

Abstract
Androgens, through their actions on the androgen receptor (AR), are required for the development of the prostate and contribute to the pathological growth dysregulation observed in prostate cancers. Consequently, androgen ablation has become an essential component of the pharmacotherapy of prostate cancer. In this study, we explored the utility of targeting processes downstream of AR as an alternate approach for therapy. Specifically, we demonstrate that the serum and glucocorticoid-regulated kinase 1 (sgk1) gene is an androgen-regulated target gene in cellular models of prostate cancer. Furthermore, functional SGK1 protein, as determined by the phosphorylation of its target Nedd4-2, was also increased with androgen treatment. Importantly, we determined that RNAi-mediated knockdown of SGK1 expression attenuates androgen-mediated growth of the prostate cancer cell line, LNCaP. Given these findings, we explored the utility of SGK1 as a therapeutic target in prostate cancer by developing and evaluating a small molecule inhibitor of this enzyme. From these studies emerged GSK650394, a competitive inhibitor that quantitatively blocks the effect of androgens on LNCaP cell growth. Thus, in addition to androgen ablation, inhibition of pathways downstream of AR are likely to have therapeutic utility in prostate cancer.
Keywords: serum and glucocorticoid-regulated kinase 1, androgen receptor, prostate cancer
Introduction
One in six men will develop prostate cancer during their lifetime and with approximately 30,000 deaths per year attributed to the disease, it is the second leading cause of cancer death in men (1). The majority of prostate cancers express the androgen receptor (AR) and rely on androgens for growth and survival. For this reason, patients with advanced prostate cancers generally undergo androgen deprivation therapy with chemical and/or surgical castration as a primary intervention. Although 80% of patients with local or metastatic prostate cancer initially respond favorably to androgen ablation therapy, most patients eventually experience a relapse of the disease, which is then considered to be hormone refractory (2, 3). Unfortunately, effective therapeutic options are limited for these patients. Although hormone refractory disease is no longer responsive to androgen deprivation therapy, AR signaling pathways remain active and necessary for cancer progression (4). Therefore, novel therapeutics that facilitate AR turnover or which target signaling pathways downstream of receptor activation may be an effective treatment option for hormone refractory disease. Consequently, there is renewed interest in the pharmacological exploitation of AR signaling pathways for prostate cancer therapeutics.
Although the phenotypic responses of the prostate and prostate cancer cells to androgens are well described, the molecular events underlying these activities are not well understood. However, the results of microarray studies performed by both our laboratory and others have led to the identification of pathways and specific genes that are likely to be important for AR biology in prostate cancer. Among the genes identified in this manner is that encoding the serum- and glucocorticoid-regulated kinase 1 (SGK1) (5–7). SGK1 belongs to the AGC family of serine/threonine protein kinases, which also includes protein kinase C and Akt. As its name implies, SGK1 expression is regulated by glucocorticoids and serum, in addition to various other types of signals (8–15). The enzymatic activity of SGK1 is regulated by specific phosphorylation events initiated by phosphoinositide-3 kinase (PI3K) (16–20). Specifically, upon PI3K activation, SGK1 is phosphorylated at serine 422 by an as yet unidentified kinase, referred to as the phosphoinositide-dependent protein kinase 2 (PDK2), followed by the phosphorylation of threonine 256 within the activation loop of the SGK1 catalytic domain by PDK1 to become fully activated (19).
Many prostate cancers rely on the PI3K signaling pathway for growth and survival. This is reflected by the high incidence of loss-of-function mutations in the gene that encodes the PI3K negative regulator phosphatase and tensin homolog (PTEN) in prostate cancers (21, 22). In fact, the prostate-specific PTEN knockout mice develop invasive prostate cancer with 100% penetrance, indicating that within the context of the murine prostate constitutive PI3K signaling is sufficient for the development of cancer (23).
A considerable number of studies have addressed the role of Akt as the mediator of PI3K-dependent stimulation of growth and survival in prostate cancer. Interestingly, however, both Akt and SGK1 phosphorylate and regulate the activity of many of the same target proteins. Recently, SGK1 has also been implicated in the regulation of cell growth and survival downstream of PI3K activation (24–27). Therefore, considering that most prostate cancers rely on the androgen signaling pathway for growth, and activation of this pathway leads to the induction of SGK1 expression, we hypothesized that SGK1 is required for androgen-mediated prostate cancer growth. This study demonstrates that inhibition of SGK1 expression or activity antagonizes androgen-induced growth of the prostate cancer cell line LNCaP, suggesting that SGK1 might be a viable target for the treatment of prostate cancer.
Materials and Methods
Reagents and antibodies
All chemicals and reagents were purchased from Sigma (St. Louis, MO) unless otherwise specified. Methyltrienolone (R1881) was purchased from Perkin Elmer (Waltham, MA) and bicalutamide (Casodex) was provided as a gift from P. Turnbull (GlaxoSmithKline, Research Triangle Park, NC). GSK650394 was synthesized by the CVU Medicinal Chemistry Department at GlaxoSmithKline (King of Prussia, PA). The goat polyclonal glyceraldehyde-3-phosphate dehydrogenase (GAPDH) V-18 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and the rabbit polyclonal SGK1 antibody (KAP-PK015) was purchased from Stressgen Bioreagents, Inc. (Victoria, BC, Canada). The mouse monoclonal AR441 antibody that recognizes AR was a gift from D. Edwards (Baylor College of Medicine, Houston, TX). The Nedd4-2 and phospho-Nedd4-2 antibodies were provided as gifts from O. Staub (University of Lausanne, Lausanne, Switzerland).
Cell culture
The human prostate carcinoma cell line LNCaP-FGC was obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in RPMI Medium 1640 (Invitrogen, Carlsbad, CA) supplemented with 8% fetal bovine serum (FBS), 0.1 mM non-essential amino acids (NEAA) and 1 mM sodium pyruvate (NaPyr) (Invitrogen). The prostate cancer cell line LAPC4 was a gift from C. Sawyers (Memorial Sloan-Kettering Cancer Center, New York, NY) and maintained in Iscove’s Modified Dulbecco’s Medium (IMDM; Invitrogen) supplemented with 15% FBS, 0.1 mM NEAA, 1 mM NaPyr and 1 nM R1881. The prostate cancer cell line VCaP was provided by K. Pienta (University of Michigan, Ann Arbor, MI) and maintained in Dulbecco’s Minimal Essential Medium (DMEM; Invitrogen) supplemented with 8% FBS, 0.1 mM NEAA and 1 mM NaPyr. The M-1 cell line (derived from SV40-transformed mouse cortical collecting duct) was obtained from ATCC and maintained in Ham’s F12/DMEM supplemented with 5% FBS, 2 mM L-glutamine, 1% antibiotic/antimycotic, 100 nM dexamethasone, and transferrin, insulin and sodium selenite (6.25 μg/mL each). The HeLa cell line was obtained from ATCC and maintained in Ham’s DMEM supplemented with 10% FBS, 2mM L-glutamine and 1% antibiotic/antimycotic. The COS-7 cell line was obtained from ATCC and maintained in DMEM (high glucose) supplemented with 10% FBS, 2 mM L-glutamine and 1% antibiotic/antimycotic. All cells were maintained in humidified incubators at 37°C with 5% CO2.
Immunoblotting
Western blots were performed as previously described (28). Membranes were probed with the appropriate primary antibody at the indicated dilutions (anti-SGK1, anti-AR, and anti-GAPDH at 1:1000, anti-Nedd4-2 at 1:500, and anti-phospho-Nedd4-2 (Ser328) at 1:200).
Transient transfection of siRNAs
Chemically synthesized Stealth siRNAs were purchased from Invitrogen and transfected using Dharmafect-1 transfection reagent (Dharmacon, Lafayette, CO) according to the manufacturer’s instructions. See Supplementary Table 1 for sequences of siRNAs used in these studies.
RNA isolation and quantitative PCR
RNA isolation and quantitative PCR (qPCR) were performed as previously described (28). See Supplementary Table 1 for sequences of qPCR primers used in these studies. In those studies utilizing actinomycin D and cycloheximide, the compounds were used at concentrations previously shown to inhibit transcription and translation in LNCaP cells (29).
Cell growth assays
LNCaP cells were plated at a density of 5,000 cells per well (for studies with GSK650394) or 20,000 cells per well (for siRNA studies) in 96-well plates in 100 μL PRF-RPMI 1640, supplemented with 8% CS-FBS, 0.1 mM NEAA, and 1 mM NaPyr. After a two-day incubation, cells were transfected with siRNAs as described above. Twenty μL of media was removed from each well and replaced with 20 μL of the siRNA:Dharmafect-1 mix to obtain a final concentration of 50 nM of siRNA. The siRNA transfection was repeated on day 6. At day three, cells were treated with hormone with or without GSK650394 by removing 50 μL of the media and replacing this with 50 μL of PRF-RPMI 1640 with 8% CS-FBS, NEAA, NaPyr containing a 2X concentration of the appropriate hormone/inhibitor treatment. At days 5 and 7, the treatment was repeated. On the tenth day, the media was removed and the relative cell number was measured using the FluoReporter Blue assay (Invitrogen) according to the manufacturer’s instructions.
Fluorescence polarization (FP) assay
SGK1 S422D cDNA (amino acids 60–431) was subcloned into pFastBAC-HTc (Invitrogen). Protein was expressed in Sf9 insect cells and purified by Ni2+-NTA agarose chromatography. SGK1 was stored in a buffer containing 50 mM Tris-HCl (pH 7.5), 270 mM sucrose, 150 mM NaCl, 0.1 mM EGTA, 0.1% β-mercaptoethanol, 0.2 mM PMSF, and 1 mM benzamidine. SGK1 was diluted to 4 μM and activated in 50 mM Tris-HCl (pH 7.5), 0.1 mM EGTA, 0.1% β-mercaptoethanol, 10 mM magnesium acetate, and 0.1 mM ATP with 3.3 μg/mL GST-PDK1 at 30°C for 30 min. SGK1 was then re-purified by Ni2+-NTA agarose chromatography. To determine the efficiency of the activation reaction, activated SGK1 (diluted in 50 mM Tris-HCl (pH 7.5), 0.1% (v/v) β-mercaptoethanol, 0.1mM EGTA, and 1 mg/mL BSA) was assayed for kinase activity using 30 μM of a CROSStide substrate peptide (a synthetic peptide consisting of the amino acids GRPRTSSFAEG; Millipore Corporation, Billerica, MA) in 50 μL of 50 mM Tris-HCl (pH 7.5), 0.1% (v/v) β-mercaptoethanol, 0.1 mM EGTA, 10 mM magnesium acetate, 100 μM γ32P-ATP (50–1000 cpm/pmole) at 30°C for 10 min. Reactions were stopped by spotting 40 μL of the assay mixture onto Whatman P81 paper, which was washed in 75 mM phosphoric acid, followed by acetone before air drying and counting. Microscint-20 liquid scintillation fluid (Packard Instruments, Meriden, CT) was added and the signal was detected by measuring for 30 sec/well in a Packard TopCount NXT Scintillation Counter.
A Rhodamine Green-labeled FP ligand (a proprietary ATP mimetic; 0.5 nM final concentration) and SGK1 (1 nM final concentration) were incubated in buffer containing 50 nM HEPES (pH 7.5), 1 mM CHAPS, 10 nM MgCl2, and 1 mM DTT for 15 min at room temperature. GSK650394 was dissolved in DMSO and diluted into buffer for concentration response curve determination. The fluorescence signal was measured using an Acquest (Molecular Devices; Sunnyvale, CA) at excitation and emission wavelengths of 485 nm and 530 nm, respectively. SGK1 inhibition IC50 was calculated from these data using GraphPad Prism 3 Software (GraphPad Software, San Diego, CA).
Scintillation proximity assay (SPA)
SGK1 S422D (60–431 aa; 0.275 μg/mL final concentration) or SGK2 (0.875 μg/mL final concentration) were activated by PDK1 (1.1 μg/mL final concentration) in a buffer consisting of 50 mM Tris (pH 7.5), 0.1 mM EGTA, 0.1 mM EDTA, 10 mM MgCl2, 0.1% β-mercaptoethanol, 1 mg/mL BSA, and ATP (final concentration of 0.15 mM) and incubated for 30 min at 30°C. SGK2 was prepared exactly as described for SGK1, except it corresponded to the full-length protein. A solution containing biotinylated CROSStide peptide at a final concentration of 75 μM and γ32P-ATP corresponding to 2×106 cpm was prepared in the reaction buffer. In a 96-well plate, 5 μL of GSK650394 was added to 25 μL of the activated enzyme mixture. To this, 20 μL of the CROSStide mixture was added and incubated for 1 h at room temperature. Next, 50 μL of a 25 mg/mL slurry of streptavidin-coated SPA beads (Amersham, Little Chalfont, Buckinghamshire, UK) in PBS with 0.1 M EDTA, pH 8.0 was added. The plate was then sealed and centrifuged for 8 min at 2000 rpm, and the signal was detected by measuring for 30 sec/well in a Packard TopCount NXT Scintillation Counter. The IC50 values of the inhibition of SGK1 and SGK2 activities by GSK650394 were calculated from these data using GraphPad Prism 3 Software.
Toxicity assays
The toxicity of GSK650394 to M-1 and HeLa cells was assessed using the Cell Proliferation Kit (XTT) following manufacturer’s instructions (Roche). Briefly, 10,000 HeLa or M-1 cells/well were plated into 96-well plates in 100μL of the appropriate maintenance media. After 48 h, media was removed and replaced with 100 μL of EMEM with Earle’s salts containing 2 mM L-glutamine and 1% antibiotic-antimycotic overnight. M-1 cells were also supplemented with 1 μg/mL insulin, 6.25 μg/mL sodium selenite, and 6.25 μg/mL transferrin. After 24 h, the media was removed and replaced with 100 μL media alone or media containing increasing concentrations of GSK650394. For HeLa cells, 50 μL of activated XTT solution was added after 4 h. For M-1 cells, 50 μL of activated XTT solution was added after 24 h. Following a 2 h incubation, absorbance was measured at 490 nm using a SpectraMAX PLUS spectrophotometer (Molecular Devices, Sunnyvale, CA) and the data analyzed to obtain IC50 values using GraphPad Prism 3 software.
Short circuit current (SCC) assay
SCC was determined in M-1 cells as previously reported (30). Cells were grown to confluence, trypsinized and plated onto 12 mm transwell membranes (0.4 μM pore diameter; Corning) at a density of 55,000 cells/well in EMEM supplemented with 5% FBS, 15 mM HEPES, 2 mM L-glutamine, 1 μg/mL insulin, 6.25 μg/mL sodium selenite, 6.25 μg/mL transferrin, and 1% antibiotic/antimycotic. After the cells reached confluency on the transwell membranes, the media was replaced with media lacking FBS. Baseline measurements were obtained to ensure monolayer confluency and viability by demonstrating adequate resistance. Voltage and resistance of the cell monolayer was measured using a EVOMX Epithelial Voltohmmeter with an STX2 electrode (WPI, Sarasota, FL). Voltage was zeroed by measuring Vte across a transwell with no cells, then Vte was determined from the basolateral chamber. The initial resistance, Rte, was determined by applying current across the cell monolayer for a period of < 1 s. The equilibrium current, Ieq, was calculated by dividing the voltage by the resistance for initial measurement and normalized by surface area of the membrane. Aldosterone (1 μM) was added to the appropriate compartment and Ieq was obtained from 6 wells. After 4 h, GSK650394 plus aldosterone was added and Ieq was obtained. At the end of the experiment, amiloride (inhibits epithelial sodium transport through the inhibition of the epithelial sodium channel ENaC) was added to a final concentration of 10 μM and the amiloride-sensitive SCC was measured and subtracted from the total SCC to obtain SGK1-induced SCC. The IC50 values were calculated from these data using GraphPad Prism 3 Software.
Results
Androgen-activated AR induces the expression of SGK1
Microarray analyses performed in our laboratory revealed that the sgk1 gene is regulated by androgens in the LNCaP prostate cancer cell line (5). To validate the microarray results and to determine if sgk1 is regulated by androgens in other AR-expressing prostate cancer cell lines, the expression levels of SGK1 were measured in the prostate cancer cell lines LNCaP, VCaP and LAPC4 in the presence and absence of the synthetic androgen R1881 using qPCR. Similar to the induction observed in our microarray study, SGK1 mRNA levels were upregulated approximately 20-fold in LNCaP cells (Figure 1A). This upregulation in response to androgens was also apparent in other AR-expressing prostate cancer cell lines, with VCaP cells exhibiting a 10-fold increase and LAPC4 cells a 2.5-fold increase in SGK1 mRNA levels. Although LAPC4 cells demonstrated a less dramatic androgen-mediated induction of SGK1 expression levels, we did note that the basal level of SGK1 expression is relatively high in these cells. Given the robust androgen-dependent induction of SGK1 expression in LNCaPs, all subsequent studies were performed in this cell line.
Figure 1. Androgens upregulate SGK1 transcript levels.
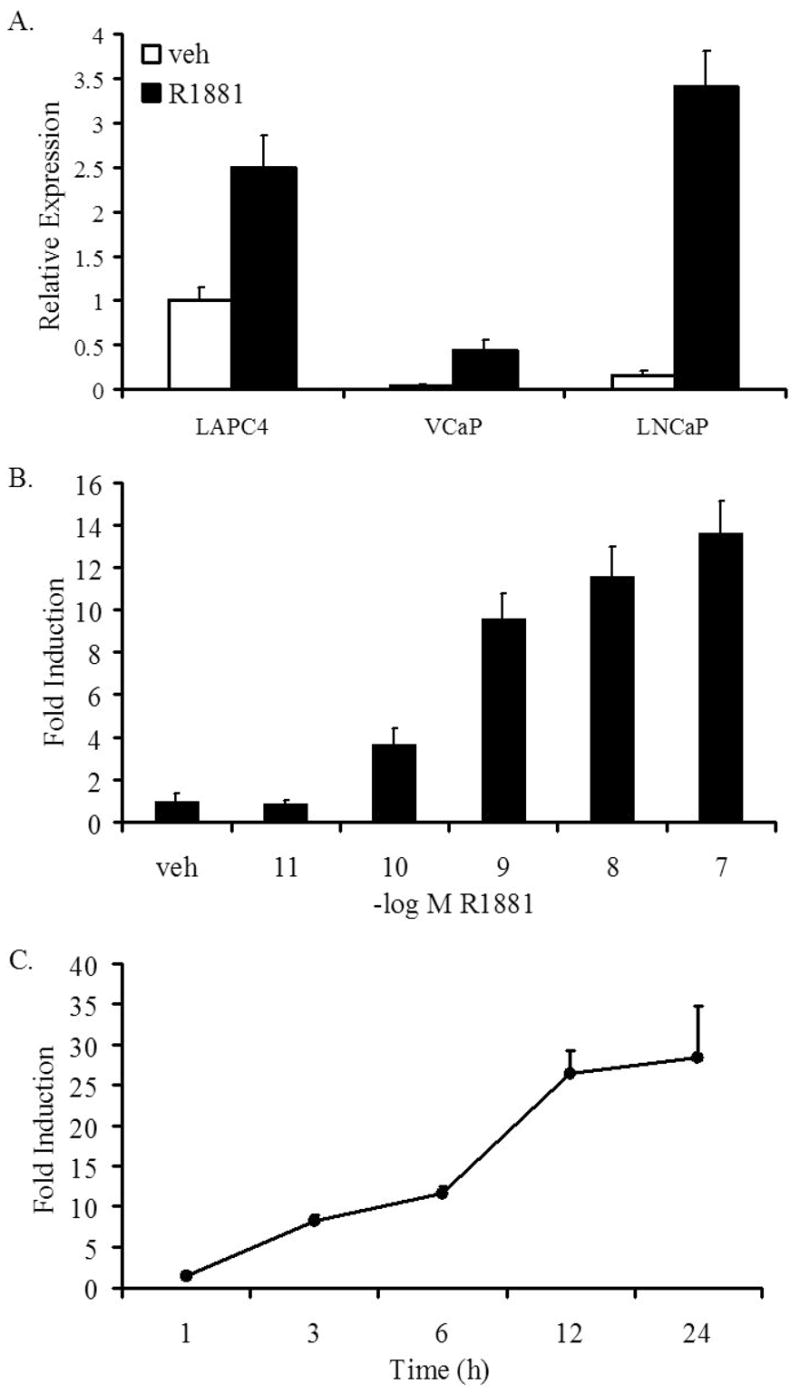
LNCaP, VCaP, and LAPC4 cells were grown in media with charcoal-stripped FBS for 3 days, prior to the addition of ethanol (veh) or R1881 (A and C, 10 nM; or B, at the indicated doses). After a 24 h incubation (A and B), or the indicated time points (C), cells were lysed, RNA isolated and reverse transcribed. The expression of SGK1 was assessed with qPCR and normalized to GAPDH expression levels. Each experiment was performed at least three times, with a representative experiment shown; bars, SD.
SGK1 mRNA levels are upregulated by androgens in a dose-dependent manner, with levels increasing at 100 pM R1881 and reaching maximal levels at 10–100 nM, which is the approximate concentration at which ligand saturates the receptor (Figure 1B). The enhancement of SGK1 mRNA levels in response to androgens is evident as early as three hours post-treatment, suggesting the sgk1 gene is a direct transcriptional target of androgen-bound AR (Figure 1C). This was confirmed by demonstrating that sgk1 mRNA was not induced in the presence of actinomycin D but was unaffected by cycloheximide (Supplementary Figure 1). Importantly, the antiandrogen Casodex (1 μM) inhibited R1881 (1 nM)-dependent increases in SGK1 transcript levels (Figure 2A). Furthermore, three AR-targeted siRNAs, each of which effectively suppressed expression of both AR mRNA and protein, suppressed the androgen-mediated upregulation of SGK1 mRNA expression (Figures 2B-D). Taken together, these studies confirm that sgk1 is a primary target of AR in prostate cancer cells.
Figure 2. The androgen-mediated upregulation of SGK1 is androgen receptor dependent.

A, LNCaP cells were grown in media with charcoal-stripped FBS for 3 days, prior to the addition of ethanol (veh) or R1881 (1 nM) in the presence or absence of Casodex (1 μM). After a 24 h incubation, cells were lysed, RNA isolated and reverse transcribed. The expression of SGK1 was assessed with qPCR and normalized to GAPDH expression levels. B-D, LNCaP cells were transiently transfected with Stealth siRNAs targeting AR (AR-A, AR-B, or AR-C) or a negative control (neg) Stealth siRNA at a final concentration of 50 nM. Cells were “mock” transfected as an additional negative control. 48 h later, cells were treated with ethanol (veh) or R1881 (10 nM) for 24 h. AR (B) and SGK1 (D) mRNA levels were detected with qPCR. SGK1 and AR mRNA levels were normalized with GAPDH mRNA levels. Each experiment was performed at least three times, with a representative experiment shown; bars, SD. C, Whole cell extracts were prepared and proteins were separated on a SDS-PAGE gel and transferred to a nitrocellulose membrane which was probed with antibodies against AR and GAPDH (loading control).
Androgen treatment increases SGK1 protein levels and activity
The upregulation of SGK1 mRNA levels in the presence of androgens was accompanied by a commensurate increase in steady-state SGK1 protein levels (Figure 3A). In the absence of androgens, SGK1 protein levels were undetectable by immunoblotting. In response to R1881 (10 nM), protein levels began to increase within three hours of treatment and were sustained up to 48 hours post-treatment (Figure 3B). This is notable considering that others have shown that SGK1 expression, in response to various stimuli, is typically rapid and transient (12). LNCaP cells are PTEN-null and therefore exhibit constitutive activation of PI3K (31). Not surprisingly therefore, the SGK1 protein is phosphorylated and migrates as multiple bands on immunoblots.
Figure 3. Androgen treatment leads to an increase in SGK1 protein levels and activity in LNCaP cells.
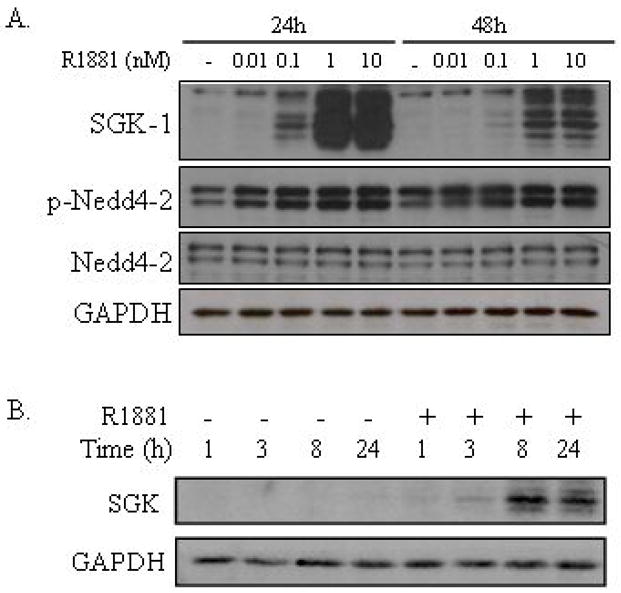
A, LNCaP cells were grown in media with charcoal-stripped FBS for 3 days, prior to the addition of ethanol (veh) or R1881 at the indicated concentrations (A) or 10 nM (B). After a 24 h or 48 h incubation (A) or an incubation for the duration indicated (B), cells were lysed and proteins were separated on a SDS-PAGE gel and transferred to a nitrocellulose membrane, which was probed with antibodies against AR, SGK1, Nedd4-2, phospho-Nedd4-2, and GAPDH (loading control). Each experiment was performed at least three times, with a representative experiment shown.
One of the most well-characterized roles for SGK1 is in hypertension where SGK1 phosphorylates and inhibits the ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 (Nedd4-2) at serine 468 (S468) which ultimately affects epithelial sodium ion transport (32). Hence, to verify that the SGK1 protein is active in LNCaP cells under our particular growth conditions, we analyzed the phosphorylation state of the SGK1 target protein Nedd4-2. Here, we demonstrate an increase in Nedd4-2 S468 phosphorylation in response to androgen treatment, confirming that androgens facilitate an enhancement of SGK1 signaling processes (Figure 3A). Interestingly, the strong enhancement of SGK1 levels in response to androgens did not produce an immediate dramatic elevation of Nedd4-2 phosphorylation. This may suggest that not only does SGK1 need to be expressed and posttranslationally activated, but that other limiting activities (i.e translocation to its target) must also occur. Alternatively, the constitutive PI3K activity in LNCaP cells due to the PTEN-null status may increase basal phosphorylation of Nedd4-2 and obscure a direct evaluation of the impact of androgens on this endpoint.
SGK1 expression is required for androgen-mediated proliferation of prostate cancer cells
Given the role of SGK1 as a growth factor-responsive kinase and the fact that it is a downstream target of PI3K, we hypothesized that its induction by androgens may be necessary for androgen-dependent growth of LNCaP cells. In order to test this hypothesis, three siRNAs, each of which effectively reduced SGK1 mRNA and protein expression, were analyzed for their effects on LNCaP cell growth (Figures 4A and 4B). As a positive control, we demonstrated that each of three different AR siRNAs completely abrogated the androgenic stimulation of growth in LNCaP cells, whereas the negative control siRNA had no effect under the same conditions (Figure 4C). More importantly, when any of the three SGK1 siRNAs were transfected into LNCaP cells, we observed a 50–60% inhibition of the 2.5-fold stimulation of growth in the presence of androgens, with no effect on the cells in the absence of androgens (Figure 4C). These studies, utilizing RNAi-mediated knockdown of SGK1, support the hypothesis that this protein is required for androgen-dependent growth of LNCaP cells.
Figure 4. SGK1 expression is required for androgen-mediated proliferation of LNCaP cells.
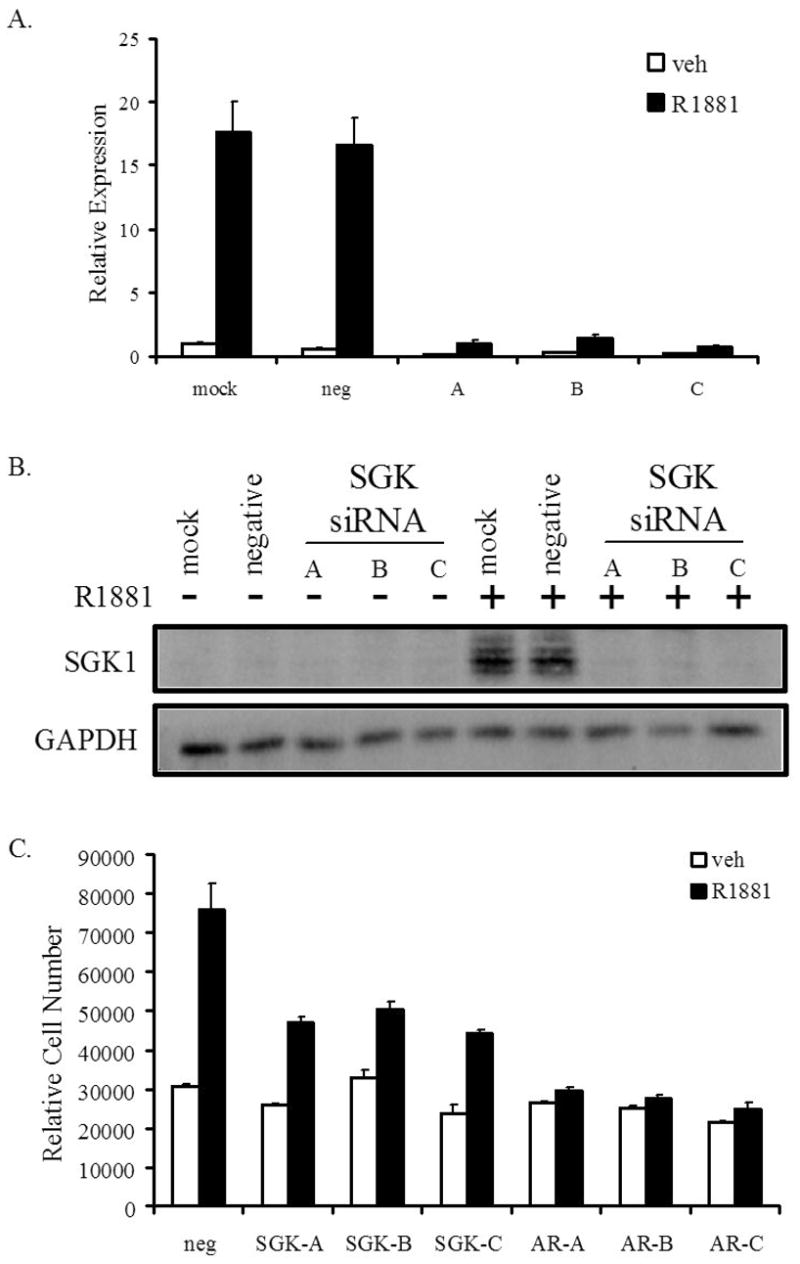
LNCaP cells were transfected with the Stealth SGK1 A, B or C siRNAs or a negative control siRNA (neg) at a final concentration of 50 nM. Cells were “mock” transfected as an additional negative control. Two days later, cells were treated with ethanol (veh) or R1881 (10 nM). A, After a 24 h incubation, cells were lysed and RNA was isolated. RNA was reverse transcribed and transcript levels of SGK1 were measured with qPCR and were normalized to GAPDH mRNA levels; bars, SD. B, Whole cell extracts were collected and proteins were separated on a SDS-PAGE gel, followed by transfer to a nitrocellulose membrane. The membrane was probed with antibodies against SGK1 or GAPDH (loading control). C, LNCaP cells were incubated in media with charcoal-stripped FBS for 2 days. Cells were transiently transfected with Stealth SGK1 (SGK-A, SGK-B, SGK-C), AR (AR-A, AR-B, AR-C) or negative control (neg) siRNAs at a final concentration of 50 nM. An additional transfection of these siRNAs was performed 4 days later. Cells were treated with ethanol (veh) or R1881 (10 nM) on days 3, 5 and 7. On day 10, cells were lysed and the relative number of cells was measured with the fluorescent DNA-binding dye FluoReporter Blue. Each sample was performed in triplicate and the experiment was performed at least three times, with a representative experiment shown; bars, SE.
Development of a novel SGK1 inhibitor, GSK650394
Given that SGK1 expression is required for androgen-dependent growth of prostate cancer cells, we hypothesized that SGK1 would be a viable target for the development of pharmacological agents for the treatment of prostate cancer. To test this, we developed a novel compound, GSK650394, that functionally inhibits SGK1 and examined the effects of this compound on cellular models of prostate cancer. The structure of GSK650394 is shown in Figure 5A and its initial characterization is described below and summarized in Supplementary Table 2.
Figure 5. GSK650394 inhibits the activity of SGK1.
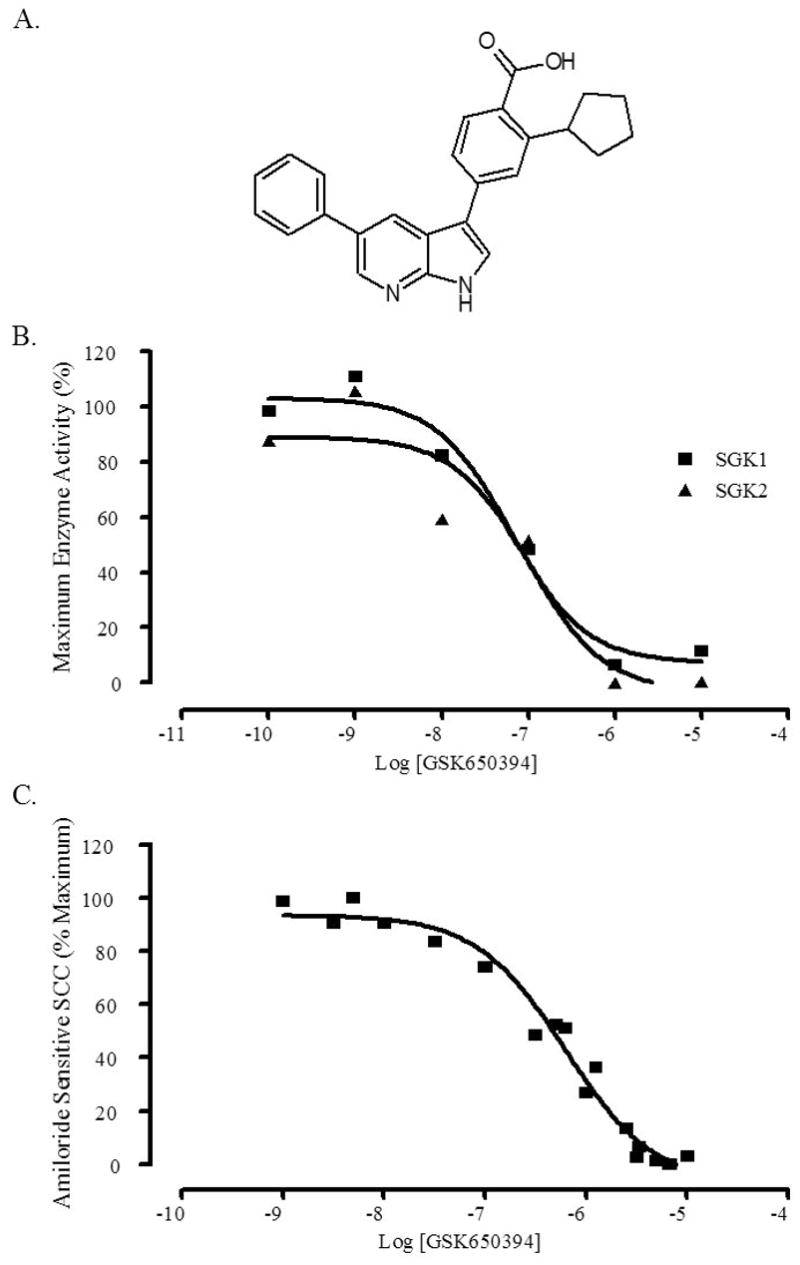
A, The chemical structure of GSK650394. B, GSK650394 quantitatively inhibits the activity of SGK1 and SGK2 in a scintillation proximity assay. Activated SGK1 or SGK2 proteins were incubated with increasing concentrations of GSK650394 and a synthetic biotinylated peptide substrate (CROSStide). Activity was assessed by detection of the radiolabeled phosphate that was incorporated into the biotinylated CROSStide, when bound to streptavidin-coated polystyrene beads containing a scintillant. The experiment was performed twice, and results were normalized to % maximum enzyme activity with a representative experiment shown. C, GSK650394 fully inhibits amiloride-sensitive short circuit current (SCC) in M-1 cells. M-1 cells were plated in EMEM with 5% FBS onto 12 mm transwell membranes with 0.4 μM pore diameter and 1.13 cm2 active surface area and maintained for 5–7 days until confluent. On the day of the experiment, media was replaced with EMEM media with no supplements and basal voltage and resistance measurements were obtained and the baseline was set. Aldosterone (1 μM) was added to the appropriate compartment, and Ieq was obtained from 6 wells. After 4 h, GSK650394 plus aldosterone was added and Ieq was obtained. At the end of the experiment, amiloride was added to a final concentration of 10 μM and the amiloride-sensitive SCC was measured and subtracted from the total SCC to obtain SGK1-induced SCC. The experiment was performed twice, and results were normalized to % maximum amiloride sensitive SCC with a representative experiment shown.
A fluorescence polarization assay was used to determine the affinity of GSK650394 for activated SGK1. The ability of GSK650394 to displace a Rhodamine Green fluorescently-labeled small molecule ATP mimetic bound to purified SGK1 protein corresponding to amino acids 60–431 (harbors a S422D mutation to mimic phosphorylation of the PDK2 site and was fully activated by recombinant PDK1) was measured. In this assay, GSK650394 inhibited activated SGK1 with an IC50 of 13 nM (data not shown).
The ability of GSK650394 to inhibit the enzymatic activity of SGK1 and SGK2 was measured using an in vitro activity-based scintillation proximity assay (SPA). This assay measures SGK1- or SGK2-mediated phosphorylation of a serine residue within a synthetic biotinylated peptide substrate. SGK1 or SGK2 phosphorylates the peptide substrate, thereby incorporating a radiolabeled phosphate, which is subsequently incubated with streptavidin-coated polystyrene beads containing a scintillant. The localization of the radiolabeled peptide within the immediate vicinity of the scintillant-containing bead generates a measurable light signal. GSK650394 inhibited the enzymatic activity of SGK1 and SGK2 in the SPA assay with IC50 values of 62 nM and 103 nM, respectively (Figure 5B).
The inhibitory effects of GSK650394 on SGK1 activity were further evaluated using cell-based assays. Importantly, GSK650394 is relatively non-toxic, with LC50 values of 41 μM in M1 cells (68 times its activity IC50) and a LC50 greater than 100 μM in HeLa cells in XTT assays, which measure mitochondrial enzymatic activity (data not shown). SGK1 has been linked to hypertension and mice lacking the sgk1 gene have higher sodium excretion and lower blood pressure than wild type mice when fed a low sodium diet (33, 34). This has been attributed to the regulation of epithelial sodium ion transport by SGK1 in response to aldosterone stimulation. GSK650394 was evaluated for its effects on this well-documented SGK1-mediated biological activity, which was measured using an aldosterone-stimulated short circuit current cellular assay (SCC). GSK650394 inhibited SGK1-mediated epithelial transport with an IC50 of 0.6 μM in the SCC assay (Figure 5C).
The specificity of GSK650394 for SGK1 over that of the most closely related AGC kinase family member, Akt, and other related kinases was measured using standard in vitro kinase assays (University of Dundee, Scotland, UK). The selectivity of GSK650394 for SGK1 over that of Akt and other related kinases proved to be greater than 30-fold, while GSK650394 was more than 60-fold selective for SGK1 over the upstream AGC kinase PDK1 (Supplementary Table 2).
GSK650394 inhibits SGK1 activity and androgen-mediated LNCaP cell growth
When tested in LNCaP cells, GSK650394 repressed the androgen-mediated enhancement of Nedd4-2 phosphorylation, suggesting GSK650394 antagonizes SGK1 activity in these cells (Figure 6A). Next, a ten-day growth assay, similar to the one described for the siRNA studies (Figure 4C), was used to measure the effects of GSK650394 on LNCaP cell growth. Of note, GSK650394 had no effect on LNCaP cell growth or survival in the absence of androgens, and is therefore not toxic at the levels used in these experiments (Figure 6B). Although GSK650394 had no effect on cell survival or growth in the absence of androgens, the compound dramatically inhibited the androgen-stimulated growth of LNCaP cells. In the LNCaP growth assay, the IC50 of GSK650394 was approximately 1 μM (which is similar to the previously measured IC50 in other cell-based assays). Furthermore, 10 μM of GSK650394 completely abrogated androgen-mediated growth, indicating that small molecule inhibitors of SGK1 may be an effective approach for the treatment of AR-driven prostate cancer.
Figure 6. GSK650394 inhibits SGK1 activity and androgen-mediated LNCaP cell growth.
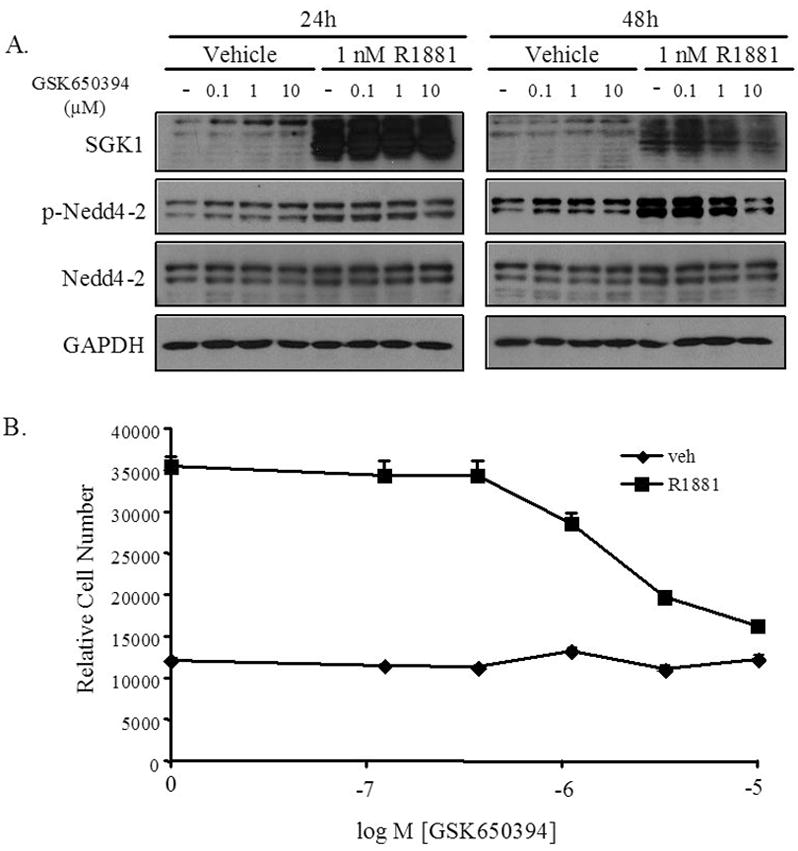
A, LNCaP cells were grown in media with charcoal-stripped FBS for 3 days, prior to the addition of ethanol (veh), R1881, DMSO (−) or GSK650394 at the indicated concentrations. Whole cell extracts were collected and proteins were separated on a SDS-PAGE gel, followed by transfer to a nitrocellulose membrane. The membrane was probed with antibodies that recognize SGK1, Nedd4-2, phospho-Nedd4-2 (Ser328), or GAPDH (loading control). B, LNCaP cells were plated in 96-well plates and grown in media with charcoal-stripped FBS for 3 days. Cells were treated with ethanol (veh) or R1881 (10 nM) and the indicated doses of GSK650394 or DMSO on days 3, 5 and 7. On day 10, cells were lysed and the relative number of cells was measured with the fluorescent DNA binding dye FluoReporter Blue. Each sample was performed in triplicate and the experiment was performed at least three times, with a representative experiment shown; bars, SE.
Discussion
These studies demonstrate that androgens upregulate SGK1 mRNA levels in an AR-dependent fashion. This observation parallels findings presented within a recent report by Shanmugam et al. (6). However, in contrast to the studies by Shanmugam et al. in which cells are grown under serum-free conditions, the experiments presented herein were performed in the presence of 8% charcoal-stripped serum, which is more reflective of physiological conditions. Therefore, our studies suggest that even in the presence of serum, which in and of itself is capable of inducing SGK1 expression, androgens are able to enhance SGK1 mRNA levels. Our studies further demonstrate that the androgen-dependent induction of SGK1 mRNA levels correlates with an increase in functionally active SGK1 protein. This observation, in combination with the demonstration that siRNA-mediated downregulation of SGK1 expression inhibits androgen-dependent growth of prostate cancer cells, suggests that the enzymatic activity of SGK1 might be responsible for androgen-stimulated prostate cancer growth and may represent a viable target for the treatment of prostate cancer.
A possible mechanism by which SGK1 might control cell cycle progression and proliferation in prostate cancer cells is through the direct or indirect regulation of the activity of the mammalian target of rapamycin (mTOR) protein. Notably, it has been demonstrated by others that androgens stimulate mTOR activity in LNCaP cells (7). Active mTOR contributes to an enhancement of the translation rates of various proteins involved in cell cycle progression, including cyclins D1 and D2 (7). In addition, Xu and colleagues indicated that SGK1 may activate mTOR through the activation of various cellular transporters and the subsequent influx of certain nutrients and amino acids (7, 35). Interestingly, the expression of many of the same cellular transporters are enhanced by androgens (7). Additionally, SGK1 may regulate mTOR activity in a more direct manner, through the phosphorylation and inactivation of TSC2, a negative regulator of mTOR, as the expression of constitutively active SGK1 in cardiomyocytes is associated with an enhancement of TSC2 phosphorylation and mTOR activity (24).
It has been suggested that SGK1 may regulate AR transcriptional activity, affecting androgen-mediated prostate cancer growth through a positive feedback mechanism (6). We have not been able to confirm this result and were unable to observe any effects of GSK650394 on the expression level of a panel of androgen-regulated genes in LNCaP cells under conditions where proliferation was completely repressed (data not shown). At this time, however, we cannot exclude the possibility that SGK1 activity is required for the regulation of a subset of androgen-regulated genes.
We have demonstrated that SGK1 expression (siRNA-mediated downregulation) and activity (GSK650394 studies) are required for the effects of androgens on prostate cancer growth. The growth inhibition observed following treatment with GSK650394 was more dramatic than that observed with the SGK1 siRNAs. This might be due to residual expression of SGK1 observed in our knockdown experiments. Given its function as an enzyme, even a relatively low level of expression of the protein could have dramatic effects on downstream targets. Alternatively, the effectiveness of GSK650394 as a growth inhibitor might also be due to nonspecific inhibition of other kinases, particularly other members of the SGK protein family, SGK2 and SGK3. Interestingly, although previous reports have suggested that SGK3 expression is constitutive and is only regulated at the protein level, we have demonstrated that SGK3 expression levels are also induced in response to androgen treatment in all the AR-positive prostate cancer cell lines that were tested (36; data not shown). Notably, the upregulation of SGK3 mRNA levels (approximately 3-fold induction) is much less dramatic than the stimulation of SGK1 expression (approximately 20-fold induction). SGK2 expression was not affected by androgens in any of the cell lines tested. While SGK1 and SGK3 have been demonstrated to phosphorylate many of the same target proteins in vitro, differences in subcellular localization suggest that these proteins regulate distinct signaling pathways (36). SGK3 is localized to endosomes where it has been suggested to play a role in endocytosis (37), unlike the defined role in growth and survival ascribed to SGK1. Therefore, although GSK650394 is able to inhibit SGK2 and possibly also inhibit SGK3, the expression patterns and subcellular localizations of these proteins suggest that inhibition of these kinases by the SGK1 antagonist will probably be of little consequence to the growth of prostate cancer cells.
GSK650394 is relatively selective for SGK1 over Akt, the most closely related AGC kinase family member, as well as a host of other related kinases indicating it should be a useful tool to dissect the relative roles of Akt and SGK1 in PI3K-regulated mechanisms. SGK1 and Akt share many of the same phosphorylation targets and Akt has been implicated in the control of cell growth and survival downstream of PI3K activation. It is unclear whether Akt activity is required for prostate cancer growth and/or survival. Transgenic mice expressing a prostate-restricted, constitutively active Akt develop prostate intraepithelial neoplasia (PIN), but never develop prostate cancer (38). Although activation of Akt by androgens via a nongenomic mechanism has been described, this phenomenon has never been reported to occur in LNCaP cells, which exhibit constitutive PI3K and Akt activation due to their PTEN-null status (31, 39, 40). Xu and colleagues were unable to observe changes in the activation state of Akt following androgen treatment of LNCaP cells (7). Furthermore, in preliminary studies performed in our laboratory, we have not observed any changes in the phosphorylation state of serine 473 of Akt in response to androgen treatment (data not shown). This suggests that, at least in these cells and possibly in all prostate cancer cells exhibiting constitutive PI3K activity, androgen-stimulated proliferation may not depend upon Akt activity, but rather, on SGK1. Therefore, the SGK1 inhibitor GSK650394 might be especially useful for the treatment of those prostate cancer tumors that have developed mutations in PTEN or PI3K, leading to constitutive activation of downstream signaling pathways.
The identification and characterization of the novel SGK1 antagonist GSK650394 can serve as a model compound to aid in the identification of other small molecule inhibitors with similar activity and ideal pharmacokinetic characteristics to be used in clinical trials. To date, GSK650394 has been formulated for in vivo use and can reach exposure levels in rats above the SCC IC50 using a 50 mg/kg BID dosing schedule. Importantly, preliminary results monitoring blood pressure and urinary metabolites as pharmacodynamic markers suggest GSK650394 is efficacious in rat models of hypertension. However, at present we have not tested GSK650394 in any animal models of prostate cancer. At a minimum, GSK650394 is a useful pharmacological tool to further identify pathways regulated by SGK1 which may permit development of more effective prostate cancer treatments. The clinical success of lapatinib for advanced/metastatic breast cancer and imantinib for chronic myelogenous leukemia and gastrointestinal tumors suggests, by analogy, that kinase activities can be selectively targeted and modulated for the treatment of prostate cancer or hormone-refractory prostate cancer.
Supplementary Material
Acknowledgments
We thank Charlene Wu (GlaxoSmithKline Pharmaceuticals) and Kendra Hightower (GlaxoSmithKline Pharmaceuticals) for technical assistance and Dr. Sir Philip Cohen (University of Dundee, Scotland, UK) for advice. We also thank Drs. Olivier Staub (University of Lausanne, Lausanne, Switzerland) and Dean Edwards (Baylor College of Medicine, Houston, TX) for antibodies, and Dr. Charles Sawyers (Memorial Sloan-Kettering Cancer Center, New York, NY) for the LAPC4 cells.
GRANT SUPPORT: Supported by DOD grant DAMD17-03-1-0569 (A. B. Sherk) andNIH grants F32 DK072794 (D. E. Frigo) and R01 DK065251 (D. P. McDonnell).
References
- 1.Cancer Facts and Figures. American Cancer Society; 2007. [Google Scholar]
- 2.Bolla M, Collette L, Blank L, et al. Long-term results with immediate androgen suppression and external irradiation in patients with locally advanced prostate cancer (an EORTC study): a phase III randomised trial. Lancet. 2002;360:103–6. doi: 10.1016/s0140-6736(02)09408-4. [DOI] [PubMed] [Google Scholar]
- 3.Lawton CA, Winter K, Murray K, et al. Updated results of the phase III Radiation Therapy Oncology Group (RTOG) trial 85-31 evaluating the potential benefit of androgen suppression following standard radiation therapy for unfavorable prognosis carcinoma of the prostate. Int J Radiat Oncol Biol Phys. 2001;49:937–46. doi: 10.1016/s0360-3016(00)01516-9. [DOI] [PubMed] [Google Scholar]
- 4.Holzbeierlein J, Lal P, LaTulippe E, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–27. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kazmin D, Prytkova T, Cook CE, et al. Linking ligand-induced alterations in androgen receptor structure to differential gene expression: a first step in the rational design of selective androgen receptor modulators. Mol Endocrinol. 2006;20:1201–17. doi: 10.1210/me.2005-0309. [DOI] [PubMed] [Google Scholar]
- 6.Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B. Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ. 2007;14:2085–94. doi: 10.1038/sj.cdd.4402227. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–92. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 8.Alliston TN, Maiyar AC, Buse P, Firestone GL, Richards JS. Follicle stimulating hormone-regulated expression of serum/glucocorticoid-inducible kinase in rat ovarian granulosa cells: a functional role for the Sp1 family in promoter activity. Mol Endocrinol. 1997;11:1934–49. doi: 10.1210/mend.11.13.0033. [DOI] [PubMed] [Google Scholar]
- 9.Bell LM, Leong ML, Kim B, et al. Hyperosmotic stress stimulates promoter activity and regulates cellular utilization of the serum- and glucocorticoid-inducible protein kinase (Sgk) by a p38 MAPK-dependent pathway. J Biol Chem. 2000;275:25262–72. doi: 10.1074/jbc.M002076200. [DOI] [PubMed] [Google Scholar]
- 10.Hollister RD, Page KJ, Hyman BT. Distribution of the messenger RNA for the extracellularly regulated kinases 1, 2 and 3 in rat brain: effects of excitotoxic hippocampal lesions. Neuroscience. 1997;79:1111–9. doi: 10.1016/s0306-4522(97)00014-6. [DOI] [PubMed] [Google Scholar]
- 11.Imaizumi K, Tsuda M, Wanaka A, Tohyama M, Takagi T. Differential expression of sgk mRNA, a member of the Ser/Thr protein kinase gene family, in rat brain after CNS injury. Brain Res Mol Brain Res. 1994;26:189–96. doi: 10.1016/0169-328x(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 12.Leong ML, Maiyar AC, Kim B, O’Keeffe BA, Firestone GL. Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003;278:5871–82. doi: 10.1074/jbc.M211649200. [DOI] [PubMed] [Google Scholar]
- 13.Maiyar AC, Phu PT, Huang AJ, Firestone GL. Repression of glucocorticoid receptor transactivation and DNA binding of a glucocorticoid response element within the serum/glucocorticoid-inducible protein kinase (sgk) gene promoter by the p53 tumor suppressor protein. Mol Endocrinol. 1997;11:312–29. doi: 10.1210/mend.11.3.9893. [DOI] [PubMed] [Google Scholar]
- 14.Waldegger S, Barth P, Raber G, Lang F. Cloning and characterization of a putative human serine/threonine protein kinase transcriptionally modified during anisotonic and isotonic alterations of cell volume. Proc Natl Acad Sci U S A. 1997;94:4440–5. doi: 10.1073/pnas.94.9.4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993;13:2031–40. doi: 10.1128/mcb.13.4.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. Embo J. 2001;20:4380–90. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. Embo J. 2003;22:4202–11. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frodin M, Antal TL, Dummler BA, et al. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. Embo J. 2002;21:5396–407. doi: 10.1093/emboj/cdf551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J. 1999;339(Pt 2):319–28. [PMC free article] [PubMed] [Google Scholar]
- 20.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. Embo J. 1999;18:3024–33. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465–74. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 22.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–83. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 23.Ma X, Ziel-van der Made AC, Autar B, et al. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 2005;65:5730–9. doi: 10.1158/0008-5472.CAN-04-4519. [DOI] [PubMed] [Google Scholar]
- 24.Aoyama T, Matsui T, Novikov M, Park J, Hemmings B, Rosenzweig A. Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation. 2005;111:1652–9. doi: 10.1161/01.CIR.0000160352.58142.06. [DOI] [PubMed] [Google Scholar]
- 25.Schoenebeck B, Bader V, Zhu XR, Schmitz B, Lubbert H, Stichel CC. Sgk1, a cell survival response in neurodegenerative diseases. Mol Cell Neurosci. 2005;30:249–64. doi: 10.1016/j.mcn.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 26.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757–64. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Cui R, Cheng X, Du J. Antiapoptotic effect of serum and glucocorticoid-inducible protein kinase is mediated by novel mechanism activating I{kappa}B kinase. Cancer Res. 2005;65:457–64. [PubMed] [Google Scholar]
- 28.Frigo DE, McDonnell DP. Differential effects of prostate cancer therapeutics on neuroendocrine transdifferentiation. Mol Cancer Ther. 2008;7:659–69. doi: 10.1158/1535-7163.MCT-07-0480. [DOI] [PubMed] [Google Scholar]
- 29.Wolf DA, Schulz P, Fittler F. Transcriptional regulation of prostate kallikrein-like genes by androgen. Mol Endocrinol. 1992;6:753–62. doi: 10.1210/mend.6.5.1376410. [DOI] [PubMed] [Google Scholar]
- 30.Nakhoul NL, Hering-Smith KS, Gambala CT, Hamm LL. Regulation of sodium transport in M-1 cells. Am J Physiol. 1998;275:F998–F1007. doi: 10.1152/ajprenal.1998.275.6.F998. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 32.Flores SY, Loffing-Cueni D, Kamynina E, et al. Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J Am Soc Nephrol. 2005;16:2279–87. doi: 10.1681/ASN.2004100828. [DOI] [PubMed] [Google Scholar]
- 33.Busjahn A, Aydin A, Uhlmann R, et al. Serum- and glucocorticoid-regulated kinase (SGK1) gene and blood pressure. Hypertension. 2002;40:256–60. doi: 10.1161/01.hyp.0000030153.19366.26. [DOI] [PubMed] [Google Scholar]
- 34.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–56. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 35.Avruch J, Hara K, Lin Y, et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25:6361–72. doi: 10.1038/sj.onc.1209882. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344(Pt 1):189–97. [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J, Liu D, Gill G, Songyang Z. Regulation of cytokine-independent survival kinase (CISK) by the Phox homology domain and phosphoinositides. J Cell Biol. 2001;154:699–705. doi: 10.1083/jcb.200105089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majumder PK, Yeh JJ, George DJ, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci U S A. 2003;100:7841–6. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baron S, Manin M, Beaudoin C, et al. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J Biol Chem. 2004;279:14579–86. doi: 10.1074/jbc.M306143200. [DOI] [PubMed] [Google Scholar]
- 40.Sun M, Yang L, Feldman RI, et al. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278:42992–3000. doi: 10.1074/jbc.M306295200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.