

Abstract
Catalytic ruthenium complexes in conjunction with an indium cocatalyst and Bronsted acid isomerize primary and secondary propargylic alcohols in good yields to provide trans enals and enones exclusively. Readily available indenylbis(triphenylphosphine)ruthenium chloride in the presence of indium triflate and camphorsulfonic acid give the best turnover numbers and reactivity with the broadest range of substrates. Deuterium labeling experiments suggest that the process occurs through propargylic hydride migration followed by protic cleavage of the resultant vinylruthenium intermediate. Application of this method to the synthesis of leukotriene B4 demonstrates its utility and extraordinary selectivity.
Introduction
The preparation of α,β-unsaturated aldehydes and ketones from propargylic alcohols has proven an important transformation, owing to the ease of preparation of the starting materials, and the broad utility of the products in organic synthesis. Normally this process is carried out by sequential reduction to the allylic alcohol followed by oxidation to the carbonyl compound, which is rather inefficient, considering that stoichiometric or even excess reductant and oxidant must be applied simply to effect reorganization of the internal oxidation pattern. Selectivity issues may introduce further inefficiencies. For the reduction step these include chemoselectivity in the presence of other unsaturation, overreduction, and control of olefin geometry. Oxidation is complicated by other alcohol functionality, which generally requires protection, further compromising the conciseness and atom economy of the synthesis. Aldol condensation followed by dehydration provides an alternative protocol, although self-condensation, oligomerization of the product, and control of olefin geometry can be problematic. Furthermore, the often harsh conditions render it of little practical importance in the preparation of unsaturated aldehydes. Phosphorous ylides likewise raise issues of atom economy and control of olefin geometry, as well as further olefination of the targeted unsaturated aldehyde.
Since a propargyl alcohol and the corresponding alkenyl carbonyl partner are simply isomers, the ideal approach would be to develop a catalyst to shuffle the hydrogens. An isomerization of propargylic alcohols could avoid the above pitfalls and offer significant advantages in economy and selectivity. Despite the potential benefits, there have been surprisingly few efforts to develop practical methods around this concept. The classical Rupe and Meyer-Schuster rearrangements effect this transformation, but with oxygen transposition.1 Of the examples that proceed with retention of the oxygen connectivity, most are highly activated substrates, which are known to rearrange through base2 or phosphine3 promoted mechanisms. Transition metal catalysts have proven effective for the isomerization of non-activated secondary propargylic alcohols to mixtures of α,β and β,γ unsaturated ketones.4 A recently reported kinetic resolution of secondary propargylic alcohols via rhodium-catalyzed redox isomerization represents an important new extension of this approach but proceeds poorly with primary propargyl alcohols and alkyl substituted secondary propargyl alcohols.5 Prior to our efforts,6 only one catalyst system had proven capable of isomerizing primary propargylic alcohols to α,β unsaturated aldehydes in reasonable yields, although it required 10-20 mol% catalyst for 30-48h at 110° and broad substrate compatibility was not demonstrated.7 Particular attention focused on generation of α,β- unsaturated aldehydes which are real tests of the mildness of the method because of their sensitivity, the failure of other catalysts for such substrates, their utility as synthetic intermediates, and the cumbersomeness of alternative modes of synthesis. This new method enhances the “alkyne strategy” for the synthesis of complex natural products and is illustrated by a concise synthesis of the delicate leukotrienes. A preliminary report of a portion of this work has appeared.6
Results and Discussion
Ruthenium complexes 1 and 2 have proven effective in the redox isomerization of allylic alcohols to saturated aldehydes and ketones.8 Under these conditions an ammonium salt was used to promote loss of chloride and generate the coordinatively unsaturated ruthenium species. Unfortunately, while direct application of these conditions to primary propargylic alcohols did promote sluggish isomerization to the corresponding unsaturated aldehydes, the moderately high temperatures and long reaction times resulted in extensive decomposition of the sensitive products. The indenyl complex 2 gave improved rates relative to its cyclopentadienyl analog, presumably due to the opening of an additional coordination site through valence tautomerization, although significant decomposition was still observed.9
Running the reaction in an alcohol solvent, intended to protect the sensitive aldehyde in situ as the acetal, gave primarily the products of conjugate addition. Previous work in our group had demonstrated the ability of indium (III) salts to promote additions to carbonyl functionality relative to alkenes.10 Indeed, inclusion of 40% indium trichloride in the catalyst system in refluxing THF was found to give rapid conversion and satisfactory yields of the sensitive products. The results summarized in Table 1 were obtained under these conditions. The ability to isolate sensitive enals in high yields attests to the overall mildness of the reaction conditions (3a – 3g). The extraordinary chemoselectivity is illustrated by the compatibility of ketones (3c), activated alcohols (3d), alkynes (3f), and alkenes (3g). Finally, the isomerization of propargylic alcohols with extended conjugation provides an attractive synthesis of dienals (3g).
Table 1.
Redox Isomerization under Indium Trichloride Cocatalysis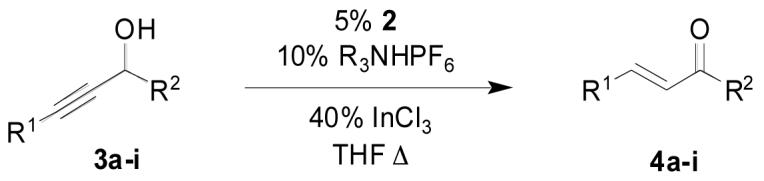
| Alcohol | R1 | R2 | Time | Yield |
|---|---|---|---|---|
| 3a | Ph(CH2)3 | H | 75 min | 88% |
| 3b | C6H11 | H | 90 min | 90% |
| 3c | PhC(=O)(CH2)3 | H | 90 min | 80% |
| 3d | PhCH(OH)(CH2)6 | H | 90 min | 86% |
| 3e | PhCH(OAc)(CH2)3 | H | 90 min | 87% |
| 3f | CH3(CH2)3C≡C(CH2)6 | H | 90 min | 83% |
| 3g | (CH3)2C=CH | H | 90 min | 67% |
| 3h | CH3(CH2)9 | CH2CH2Ph | 24 h | 86% |
| 3i | CH3(CH2)3 | (CH2)8CH=CH2 | 24 h | 83% |
Reactions were performed on 1 mmol scale at 0.25M with 5% catalyst 2 and 40% InCl3 in THF at reflux. For primary alcohols 5% NH4PF6 and 5% Et3NHPF6 was added. For secondary alcohols 10% Et3NHPF6 was added.
Mechanism
The stability of benzylic alcohols and isolated alkenes to the reaction conditions suggested that intermolecular hydrogen transfer, mediated through a ruthenium hydride species, was not the mechanism of the reaction. This was further probed through deuterium labeling experiments. Subjecting labeled propargylic alcohol 5 to the standard reaction conditions resulted in complete deuterium incorporation at the α carbon as determined by proton NMR (hydride shift pathway) in complete contrast to the isomerization of allyl alcohols wherein the propargylic hydrogen is transferred exclusively to the β- carbon.
This result suggests a mechanism as depicted in Scheme 2 wherein the product is formed by a net anti hydrometallation. This proposal is consistent with the observation that the trans products are obtained exclusively and is supported by the report of an analogous aryl shift in the literature.11 The intramolecular nature of the hydride migration was demonstrated by crossover experiment involving equimolar quantities of propargylic alcohol 3a and bisdeuterated substrate 5 to give 6 and no appearance of label in enal 4a. Addition of deuterium oxide to the reaction mixture resulted in roughly 50% deuterium incorporation at the β carbon by proton NMR. This suggests liberation of the catalyst by protonolysis of the vinylruthenium intermediate. Similar protic cleavages of vinylmetal species have been invoked previously.12 In combination, these results suggest the following catalytic cycle, which predicts both the remarkable chemoselectivity of the reaction, and the stereospecific formation of the trans product.
Scheme 2.

Proposed Catalytic Cycle for Redox Isomerization
Not addressed by this proposal is the effect of the indium cocatalyst, although formation of the active cationic ruthenium species by scavenging chloride is one probable role. Two possible modes for this activation can be envisaged: reversible sequestration of chloride by formation of an ion pair with the indiate complex (9) or an indium-bridged species (10). The latter proposal has the additional advantage of relieving the strain associated with complex 8b in the catalytic cycle above.
Reaction Optimization
The above isomerization conditions, while providing good yields under mild conditions, require numerous reaction components and high loads of indium trichloride. It was desirable to improve upon these results in terms of both reaction simplicity and atom economy. The proposed mechanism suggests that inclusion of a stronger proton source could enhance the reaction rate by promoting the cleavage of the vinylruthenium intermediate. Indeed, replacing the weakly acidic ammonium hexafluorophosphate salts with camphorsulfonic acid (CSA) allowed some reduction in the indium cocatalyst load without increasing the reaction time. For simple substrates, inclusion of large quantities of CSA even allowed complete conversion in the absence of indium salts, although reaction times were longer and yields correspondingly lower.
Another issue was the high concentration of chloride introduced by the indium cocatalyst, and the likely detrimental effect in terms of available coordination sites on the ruthenium catalyst. Indeed indium triflate had proven superior to indium chloride in the ruthenium catalyzed addition of alkynes to vinyl ketones.13 In the isomerization system, switching from indium chloride to the triflate likewise resulted in more rapid reaction, and allowed drastic reduction of the cocatalyst load. In conjunction with camphorsulfonic acid, further rate improvements and ultimately higher turnover numbers were observed. It was noted that large indium to ruthenium ratios gave more rapid initial reaction but ultimately lower turnover numbers. Use of stoichiometric indium relative to ruthenium generally provided an excellent balance between turnover number and reaction time for primary alcohols. For secondary alcohols the use of higher indium to ruthenium ratios (10% In and 3% Ru) increased the reaction rate roughly tenfold. In contrast to our earlier observations in the indium trichloride system, a very small amount (less than 4% by gc analysis) migration of the terminal alkene was observed when secondary alcohol 3i was isomerized under these conditions. These modifications now constitute our standard conditions.
A wide variety of other catalysts and cocatalysts were explored, and while none proved as practical as the above conditions, several interesting observations were made. The putative role of the indium cocatalyst, namely generation of the active cationic ruthenium complex through chloride abstraction, might be even better served by either silver or thallium, both of which form insoluble chloride salts.14 Indeed use of one equivalent of sliver or thallium triflate relative to catalyst 2 resulted in rapid isomerization at room temperature, although turnover numbers were ultimately lower than those obtained with indium, possibly due to poor stabilization of the active complex as the concentration of alkyne decreases.
The cyclopentadienyl catalyst 1 had proven significantly less active than the indenyl analog 2 under indium trichloride cocatalysis. While the new conditions actually widened the discrepancy, the absolute level of activity for the cyclopentadienyl catalyst nonetheless increased dramatically, making it an acceptable alternative to the indenyl complex. For example 5% of complex 1 in conjunction with 5% indium triflate and 20% CSA gave complete conversion of model compound 3a after 4 hours. By comparison only 3% of the indenyl catalyst 2 in conjunction with 3% indium triflate and 5% CSA gave complete conversion in just 30 minutes. Both complexes are now commercially available.
The modified cyclopentadienyl catalyst 11 was prepared for use in the constitutive condensation reaction of terminal alkynes and allylic alcohols.15 Ultimately the catalyst proved unsuitable for that purpose because of its propensity to isomerize the allylic alcohol. For the isomerization of propargylic alcohols it proved to be roughly as active as the indenyl catalyst, giving complete isomerization after 90 minutes under the indium trichloride conditions. This higher reactivity as compared to the unsubstituted cyclopentadienyl catalyst can be rationalized by invoking more facile loss of phosphine and opening of an additional coordination site induced by the bulky o-tolyl substituent.
The methallyl catalyst 12 was investigated with the expectation that the elimination of the chloride and one equivalent of phosphine would result in an activated isomerization catalyst that would exhibit reactivity on the order of the silver cocatalyzed system, that is rapid conversion at room temperature, without the addition of a metal cocatalyst.16 However this hypothesis was not borne out experimentally. No reaction was observed at room temperature and conversion was low even in THF at reflux. Addition of indium triflate dramatically improved the situation with 60% conversion after 60 minutes at reflux, but the reaction was still incomplete after 4 hours. The activity of indium on this catalyst suggests that sequestration of chloride ion may not be the sole mechanism of its cocatalysis.
Additional Substrates
The new isomerization conditions allowed expansion of its scope to substrates which had proven unreactive under the previously reported protocol. In particular, the preparation of polyconjugated carbonyl compounds from propargylic alcohols would dramatically demonstrate the selectivity of the isomerization. The indium trichloride conditions gave acceptable results for alcohol 3g as mentioned previously. The indium triflate conditions gave the desired dienal and dienone from alcohols 3j and 3k in good yields, although turnover numbers were lower than for nonconjugated substrates, particularly for the primary alcohol. This problem was alleviated by using substoichiometric indium relative to ruthenium, which predictably extended the reaction time, but ultimately allowed good conversion to the dienal. Propargylic alcohol 3l reacted even more sluggishly and suffered from even lower turnover numbers. Increasing the concentration of substrate improved the turnover numbers in this case, and provided modest yields of the desired ketone.
Unsaturated aldehydes bearing 4-hydroxy substituents would provide entry into several interesting natural product substructures.17 Unfortunately attempts to access these compounds by redox isomerization of monoprotected butynediols were unsuccessful under the indium trichloride cocatalyzed conditions. Diol 3m gave only low yields of the expected ketoaldehyde, and protection of the secondary hydroxyl as the acetate or PMB ether resulted in unreactive substrates. Under the higher activity of the indium triflate cocatalyst, diol 3m rapidly underwent dual isomerization to the corresponding ketoaldehyde in 80% yield. Protection of the secondary hydroxyl group as the PMB ether dramatically curtailed the reactivity, but the desired aldehyde could be isolated from 3n in an acceptable 62% yield. The corresponding acetate was essentially unreactive, while the t-butyldimethylsilyl ether rapidly succumbed to hydrolysis.
Hydrolysis of the protecting group under the acidic conditions was an even greater issue for monoprotected 2-butyn-1,4-diols. Fortunately the TBDPS ether provided adequate resistance, and the desired aldehyde could be isolated from propargylic alcohol 3o in 62% yield. Lowering the concentration of indium triflate helped to curtail the hydrolysis despite the somewhat longer reaction time.
Synthesis of Leukotriene B4
The arachidonic acid metabolite leukotriene B4 has attracted significant interest due to its broad biological activity, with its varied effects including the stimulation of leukocyte formation, increased vascular permeability, and smooth muscle contraction. Numerous total syntheses have been reported since its structure was elucidated nearly two decades ago.18 The challenging triene subunit, with its flanking allylic alcohols, makes the molecule an attractive proving ground for methodologies that facilitate the preparation of these functionalities. Successful application of our ruthenium-catalyzed redox isomerization of propargylic alcohols to this target would demonstrate various aspects of its scope, and testify to the mildness and selectivity of the method. The retrosynthesis outlined below capitalizes on the potential of generating both trans alkenes of the triene unit via ruthenium-catalyzed redox isomerization (Figure 4). In devising this synthetic approach, it was established from the outset that the objective would be to explore the chemoselectivity of the isomerization reaction, and that efforts to address stereochemical issues would be put aside.
Figure 4.

Retrosynthesis of Leukotriene B4
Isomerizations of model diynol 13 and monoprotected 2,4-hexadiyn-1,6-diol 15 proceeded cleanly, (Scheme 3), but the rate of conversion was slow. The source of the slow isomerization rate for conjugated diynes and has not been fully elucidated, and, as such, this class of substrate presents an opportunity for further catalyst design and system optimization. Alternatively, sequential reduction and oxidation under classical conditions proceeded normally (Scheme 4). While this approach required multiple steps and proceeded with low atom economy, it offered higher throughput during the initial steps of the synthesis. The monoprotected 2,4-hexadiyn-1,6-diol starting material could be prepared by one of two methods. Copper-catalyzed coupling of the TBDMS ether of 3-iodo-2-propyn-1-ol with propargyl alcohol in pyrrolidine gave the desired monoprotected diynol in 50% yield over two steps.19 However, due to thermochemical safety concerns associated with this approach, an alternative was sought for use on large scale. The commercially available and easily prepared 2,4-hexadiyn-1,6-diol could be silylated to give 62% yield of the monoprotected substrate, with bis-silylated material comprising the balance.20 Regioselective reduction of the unprotected propargylic moiety was achieved using Red-Al in 88% yield. Oxidation of the resultant allylic alcohol under Mofatt-Swern conditions gave the required aldehyde 17 in 84% yield.21
Scheme 3.

Isomerization of 2,4-Diyn-1-ols
Scheme 4.

Synthesis of Leukotriene B4a a Reaction conditions: a) NaH, TBDMSCl, 62%; b) Red-Al, 88%; c) (COCl)2, DMSO, Et3N, 84%; d) borane 18, then NaOH, H2O2, 67%; e) TBDPSCl, imidazole, DMAP; f) CSA, MeOH, 81% over 2 steps; g) 2, In(OTf)3, CSA, 92%; h) [(Ph3PCH2Br]Br, t-BuOK, 65%; i) n-BuLi, OHC(CH2)3COOCH3, 27%, 67%brsm; j) TBDPSCl, imidazole, DMAP; k) H2, Lindlar, quinoline, 92%; l) TBAF, 87%.
The next challenge was introduction of the homopropargylic alcohol moiety by addition of a propargyl metal equivalent to aldehyde 17. Propargylic lithium and zinc species are known to exist as equilibrating mixtures of propargyl and allenyl species, and hence their addition to aldehydes typically yields mixtures of allenyl and homopropargylic alcohols, the selectivity being dependent on both the metal species and the steric requirements of the substrate.22 Zweifel has shown that addition of trialkylboranes to lithiated propargyl chloride generates substituted allenylboranes, which can subsequently undergo addition to aldehydes with outstanding selectivity for the homopropargylic alcohol, obtained after standard oxidative workup.23 The required tripentylborane was readily prepared from 1-pentene and borane dimethylsulfide complex. Treatment with lithiated propargyl chloride and addition of the resultant allenylborane 18 to aldehyde 17 gave greater than 20:1 selectivity for homopropargylic alcohol over the allene by gas chromatography. These regioisomers could be separated by silica gel chromatography to give the key intermediate 19 in 67% yield on multigram scale.
Protection of the secondary alcohol as the TBDPS ether and selective removal of the TBDMS group with CSA in methanol gave isomerization substrate 20 in 81% yield over two steps. Redox isomerization using 5% of all components was complete in 20 minutes and on large scale to provide dienal 21 in 92% yield. This result demonstrates the extraordinary selectivity of the system, with isomerization proceeding cleanly in the presence of the conjugated alkene, isolated alkyne, silyl protecting group, and the sensitive dienal product. The high yield and rapid rate of conversion contrasts markedly with the result obtained previously for the conjugated diynol, suggesting that the slow kinetics and low turnover observed in that system are the exception rather than the rule.
In light of our decision to set aside issues of stereochemical control, the most direct route to the eicosanoate from the aldehyde was by homologation to the alkyne and addition to an appropriate glutaric moiety. The literature describes few reliable methods for the conversion of enals, let alone dienals, to the corresponding alkynes. Ultimately acceptable results were obtained by one-pot homologation to the vinyl bromide followed by elimination to give the dienyne in 65% yield.24 This marginally stable material contained roughly 15% of remaining vinyl bromide and was taken to the subsequent addition immediately following isolation. In what proved the most problematic transformation of the synthesis, lithiation of the dienyne and addition to methyl 5-oxopentanoate yielded only 27% of the eicosanoate backbone 22 (67% based on recovered alkyne). Similar attempts described in the literature have also been met with modest yields and significant recovery of starting material.25 While no alternatives to the lithium acetylide were explored, there is precedent to suggest that the use of the more nucleophilic magnesium or cerium acetylides might give superior results.26
Attempted semihydrogenation of the two alkyne moieties of eicosanoate 22 under Lindlar conditions resulted in rapid reduction of the propargylic alcohol to the desired cis allylic alcohol. Reduction of the more sterically encumbered alkyne, however, was competitive with over-reduction of the newly formed triene unit. Protection of the hydroxyl group as the TBDPS ether evened the steric accessibility and allowed the double hydrogenation to proceed selectively in 92% yield. Finally, global deprotection with tetrabutylammonium fluoride gave the product in 87% yield.18f Although the product consisted of four stereochemical isomers, the stereo centers were sufficiently remote that spectroscopic characterization of the material was not rendered unduly troublesome. Reverse phase HPLC showed the expected 1:1 mixture of diastereomers and indicated greater than 94% chemical purity. Overall the synthesis proceeded in 12 steps from commercially available 2,4-hexadiyn-1,6-diol to give the target in 7% overall yield.
Conclusions
This new catalytic system currently constitutes the most effective method to perform redox isomerization of both primary and secondary propargyl alcohols with catalyst loadings as low as 1%. Further application of this method to the synthesis of sphingofungins E and F27 and more recently to the enantioselective synthesis of adociacetylene B28 further attests to the potential scope of its synthetic utility. While some of the above systems demonstrate the current limitations of the method with regard to catalyst turnover, the extraordinary selectivity of the system sets it apart from other methods of preparing enals and enones from propargylic precursors. Particularly noteworthy is the stability of alkenes, alkynes, aldehydes, ketones, and isolated alcohols, all of which are in principal susceptible to redox side reactions proceeding through ruthenium-catalyzed mechanisms. Furthermore, the accelerating influence of electrophilic indium (III) complexes on some ruthenium catalyzed reactions, first demonstrated in this system, has been proven operative in a number of other applications.13 The recently reported rhodium-catalyzed kinetic resolution of secondary propargylic alcohols via redox isomerization, while apparently proceeding through an allyl-metal intermediate, represents an attractive potential extension of this method.5 However, it should be noted that this method isomerizes primary propargyl alcohols in poor yields. Furthermore, the cost of ruthenium vs rhodium makes catalysts based upon ruthenium more attractive. Finally, the operative hydride shift mechanism, in addition to providing the extraordinary chemoselectivity observed, suggests a number of related reaction manifolds, several of which remain under active investigation.
The mildness of this method is highlighted by its effectiveness for the isomerization of eneyne 20 to dienal 21. It also vividly illustrates the effectiveness of the “alkyne strategy” for complex molecule synthesis. The ease of access of eneyne 20 via a three component coupling provides most of the carbon skeleton of the leukotrienes in simple fashion.
Experimental Section
Representative Isomerization Procedures
Glassware was not flame dried but all solvents were freshly distilled under nitrogen to ensure deoxygenation. The apparatus was flushed with nitrogen or argon immediately following setup and the reactions were continued under an inert atmosphere. For particularly sensitive systems it was helpful to briefly sparge the solution with either nitrogen or argon prior to heating. Reactions were typically sampled by glass capillary under positive nitrogen pressure. Samples were washed through a small plug of silica with ether and analyzed by thin layer or gas chromatography.
Aldehyde 4a by Indium Trichloride Cocatalyzed Isomerization
Indium trichloride (88.5 mg, 0.40 mmol), indenyl catalyst 2 (28.8 mg, 0.05 mmol), triethylammonium hexafluorophosphate (12.4 mg, 0.05 mmol), and ammonium hexafluorophosphate (8.3 mg, 0.05 mmol) were combined sequentially. The system was purged with nitrogen and 6-phenyl-2-hexyn-1-ol (174 mg, 1.00 mmol) was introduced by syringe followed by THF (4 mL). The red solution was stirred for several minutes at room temperature and then gradually heated to gentle reflux. Thin layer and gas chromatography indicated that the reaction was complete within 75 min. The solvent was removed under reduced pressure and the residue was filtered through florisil with ether to remove the catalyst. Removal of the solvent under reduced pressure and flash chromatography on silica gel (10:1 hexanes:ethyl acetate) gave 153 mg of clear oil (88% yield). All spectral data compared favorably to those reported in the literature.29 IR (film): 3027, 2933, 2859, 2733, 1649, 1636, 1603, 1496, 1455, 1124, 784, 700; 1H NMR (300 MHz, CDCl3): δ 9.51 (d, J = 7.9 Hz, 1H), 7.33-7.14 (m, 5H), 6.85 (dt, J = 15.6, 6.7 Hz, 1H), 6.13 (dd, J = 15.7, 7.9 Hz, 1H), 2.68 (t, J = 7.6 Hz, 2H), 2.37 (q, J = 6.7 Hz, 2H), 1.86 (quint, J = 7.6 Hz, 2H).
Aldehyde 4a by Indium Triflate Cocatalyzed Isomerization
6-Phenyl-2-hexyn-1-ol (174 mg, 1.00 mmol), indenyl catalyst 2 (23.3 mg, 0.03 mmol), and camphorsulfonic acid (11.6 mg, 0.05 mmol) were combined in THF (5 mL) and the solution was stirred under an inert atmosphere for several minutes. Indium triflate (16.9 mg, 0.03 mmol) was added and the solution was brought to reflux in a preheated oil bath. Thin layer and gas chromatography indicated that the reaction was complete within 30 min. The mixture was diluted with ether and filtered through florisil to remove the catalyst. Removal of the solvent under reduced pressure and flash chromatography on silica gel (10:1 pet ether:ethyl acetate) gave 150 mg of clear oil (86% yield).
Ketone 4i by Indium Trichloride Cocatalyzed Isomerization
16-Heptadecen-5-yn-7-olError! Bookmark not defined. (250 mg, 1.00 mmol) was subjected to the indium trichloride isomerization conditions described above except that 10 mol% triethylammonium hexafluorophosphate and no ammonium hexafluorophosphate was used. After 24 h standard workup and purification of the product by flash chromatography on silica gel (20:1 hexanes:ethyl acetate) gave 207 mg of clear oil (83% yield). IR (film): 2928, 2855, 1698, 1676, 1631, 1465, 981, 909 cm-1; 1H NMR(300 MHz, CDCl3): δ 6.83 (dt, J = 15.9, 6.9 Hz, 1H), 6.09 (dt, J = 15.9,1.5 Hz, 1H), 5.86-5.77 (m, 1H), 5.03-4.90 (m, 2H), 2.52 (t, J = 7.4 Hz, 2H), 2.22 (dq, J = 7.1, 1.4 Hz, 2H), 2.03 (q, J = 7.0 Hz, 2H), 1.62-1.29 (m, 16H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 200.9, 147.2, 139.1, 130.3, 114.1, 40.0, 33.7, 32.1, 30.2, 29.32, 29.25 (2), 29.0, 28.8, 24.3, 22.2, 13.8; HRMS Calcd for C17H30O (M+): 250.2297. Found 250.2292.
(E)-3-(1-Cyclohexenyl)-2-propenal (4j)
3-(1-Cyclohexenyl)-2-propyn-1-ol (136 mg, 1.00 mmol), indenyl catalyst 2 (23.3 mg, 0.03 mmol), and camphorsulfonic acid (11.6 mg, 0.05 mmol) were combined in THF (5 mL) and the solution was stirred under an inert atmosphere for several minutes. Indium triflate (5.6 mg, 0.01 mmol) was added and the solution was brought to reflux in a preheated oil bath. After 2 h the mixture was diluted with ether and filtered through florisil to remove the catalyst. Removal of the solvent under reduced pressure and flash chromatography on silica gel (10:1 pet ether:ethyl acetate) gave 115 mg of clear oil (85% yield). All spectral data compared favorably with those reported in the literature.30 IR (film): 2934, 2862, 2824, 2727, 1679, 1624, 1603, 1129, 970 cm-1; 1H NMR (300 MHz, CDCl3): δ 9.52 (d, J = 8.1 Hz, 1H), 7.06 (d, J = 15.56 Hz, 1H), 6.28 (s, 1H), 6.05 (dd, J = 15.6, 7.8 Hz, 1H), 2.24-2.15 (m, 4H), 1.74-1.58 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 194.3, 156.2, 141.3, 135.8, 125.7, 26.7, 24.2, 21.9, 21.8.
(E)-1-(1-Cyclohexenyl)-1-nonen-3-one (4k)
1-(1-Cyclohexenyl)-1-nonyn-3-ol (220 mg, 1.00 mmol), indenyl catalyst 2 (23.3 mg, 0.03 mmol), and camphorsulfonic acid (11.6 mg, 0.05 mmol) were combined in THF (5 mL) and the solution was stirred under an inert atmosphere for several minutes. Indium triflate (16.9 mg, 0.03 mmol) was added and the solution was brought to reflux in a preheated oil bath. After 2 h the mixture was diluted with ether and filtered through florisil to remove the catalyst. Removal of the solvent under reduced pressure and flash chromatography on silica gel (20:1 pet ether:ethyl acetate) gave 163 mg of clear oil (74% yield). The product appeared to decompose via electrocyclic rearrangement upon standing at room temperature for several days. IR (film): 2930, 2859, 1688, 1662, 1626, 1597, 1458, 1317, 1190, 1072, 974 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.15 (d, J = 15.9 Hz, 1H), 6.21 (s, 1H), 6.07 (d, J = 16.1 Hz, 1H), 2.56 (t, J = 7.7 Hz, 2H), 2.23-2.15 (m, 4H), 1.72-1.60 (m, 6H), 1.34-1.29 (m, 6H), 0.88 (t, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 201.4, 145.9, 139.7, 135.2, 123.2, 40.5, 31.6, 29.0, 26.6, 24.5, 24.1, 22.5, 22.0 (2), 14.0; HRMS Calcd for C15H24O (M+): 220.1827. Found 220.1829.
(E)-6-(t-Butyldimethylsilyl)oxy-2-hexen-4-yn-1-ol
To 6-(t-butyldimethylsilyl)oxy-2,4-hexadiyn-1-ol (16.43 g, 73.35 mmol) in THF (150 mL) was added Red-Al (65% solution in toluene, 33.03 mL, 34.22 g, 110 mmol) at 0 °C. Reduction was complete after stirring 1 h at room temperature and the reaction was quenched at 0 °C with ethyl acetate (50 mL). The reaction was worked up with 3M sulfuric acid (300 mL) and ether (4×250 mL). The combined extracts were washed with saturated aqueous sodium bicarbonate (100 mL) and brine (100 mL) and dried over magnesium sulfate. Removal of the solvent under reduced pressure and flash chromatography on silica gel (5:1 pet ether:ethyl acetate) gave 14.53 g of clear oil (88% yield). IR (film): 3360, 2956, 2930, 2858, 2218, 1635, 1472, 1363, 1256, 1086, 835, 779 cm-1; 1H NMR (300 MHz, CDCl3): δ 6.23 (dt, J = 15.9, 5.1 Hz, 1H), 5.76 (dt, J = 15.9, 1.6 Hz, 1H), 4.43 (d, J = 1.7 Hz, 2H), 4.20 (br, 2H), 1.96 (br, 1H), 0.91 (s, 9H), 0.13 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 141.8, 109.9, 88.4, 82.7, 62.7, 52.1, 25.8 (3), 18.3, -5.2 (2); HRMS Calcd for C12H21O2Si (M+ - H): 225.1311. Found 225.1308.
(E)-6-(t-Butyldimethylsilyl)oxy-2-hexen-4-ynal (17)
To oxalyl chloride (6.28 mL, 9.14 g, 72 mmol) in methylene chloride (150 mL) at -78 °C was slowly added DMSO (6.39 mL, 7.03 g, 90 mmol). After stirring 10 min, a precooled solution of the above alcohol (13.56 g, 60 mmol) in methylene chloride (50 mL) was added via cannula. After 10 min, triethylamine (25.09 mL, 18.21 g, 180 mmol) was slowly added and stirring was continued at -78 °C for 30 min. After warming and stirring at room temperature for 30 min, the mixture was diluted with methylene chloride (200 mL) and washed with 10% sodium bisulfate. After removal of the solvent under reduced pressure, the residue was dissolved in ether (250 mL) and washed with saturated sodium bicarbonate (100 mL) and water (2×100 mL). Drying over magnesium sulfate and removal of the solvent under reduced pressure followed by filtration through silica gel (10:1 pet ether:ethyl acetate) gave 11.31 g of clear oil (84% yield). IR (film): 2956, 2930, 2858, 2729, 2213, 1689, 1606, 1256, 1089, 837, 779 cm-1; 1H NMR (300 MHz, CDCl3): δ 9.57 (d, J = 7.6 Hz, 1H), 6.63 (dt, J = 15.9, 1.8 Hz, 1H), 6.44 (dd, J = 15.9, 7.8 Hz, 1H), 4.53 (d, J = 1.7 Hz, 2H), 0.92 (s, 9H), 0.14 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 193.0, 139.6, 132.1, 103.2, 81.3, 52.2, 25.7 (3), 18.3, -5.2 (2); Anal. Calcd for C12H20O2Si: C, 64.23; H, 8.99. Found C, 64.06; H, 9.16.
(E)-1-(t-Butyldimethylsilyl)oxy-4-tetradecen-2,8-diyn-6-ol (19)
Tripentylborane was prepared by slow addition at 0 °C of borane dimethyl sulfide complex (30 mL, 2M in THF, 60.0 mmol) to 1-pentene (19.73 mL, 12.63 g, 180 mmol) in THF (100 mL). To propargyl chloride (4.34 mL, 4.47 g, 60 mmol) in ether (100 mL) at -90 °C was added n-butyllithium (37.5 mL, 1.6M in hexanes, 60 mmol). After stirring 10 min, the mixture was diluted with THF (150 mL) and the tripentylborane solution precooled to -78 °C was transferred via cannula over 30 min. After stirring 30 min at -78 °C a precooled solution of aldehyde 17 (11.31 g, 50.5 mmol) was transferred via cannula and stirring was continued 30 min. After warming to room temperature, the mixture was treated with methanol (30 mL) and 3N NaOH (21.6 mL) and oxidized by slow addition of 30% hydrogen peroxide (14.4 mL). Gas chromatography showed roughly 20:1 selectivity for the desired isomer. The mixture was worked up with water (400 mL) and ether (4×100 mL) and the combined extracts were dried over magnesium sulfate. Removal of the solvent under reduced pressure and flash chromatography on silica gel (10:1 pet ether:ethyl acetate) gave 13.39 g of clear oil (67% yield). IR (film): 3416, 2957, 2931, 2859, 2218, 1636, 1255, 1085, 837, 779 cm-1; 1H NMR (300 MHz, CDCl3): δ 6.16 (dd, J = 15.9, 5.5 Hz, 1H), 5.80 (dd, J = 15.9, 1.6 Hz, 1H), 4.43 (d, J = 1.7 Hz, 2H), 4.27 (t, J = 5.4 Hz, 1H), 2.51-2.35 (m, 2H), 2.22 (br, 1H), 2.19-2.13 (m, 2H), 1.54-1.40 (m, 2H), 1.38-1.12 (m, 4H), 0.91 (s, 9H), 0.90 (t, J = 6.7 Hz, 3H), 0.13 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 143.5, 110.3, 88.8, 83.9, 82.6, 74.8, 70.2, 52.1, 31.0, 28.6, 27.8, 25.8 (3), 22.2, 18.6, 18.3, 13.9, -5.2 (2); Anal. Calcd for C20H34O2Si: C, 71.80; H, 10.24. Found C, 71.68; H, 9.96.
(2E,4E)-6-(t-Butyldiphenylsilyl)oxy-2,4-tetradecadien-8-ynal (21)
To (E)-6-(tbutyldiphenylsilyl)oxy-4-tetradecen-2,8-diyn-1-ol (916 mg, 2.00 mmol) in THF (10 mL) was added indenyl catalyst 2 (77 mg, 0.10 mmol) and camphorsulfonic acid (32 mg, 0.10 mg). The solution was sparged with argon for several minutes and indium triflate (56 mg, 0.10 mmol) was added. The solution was brought to reflux in a preheated oil bath and the reaction was monitored by thin layer chromatography. Isomerization was complete within 20 min, at which point the solution was filtered through florisil with ether to remove the catalyst. Removal of the solvent under reduced pressure and flash chromatography on silica gel (10:1 pet ether:ethyl acetate) gave 845 mg of clear oil (92% yield). IR (film): 3072, 2958, 2932, 2859, 2732, 1688, 1646, 1428, 1106, 1009, 988, 741, 702 cm-1; 1H NMR (300 MHz, CDCl3): d 9.53 (d, J = 8.1 Hz, 1H), 7.73-7.60 (m, 4H), 7.46-7.32 (m, 6H), 7.06-6.98 (m, 1H), 6.36-6.32 (m, 2H), 6.03 (dd, J = 15.1, 8.1 Hz, 1H), 4.38 (dt, J = 9.5, 4.8 Hz, 1H), 2.46 -2.28 (m, 2H), 2.10-2.05 (m, 2H), 1.44-1.21 (m, 6H), 1.08 (s, 9H), 0.86 (t, J = 7.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): d 193.8, 151.6, 146.0, 135.8 (4), 133.5, 133.3, 131.7, 129.8 (2), 128.0, 127.6 (4), 83.1, 75.3, 72.1, 31.0, 28.5, 28.0, 26.9 (3), 22.2, 19.3, 18.6; HRMS Calcd for C26H29O2Si (M+ - C4H9): 401.1937. Found 401.1939.
(6Z,8E,10E,14Z)-Methyl 5,12-bis[(t-butyldiphenylsilyl)oxy]-6,8,10,14-eicosatetraenoate
(8E,10E)-Methyl 5,12-bis[(t-butyldiphenylsilyl)oxy]-8,10-eicosadiene-6,14-diynoate (207 mg, 0.25 mmol) and Lindlar catalyst (50 mg) were suspended in ethyl acetate (50 mL) containing quinoline (590 mL, 646 mg, 5.00 mmol). The mixture was stirred under an atmosphere of hydrogen and reaction progress was carefully monitored by thin layer chromatography. The first semihydrogenation was complete within 1 h (monohydrogenated Rf = 0.34 in 20:1 pet ether:ethyl acetate) but the second required 4 h to reach completion (dihydrogenated Rf = 0.39 in 20:1 pet ether:ethyl acetate). The mixture was filtered through florisil with ethyl acetate to remove the catalyst and the solution was diluted with pet ether (50 mL). The solution was washed with 1M aqueous sulfuric acid (25 mL) and saturated aqueous sodium bicarbonate (50 mL). The extracts were dried over magnesium sulfate and the solvent was removed under reduced pressure. Flash chromatography on silica gel (20:1 pet ether:ethyl acetate) gave 191 mg of clear oil (92 % yield). All spectral data compared favorably with those reported in the literature.18f IR (film): 3015, 2931, 2858, 1742, 1599, 1472, 1428, 1362, 1112, 1074, 998, 822, 740, 702 cm-1; 1H NMR (300 MHz, CDCl3): d 7.70-7.58 (m, 8H), 7.40-7.29 (m, 12H), 5.96-5.56 (m, 5H), 5.42-5.20 (m, 3H), 4.47 (br, 1H), 4.15 (br, 1H), 3.63 & 3.61 (s, 3H, diastereomers), 2.28-2.04 (m, 4H), 1.81-1.77 (m, 2H), 1.60-1.42 (m, 4H), 1.28-1.14 (m, 6H), 1.07 & 1.06 (s, 9H, diastereomers), 1.03 & 1.02 (s, 9H, diastereomers), 0.85 (t, J = 6.1 Hz, 3H).
Leukotriene B4
To the above eicosatetraenoate (135 mg, 0.16 mmol) in THF (1.6 mL) at 0 °C was added tetrabutylammonium fluoride (0.16 mL, 1M in THF, 1.6 mol). After stirring at room temperature overnight the mixture was poured into vigorously stirred ether (10 mL) and pH 5 phosphate buffer (10 mL). The aqueous layer was extracted with ether (3×10 mL) and the combined extracts were dried over magnesium sulfate. Removal of the solvent under reduced pressure and flash chromatography on silica gel (20:1 to 10:1 ether:methanol) gave the product as 49 mg of clear oil (89% yield). Analysis by reverse phase HPLC (300:100:1 methanol:water:acetic acid) showed the expected 1:1 mixture of diastereomers and greater than 94% purity. All spectral data compared favorably with those reported in the literature.18f IR (film): 3382, 3013, 2959, 2928, 2858, 1716, 1457, 1243, 1038, 966 cm-1; 1H NMR (500 MHz, CDCl3): d 6.51 (t, J = 13.0 Hz, 1H), 6.37-6.24 (m, 2H), 6.11 (t, J = 11.2 Hz, 1H), 5.82 (dd, J = 14.8, 6.0 Hz, 1H), 5.63-5.58 (m, 1H), 5.47- 5.38 (m, 3H), 4.64 (br, 1H), 4.25 (br, 1H), 2.42-2.26 (m, 4H), 2.08-2.05 (m, 2H), 1.77-1.29 (m, 12H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3): d 178.2, 136.8, 134.1 (2), 133.5, 130.2 (2), 127.5, 124.1, 71.9, 67.6, 36.5, 35.4, 33.7, 31.5, 29.3, 27.5, 22.6, 20.6, 14.2.
Supplementary Material
Figure 1.
Commercial Catalysts for Redox Isomerization of Propargylic Alcohols
Scheme 1.

Deuterium Labeling Experiments
Figure 2.
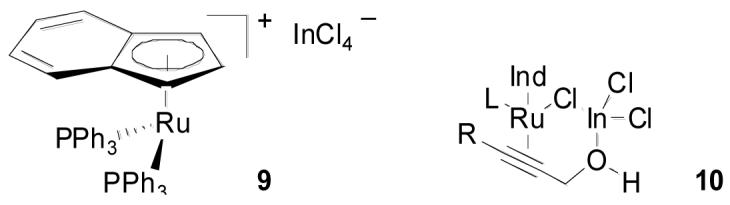
Indium Activation of Cationic Ruthenium Species
Figure 3.

Other Ruthenium Complexes for Redox Isomerization
Scheme 5.

Preparation of Allenylborane 18
Table 2.
Isomer ization under Indium Triflate Cocatalysis
| Alcohol | Propargylic Alcohol | % 2 | % In | Substrate [] | Time | Yielda) |
|---|---|---|---|---|---|---|
| 3a | 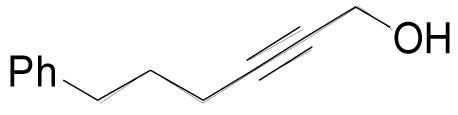 |
1 | 1 | 0.25 M | 2 h | 83% |
| 3i | 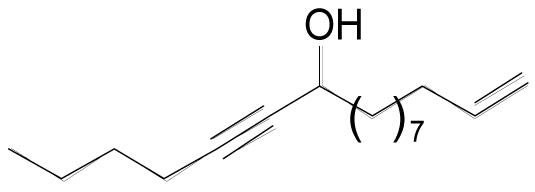 |
3 | 10 | 0.20 M | 20 min | 82% |
| 3j | 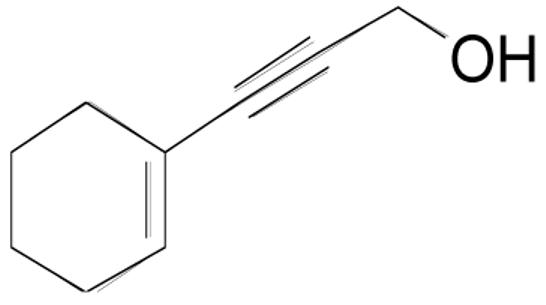 |
3 | 1 | 0.20 M | 2 hr | 85% |
| 3k | 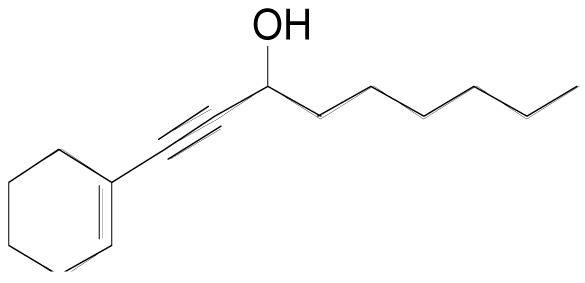 |
3 | 3 | 0.20 M | 2 hr | 74% |
| 3l | 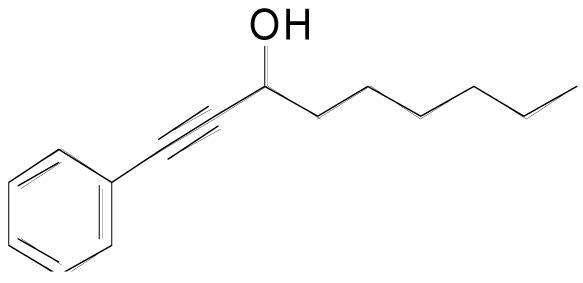 |
5 | 5 | 1.0 M | 90 min | 60% |
| 3m | 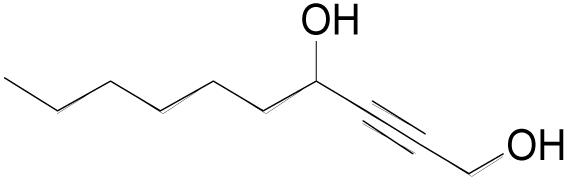 |
3 | 3 | 0.20 M | 30 min | 80%b) |
| 3n | 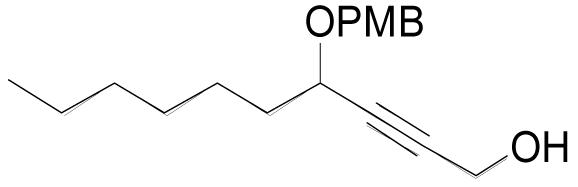 |
3 | 3 | 0.20 M | 3 h | 62% |
| 3o | 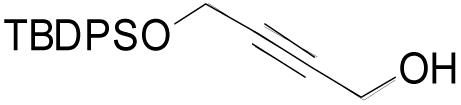 |
5 | 2 | 0.20 M | 45 min | 62% |
Reactions were performed on 1 mmol scale in THF at reflux. Yield refers to isolated yields.
The product in this case is 4- keto-n-decanal.
Acknowledgment
We thank the National Science Foundation and the National Institutes of Health (GM-13598) for their generous support of our programs. Mass spectra were provided by the Mass Spectrometry Regional Center of the University of California, San Francisco, supported by the NIH Division of Research Resources.
Footnotes
Supporting Information Available: Experimental procedures, characterization, and spectral data for all new intermediates and products, including preparation of isomerization substrates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Swaminathan S, Narayanan KV. Chem. Rev. 1971;71:429.For recent metal catalyzed versions see: Bustelo E, Dixnef PH. Adv. Synth. Catal. 2007;349:933.Cadierno V, Garcia-Garrido SE, Gimeno J. Adv. Synth. Catal. 2006;348:101.Lopez SS, Engel DA, Dudley GB. Synlett. 2007;949
- 2.For recent examples see: Sonye JP, Koide K. J. Org. Chem. 2007;72:1846. doi: 10.1021/jo0623944.2006. p. 6254.Org. Lett. 2006;8:199.Erenler R, Biellmann JF. Tetrahedron Lett. 2005;46:5683.Bernotas RC. Synlett. 2004:2165.Coelho A, Sotelo E, Raviña E. Tetrahedron. 2003;59:2477.Ishikawa T, Mizuta T, Hagiwara K, Aikawa T, Kudo T, Saito S. J. Org. Chem. 2003;68:3702. doi: 10.1021/jo026592g.
- 3.Lu X, Zhang C, Xu Z. Acc. Chem. Res. 2001;34:535. doi: 10.1021/ar000253x.Other examples reported as metal-catalyzed are possibly phosphine mediated: Saiah MKE, Pellicciari R. Tetrahedron Lett. 1995;36:4497.
- 4.Guo C, Lu X. J. Chem. Soc., Perkin Trans. 1. 1993:1921. [Google Scholar]; Lu X, Ji J, Guo C, Shen W. J. Organomet. Chem. 1992;428:259. [Google Scholar]; Lu X, Ji J, Ma D, Shen W. J. Org. Chem. 1991;56:5774. [Google Scholar]; Ma D, Lu X. Tetrahedron Lett. 1989;30:2109. [Google Scholar]
- 5.Tanaka K, Shoji T, Hirano M. Eur. J. Org. Chem. 2007:2687. [Google Scholar]; Tanaka K, Shoji T. Org. Lett. 2005;7:3561. doi: 10.1021/ol051335u. [DOI] [PubMed] [Google Scholar]
- 6.Ma D, Lu X. J. Chem. Soc., Chem. Commun. 1989:890. [Google Scholar]
- 7.Trost BM, Livingston RC. J. Am. Chem. Soc. 1995;117:9586. [Google Scholar]
- 8.Trost BM, Kulaweic RJ. J. Am. Chem. Soc. 1993;115:2027. [Google Scholar]; Trost BM, Kulaweic RJ. Tetrahedron Lett. 1991;32:3039. [Google Scholar]
- 9.Marder TB, Roe DC, Milstein D. Organometallics. 1988;7:1451. [Google Scholar]; Casey CP, O’Connor JM. Organometallics. 1985;4:384. [Google Scholar]; Bönneman H. Angew. Chem. Int. Ed. Engl. 1985;24:248. [Google Scholar]; Rerek ME, Basolo F. J. Am. Chem. Soc. 1984;106:5908. [Google Scholar]
- 10.Trost BM, Sharma S, Schmidt T. Tetrahedron Lett. 1993;34:7183. [Google Scholar]; J. Am. Chem. Soc. 1992;114:7903. [Google Scholar]
- 11.Pilette D, Moreau S, Le Bozec H, Dixneuf PH, Corrigan J, Carty AJ. J. Chem. Soc. Chem. Commun. 1994:409. [Google Scholar]
- 12.Kennedy-Smith JJ, Staben ST, Toste FD. J. Am. Chem. Soc. 2004;126:4526. doi: 10.1021/ja049487s. [DOI] [PubMed] [Google Scholar]; Drent E, Arnoldy P, Budzelaar PHM. J. Organomet. Chem. 1994;475:57. [Google Scholar]; 1993. p. 247.; Komiya S, Ito T, Cowie M, Yamamoto A, Ibers J. J. Am. Chem. Soc. 1976;98:3874. [Google Scholar]
- 13.Trost BM, Portnoy M, Kurihara H. J. Am. Chem. Soc. 1997;119:836. [Google Scholar]; Trost BM, Krause L, Portnoy M. J. Am. Chem. Soc. 1997;119:11319. [Google Scholar]
- 14.Evans DA, Burgey CS, Kozlowski MC, Tregay SW. J. Am. Chem. Soc. 1999;121:686. [Google Scholar]
- 15.Trost BM, Thommen M. Unpublished results.cf Trost BM, Vidal B, Thommen M. Chem. Eur. J. 1999;S:1055.
- 16.Lehmkuhl H, Mauermann H, Benn R. Liebigs Ann. Chem. 1980:754. [Google Scholar]
- 17.For some recent examples see: Yadav JS, Prathap I, Tadi BP. Tetrahedron Lett. 2006;47:3773.Schneider C, Porter N, Brash AR. Chem. Res. Toxicol. 2004;17:937. doi: 10.1021/tx049913n.Sun M, Salomon RG. J. Am. Chem. Soc. 2004;126:5699. doi: 10.1021/ja038756w.Chakraborty TK, Purkait S, Das S. Tetrahedron. 2003;59:9127.
- 18 (a).Corey EJ, Marfat A, Goto G, Brion F. J. Am. Chem. Soc. 1980;102:7984. [Google Scholar]; (b) Corey EJ, Marfat A, Munroe J, Kim KS, Hopkins PB, Brion F. Terahedron Lett. 1981;22:1077. [Google Scholar]; (c) Guindon Y, Zamboni R, Lau CK, Rokach J. Tetrahedron Lett. 1982;23:739. [Google Scholar]; (d) Zamboni R, Rokach J. Tetrahedron Lett. 1982;23:2631. [Google Scholar]; (e) Mills LS, North PC. Tetrahedron Lett. 1983;24:409. [Google Scholar]; (f) Nicolaou KC, Zipkin RE, Dolle RE, Harris BD. J. Am. Chem. Soc. 1984;106:3548. [Google Scholar]; (g) Han C-Q, DiTullio D, Wang Y-F, Sih CJ. J. Org. Chem. 1986;51:1253. [Google Scholar]; (h) Le Merrer Y, Gravier C, Languin-Micas D, Depezay JC. Tetrahedron Lett. 1986;27:4161. [Google Scholar]; (i) Guindon Y, Delorme D, Lau CK, Zamboni R. J. Org. Chem. 1988;53:267. [Google Scholar]; (j) Le Merrer Y, Gravier-Pelletier C, Micas-Languin D, Mestre F, Dureault A, Depezay JC. J. Org. Chem. 1989;54:2409. [Google Scholar]; (k) Avignon-Tropis M, Treilhou M, Lebreton J, Pougny JR, Frechard-Ortuno I, Huynh C, Linstumelle G. Tetrahedron Lett. 1989;30:6335. [Google Scholar]; (l) Kobayashi Y, Shimazaki T, Taguchi H, Sato F. J. Org. Chem. 1990;55:5324. [Google Scholar]; (m) Solladie G, Hamdouchi C, Ziani-Cherif C. Tetrahedron Asymmetry. 1991;2:457. [Google Scholar]; (n) Avignon-Tropis M, Treilhou M, Pougny JR, Frechard-Oturno I, Linstumelle G. Tetrahedron. 1991;47:7279. [Google Scholar]; (o) Cremin D, Linstumelle G. Tetrahedron. 1992;48:1943. [Google Scholar]; (p) Kerdesky FA, Schmidt SP, Brooks DW. J. Org. Chem. 1993;58:3516. [Google Scholar]; (q) Solladie G, Urbano A, Stone GB. Tetrahedron Lett. 1994;34:6489. [Google Scholar]; (r) Rodriguez A, Nomen M, Spur BW, Godfroid JJ, Lee TH. Tetrahedron. 2001;57:25. [Google Scholar]
- 19.Alami M, Ferri F. Tetrahedron Lett. 1996;37:2763. The reaction was highly exothermic when performed on 30 mmol scale.
- 20.Bierer DE, Dener JM, Dubenko LG, Gerber RE, Litvak J, Paterli S, Peterli-Roth P, Truong TV, Mao G, Bauer BE. J. Med. Chem. 1995;38:2628. doi: 10.1021/jm00014a016. [DOI] [PubMed] [Google Scholar]
- 21.Dupradeau F-Y, Prandi J, Beau J-M. Tetrahedron. 1995;51:3205. [Google Scholar]
- 22.Suzuki M, Morita Y, Noyori R. J. Org. Chem. 1990;55:441. [Google Scholar]
- 23.Zweifel G, Backlund SJ, Leung T. J. Am. Chem. Soc. 1978;100:5561. [Google Scholar]
- 24.Matsumoto M, Kuroda K. Tetrahedron Lett. 1980;21:4021. [Google Scholar]
- 25.Morris J, Wishka DG. Tetrahedron Lett. 1986;27:803. [Google Scholar]; Charoenying P, Davies DH, MeKerrecher D, Taylor RJK. Tetrahedron Lett. 1996;37:1913. [Google Scholar]
- 26.Krasovskiy A, Kopp F, Knochel P. Angew. Chem. Int. Ed. 2006;45:497. doi: 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]
- 27.Trost BM, Lee C. J. Am. Chem. Soc. 2001;123:12191. doi: 10.1021/ja0118338. [DOI] [PubMed] [Google Scholar]
- 28.Trost BM, Weiss AH. Org. Lett. 2006;8:4461. doi: 10.1021/ol0615836. [DOI] [PubMed] [Google Scholar]
- 29.Takayama H, Koike T, Aimi N, Sakai S. J. Org. Chem. 1992;57:2173. [Google Scholar]
- 30.Zimmerman B, Lerche H, Severin T. Chem. Ber. 1986;119:2848. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.