise IL-4Rα−/− BALB/c T cells (black hollow peak) did not up-regulate CXCR3 more efficiently than WT BALB/c T cells (gray hollow peak) (B). Isotype controls are represented by a solid black peak. Also C57BL/6 derived T cells were unaffected by IL-4 neutralization (C). Shown are representative histograms from one of three independent trials and MFI values for CXCR3-PE staining from at least three experiments are indicated as are the averaged result for isotype control treated cells.
Blockade of IL-10R partially restores expression of CXCR3 on T cells from BALB/c mice
Because T cells from BALB/c mice also produced significantly more IL-10 than C57BL/6 T cells after stimulation, we investigated whether the cytokine was involved in preventing up-regulation of CXCR3 on BALB/c T cells. To this end we blocked IL-10 signaling by including anti-IL-10 receptor antibodies in the culture media of BALB/c- and C57BL/6-derived T cells and examined its effect on CXCR3 expression. Additionally, we activated T cells from WT BALB/c and IL-10−/− BALB/c mice in vitro with anti-CD3/CD8 and compared levels of CXCR3. Flow cytometric analysis showed that CXCR3 expression by anti-IL-10 treated BALB/c T cells was greater than that of isotype control treated cells yet still lower than C57BL/6 T cells (Figure 7A). Not surprisingly, C57BL/6 derived T cells were unaffected by IL-10R blockade (data not shown). Furthermore, activated T cells from IL-10−/− BALB/c mice expressed more CXCR3 than WT controls (Figure 7B). Additionally, increased CXCR3 expression in the absence of IL-10 was also noted on T cells in the dLN of IL-10−/− mice infected with L. major (Figure 7C). Collectively, these results indicate that IL-10 is partially responsible for inhibiting CXCR3 expression on T cells in BALB/c mice.
Figure 7.

IL-10 suppresses CXCR3 expression by BALB/c T cells, but not those of C57BL/6. Blocking IL-10R with monoclonal antibodies (clone 1B1.3A, 10μg/ml) allowed moderate surface CXCR3 expression by BALB/c T cells activated in vitro (hollow gray peak). Control Ig-treated cells are represented by a solid black peak (A). Likewise, IL-10−/− BALB/c T cells (black hollow peak) induce CXCR3 more readily than WT controls (gray hollow peak). A solid black peak represents isotype controls (B). Treatment of C57BL/6 T cells with rIL-10 (10μg/ml) (hollow gray peak) did not suppress CXCR3 expression compared to untreated controls (solid black peak) (C). IL-10 deficiency also results in higher CXCR3 expression in vitro. The dLNs of IL-10−/− BALB/c mice infected with L. major were analyzed by flow cytomtry three weeks post infection. IL-10−/− mice contained more CXCR3+/CD3+ cells in their dLN than wild type BALB/c mice (D). Shown are representative results from at least three independent experiments. Shown are MFI values for CXCR3-PE staining (+/−SEM) from at least three experiments and the averaged result for isotype control treated cells. *= a P value < 0.05.
T cells from C57BL/6 mice are resistant to IL-10 mediated CXCR3 suppression and express significantly lower levels of IL-10R
Since we found that IL-10 was partially involved in preventing up-regulation of CXCR3 on BALB/c T cells, we determined whether IL-10 can block induction of CXCR3 on T cells in resistant mice. Naive T cells from C57BL/6 mice were stimulated in vitro with anti-CD3/CD28 as described above in the presence of rIL-10 and expression of CXCR3 was analyzed by flow cytometry. Interestingly, rIL-10 failed to suppress expression of CXCR3 on C57BL/6 T cells (Figure 4C) suggesting that these T cells were refractory to IL-10-induced suppression of CXCR3 expression. We therefore compared levels of IL-10R on activated T cells from BALB/c and C57BL/6 mice using real time RT-PCR as well as flow cytometry. Anti-CD3/CD28-activated CD4+ and CD8+ T cells from C57BL/6 mice displayed less induction of IL-10R mRNA and expressed less IL-10R as compared to BALB/c derived T cells (Figure 8A and 8B). These data suggest that low levels of IL-10R on C57BL/6 mice may be responsible for their refractoriness to IL-10-mediated down regulation of CXCR3.
Figure 8.

T cells of BALB/c mice are more receptive to IL-10 signaling than those of C57BL/6 mice. Real-time PCR (A) and flow cytometric analysis (B) of IL-10R expression revealed that BALB/c derived T cells (white bars and hallow gray peaks) express more IL-10R mRNA and surface protein than C57BL/6 T cells (black bars and solid black peaks). In panel A the averaged results of three independent trials (+/−SEM) are shown. Panel B represents the results from one of three independent trials. *= a P value < 0.05.
Discussion
We had previously shown that chemokine receptor CXCR3 plays a critical role in immunity against L. major by controlling T cell recruitment and IFN-γ levels at the site of infection. The novel finding in the present study is that CD4+ and CD8+ T cells from L. major-susceptible BALB/c, but not resistant C57BL/6 mice have a defect in efficient up-regulation of CXCR3 up on activation. It is well documented that Th1-associated cytokines IL-12 and IFN-γ are crucial for resolution of L. major infection in C57BL/6 mice whereas Th2-associated cytokines IL-4, IL-10 and IL-13 have been implicated as susceptibility factors in BALB/c mice. However, a previous study has documented that both BALB/c and C57BL/6 mice contain comparable frequencies of IFN-γ-producing T cells in their dLNs during early phase of L. major infection, yet BALB/c mice fail to control infection. One explanation for this observation could be that IFN-γ-producing T cells in the lymph nodes of BALB/c mice migrate less efficiently to the infected skin because BALB/c mice produce less CXCR3 ligand CXCL10 than C57BL/6 mice during L. major infection (13). However, intralesional administration of recombinant IP-10 (CXCL10) to BALB/c mice shortly after L. major infection increases NK cell cytotoxicity in the lymph nodes but fails to limit disease progression (13). These data suggest that IFN-γ-producing T cells in BALB/c mice may not be responsive to CXCL10 and fail to migrate to lesion. In the present study, we found that resolution of L. major infection in C57BL/6 mice was associated with an influx CXCR3+ T cells in the lymph nodes as well as lesions whereas disease progression in BALB/c mice was associated with reduced frequency of CXCR3+ T cells at both these sites. We also found that naïve T cells from BALB/c mice up-regulated CXCR3 less efficiently than C57BL/6 T cells following in vitro activation with anti-CD3/CD28. Furthermore, low CXCR3 expression on BALB/c T cells also correlated with low CXCR3 mRNA levels suggesting that suppression of CXCR3 was at the level of transcription. In vitro stimulation studies performed using purified CD4+ and CD8+ T cells revealed that both these cell types in BALB/c mice had a defect in up-regulating CXCR3 (data not shown). Taken together, these findings suggest that a defect in CXCR3 up-regulation on T cells could contribute to disease progression in BALB/c mice. In addition, they indicate that the inability of activated T cells from BALB/c mice to up-regulate CXCR3 is not due to lack of IFN-γ.
Previous studies have shown that IFN-γ, STAT1 and the transcription factor T-bet are required for efficient induction of CXCR3 on certain T cell subsets (16-18). IFN-γ, which signals via IFN-γR/STAT1 pathway, induces expression of T-bet (19), which in turn enhances expression of CXCR3 on T cells (17, 18). To determine whether reduced expression of CXCR3 on BALB/c T cells is due to lack of IFN-γ production, we measured levels IFN-γ as well as Th2-associated IL-4 and IL-10 in culture supernatant of activated T cells from BALB/c and C57BL/6 mice. In addition, we quantified mRNA levels of T-bet as well as IFN-γR by real-time RT-PCR. Upon activation with anti-CD3/CD28, T cells from C57BL/6 and BALB/c mice produced significant and comparable amounts of IFN-γ, but later produced significantly more IL-4 and IL-10. Both groups showed significant induction of T-bet and comparable levels of T-bet mRNA. Levels of IFN-γR mRNA were also comparable in T cells from BALB/c and C57BL/6 mice and correlated with surface expression of IFN-γR as assessed by flow cytometry (data not shown). Collectively, these findings show that lack of IFN-γ production or impaired expression of T-bet or IFN-γR are not responsible for failure of CXCR3 up-regulation in T cells from BALB/c mice. In addition they suggest that Th2-associated cytokines may be involved antagonizing IFN-γ and T-bet-induced expression of CXCR3 in BALB/c T cells.
Th2 associated cytokines IL-4 and IL-10 that mediate susceptibility to L. major, also regulate expression of several chemokine receptors and their ligands. However, previous studies have reported that IL-4 and IL-10 play distinct roles in regulation of expression of CXCR3 and its ligands CXCL9 and CXCL10. For example, Albanei et al. found that IL-4 enhances CXCL9 and CXCL10 production in keratinocytes and induces migration of CXCR3+ T cells in murine model of allergic contact dermatitis (20). IL-4 treatment also exacerbated disease in a Th1 cells transfer model of colitis which was associated with up-regulation of CXCR3 and its ligands in the colon (21). In contrast, IL-10 was found to inhibit expression of CXCR3 in microglial cells as well as human eosinophils (22). In the present study, a decrease in the levels of IL-4 in the culture supernatants from BALB/c mice was associated with a modest increase in levels of CXCR3 suggesting that IL-4 may be involved in suppressing expression of CXCR3. However, blockade of IL-4 using neutralizing antibodies failed to restore expression of CXCR3 to levels observed on C57BL/6 T cells. Additionally, T cells from IL-4Rα−/− BALB/c mice were deficient in up-regulating CXCR3 upon activation. On the other hand, blockade of IL-10R using anti-IL-10R Ab enhanced expression of CXCR3 on BALB/c derived T cells. Furthermore, in vitro activated T cells from IL-10−/− BALB/c mice expressed CXCR3 more readily than WT controls. Expression of CXCR3 on the T cells of C57BL/6 mice were unaffected by IL-4 neutralization or IL-10R blockade or rIL-4 treatment. Together, these results demonstrate that IL-10 is partly responsible for preventing up-regulation of CXCR3 on T cells in BALB/c mice. Furthermore, they show that IL-4 is not involved in inducing CXCR3 expression on T cells. It is interesting to note that levels of IL-4 in culture supernatants dropped rapidly when activated T cells of BALB/c mice were rested. This may be due to rapid consumption of IL-4 by these cells or simply due to degradation of this cytokine. Nonetheless, despite the presence of abundant ‘pro-Th1 factors’ (i.e. T-bet and IFN-γ) and decrease in IL-4 levels, BALB/c derived T cells failed to up-regulate CXCR3 levels. Furthermore, blockade of IL-10R or lack of IL-10 only partially restored expression of CXCR3 on BALB/c T cells indicating that other molecular mechanisms may be involved in negatively regulating induction of CXCR3 on these cells. One such potential mechanism is that a Th2-associated transcription factor GATA3, which directly binds to T-bet, may be involved in preventing T-bet-induced induction of CXCR3 in BALB/c T cells. We are currently investigating this possibility in ongoing studies in our laboratory.
Because IL-10/IL-10R pathway was found to be involved in suppressing CXCR3 expression on BALB/c T cells, we investigated whether IL-10 can block induction of CXCR3 on T cells in L. major-resistant mice. In addition, we analyzed expression of IL-10R and measured IL-10R mRNA levels in activated T cells from both strains. We found that rIL-10 failed to prevent up-regulation of CXCR3 on C57BL/6 T cells suggesting that these cells were resistant to IL-10-mediated CXCR3 suppression. Furthermore, BALB/c T cells expressed higher levels of IL-10R and contained more IL-10R mRNA than C57BL/6 T cells. Taken together, these results suggest that lower levels of IL-10R on C57BL/6 T cells may render them resistant to IL-10-mediated CXCR3 suppression and aid in up-regulation of CXCR3. Our finding are supportive of a previous study which showed that C57BL/6 leukocytes are more resistant to the IL-10 mediated immune-suppressive effect of regulatory T cells than those of BALB/c mice (23).
While IL-10 has been observed to inhibit CXCR3 expression by mouse microglia and human eosinophils (24), no such role is known for T cells. Interestingly, in the murine colitis model, forced expression of IL-10 results in amelioration of this Th1- (and CXCL10-) mediated disease which is associated with reduced expression of CXCR3 and its ligands (25). Our observations are particularly interesting in light of recent studies revealing the IL-10 producing capacity of Th1 cells as a means of limiting their own inflammatory potential (26). One such consequence of T cell-derived IL-10 may be to limit CXCR3-mediated cell homing – a recruitment axis critical for Th1 trafficking in numerous disease models.
In conclusion, resolution of L. major infections by resistant C57BL/6 mice is associated with an increase in CXCR3 expressing T cells in their lymph nodes and lesions whereas disease progression in susceptible BALB/c mice correlates with fewer CXCR3+ T cells in these tissues. . Certainly extrapolating the results of this and other mouse model intensive studies to human leishmaniasis should be done with caution and further study is needed to confirm the protective role of CXCR3 expression and signaling in the human response to Leishmania infection. Nevertheless, our present study has elucidated some previously unrecognized aspects of CXCR3 regulation and its dependence on elements of the Th1 and Th2 response. Naïve T cells from BALB/c mice and C57BL/6 mice markedly differ in their abilities to up-regulate CXCR3 upon activation. Anti-CD3/CD28 activated T cells from BALB/c mice produce as much IFN-γ as T cells from C57BL/6 mice but they secrete more IL-4 and IL-10 and fail to up-regulate CXCR3 efficiently. Blockade of IL-4 or lack of IL-4Rα fails to increase CXCR3 levels on BALB/c T cells but blockade of IL-10R or IL-10 deficiency partially restores CXCR3 expression. These results indicate that L. major-susceptible BALB/c mice, but not resistant C57BL/6 mice have a defect in efficiently up-regulating CXCR3 on T cells, which is partly mediated by IL-10. Yoon et al have described differences in CXCR3 transcript level in the stressed ocular tissues of C57BL/6 and BALB/c mice (14). Furthermore, a recent study has found that C57BL/6 mice, which are susceptible to cerebral malaria (CM), contain more CXCR3+ T cells in spleens compared to CM-resistant BALB/c mice during infection (15). Given our findings, differential expression of CXCR3 on T cells of BALB/c and C57BL/6 mice may explain the observations reported in above studies. Further studies are needed to determine whether this defect in CXCR3 up-regulation in BALB/c mice contributes to susceptibility to L. major as well as their phenotype in above models
Acknowledgements
We thank Sarah E. Walker for critical review of this manuscript and Dr. Alison Finnegan for kindly providing IL-4Rα−/− BALB/c mice.
Abbreviations
- DC
dendritic cell
- NK cell
natural killer cell
- CXCR3
CXC chemokine receptor 3
- IFNγ
interferon gamma
Footnotes
This work was supported by grant RO1 AI A151823 to ARS.
References
- 1.Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- 2.Reiner SL, Locksley RM. The regulation of immunity to Leishmania major. Annu.Rev.Immunol. 1995;13:151–177. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- 3.Moll H, Rollinghoff M. Resistance to murine cutaneous leishmaniasis is mediated by TH1 cells, but disease-promoting CD4+ cells are different from TH2 cells. Eur.J.Immunol. 1990;20:2067–2074. doi: 10.1002/eji.1830200927. [DOI] [PubMed] [Google Scholar]
- 4.Scott P. IFN-gamma modulates the early development of Th1 and Th2 responses in a murine model of cutaneous leishmaniasis. J.Immunol. 1991;147:3149–3155. [PubMed] [Google Scholar]
- 5.Lohoff M, Sommer F, Solbach W, Rollinghoff M. Coexistence of antigen-specific TH1 and TH2 cells in genetically susceptible BALB/c mice infected with Leishmania major. Immunobiology. 1989;179:412–421. doi: 10.1016/S0171-2985(89)80045-2. [DOI] [PubMed] [Google Scholar]
- 6.Sommer F, Meixner M, Mannherz M, Ogilvie AL, Rollinghoff M, Lohoff M. Analysis of cytokine patterns produced by individual CD4+ lymph node cells during experimental murine leishmaniasis in resistant and susceptible mice. Int.Immunol. 1998;10:1853–1861. doi: 10.1093/intimm/10.12.1853. [DOI] [PubMed] [Google Scholar]
- 7.Noben-Trauth N. Susceptibility to Leishmania major infection in the absence of IL-4. Immunol.Lett. 2000;75:41–44. doi: 10.1016/s0165-2478(00)00280-7. [DOI] [PubMed] [Google Scholar]
- 8.Mohrs M, Holscher C, Brombacher F. Interleukin-4 receptor alpha-deficient BALB/c mice show an unimpaired T helper 2 polarization in response to Leishmania major infection. Infect.Immun. 2000;68:1773–1780. doi: 10.1128/iai.68.4.1773-1780.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosas LE, Barbi J, Lu B, Fujiwara Y, Gerard C, Sanders VM, Satoskar AR. CXCR3−/− mice mount an efficient Th1 response but fail to control Leishmania major infection. Eur.J.Immunol. 2005;35:515–523. doi: 10.1002/eji.200425422. [DOI] [PubMed] [Google Scholar]
- 10.Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J.Exp.Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbi J, Oghumu S, Lezama-Davila CM, Satoskar AR. IFN-gamma and STAT1 are required for efficient induction of CXC chemokine receptor 3 (CXCR3) on CD4+ but not CD8+ T cells. Blood. 2007;110:2215–2216. doi: 10.1182/blood-2007-03-081307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbi J, Oghumu S, Rosas LE, Carlson T, Lu B, Gerard C, Lezama-Davila CM, Satoskar AR. Lack of CXCR3 delays the development of hepatic inflammation but does not impair resistance to Leishmania donovani. J.Infect.Dis. 2007;195:1713–1717. doi: 10.1086/516787. [DOI] [PubMed] [Google Scholar]
- 13.Yoneyama H, Narumi S, Zhang Y, Murai M, Baggiolini M, Lanzavecchia A, Ichida T, Asakura H, Matsushima K. Pivotal role of dendritic cell-derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J.Exp.Med. 2002;195:1257–1266. doi: 10.1084/jem.20011983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon KC, De Paiva CS, Qi H, Chen Z, Farley WJ, Li DQ, Pflugfelder SC. Expression of Th-1 chemokines and chemokine receptors on the ocular surface of C57BL/6 mice: effects of desiccating stress. Invest Ophthalmol.Vis.Sci. 2007;48:2561–2569. doi: 10.1167/iovs.07-0002. [DOI] [PubMed] [Google Scholar]
- 15.Van den Steen PE, Deroost K, Aelst IV, Geurts N, Martens E, Struyf S, Nie CQ, Hansen DS, Matthys P, Damme JV, Opdenakker G. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. Eur.J.Immunol. 2008;38:1082–1095. doi: 10.1002/eji.200737906. [DOI] [PubMed] [Google Scholar]
- 16.Nakajima C, Mukai T, Yamaguchi N, Morimoto Y, Park WR, Iwasaki M, Gao P, Ono S, Fujiwara H, Hamaoka T. Induction of the chemokine receptor CXCR3 on TCR-stimulated T cells: dependence on the release from persistent TCR-triggering and requirement for IFN-gamma stimulation. Eur.J.Immunol. 2002;32:1792–1801. doi: 10.1002/1521-4141(200206)32:6<1792::AID-IMMU1792>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 17.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taqueti VR, Grabie N, Colvin R, Pang H, Jarolim P, Luster AD, Glimcher LH, Lichtman AH. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. J.Immunol. 2006;177:5890–5901. doi: 10.4049/jimmunol.177.9.5890. [DOI] [PubMed] [Google Scholar]
- 19.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O'Shea JJ. T-bet is rapidly induced by interferon-γ in lymphoid and myeloid cells. Proc. Natl. Acad. Sci. USA. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albanesi C, Scarponi C, Sebastiani S, Cavani A, Federici M, De Pità O, Puddu P, Girolomoni G. IL-4 enhances keratinocyte expression of CXCR3 agonistic chemokines. J Immunol. 2000;165:1395–402. doi: 10.4049/jimmunol.165.3.1395. [DOI] [PubMed] [Google Scholar]
- 21.Fort M, Lesley R, Davidson N, Menon S, Brombacher F, Leach M, Rennick D. IL-4 exacerbates disease in a Th1 cell transfer model of colitis. J Immunol. 2001;166:2793–800. doi: 10.4049/jimmunol.166.4.2793. [DOI] [PubMed] [Google Scholar]
- 22.Kremlev SG, Palmer C. Interleukin-10 inhibits endotoxin-induced pro-inflammatory cytokines in microglial cell cultures. J Neuroimmunol. 2005;162:71–80. doi: 10.1016/j.jneuroim.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Roque S, Nobrega C, Appelberg R, Correia-Neves M. IL-10 underlies distinct susceptibility of BALB/c and C57BL/6 mice to Mycobacterium avium infection and influences efficacy of antibiotic therapy. J.Immunol. 2007;178:8028–8035. doi: 10.4049/jimmunol.178.12.8028. [DOI] [PubMed] [Google Scholar]
- 24.Jinquan T, Jing C, Jacobi HH, Reimert CM, Millner A, Quan S, Hansen JB, Dissing S, Malling HJ, Skov PS, Poulsen LK. CXCR3 expression and activation of eosinophils: role of IFN-gamma-inducible protein-10 and monokine induced by IFN-gamma. J.Immunol. 2000;165:1548–1556. doi: 10.4049/jimmunol.165.3.1548. [DOI] [PubMed] [Google Scholar]
- 25.Chen D, Ding Y, Zhang N, Schroppel B, Fu S, Zang W, Zhang H, Hancock WW, Bromberg JS. Viral IL-10 gene transfer inhibits the expression of multiple chemokine and chemokine receptor genes induced by inflammatory or adaptive immune stimuli. Am.J.Transplant. 2003;3:1538–1549. doi: 10.1046/j.1600-6135.2003.00263.x. [DOI] [PubMed] [Google Scholar]
- 26.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(-)Foxp3(-) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J.Exp.Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]