

Abstract
The 4 mammalian arrestins serve as almost universal regulators of the largest known family of signaling proteins, G-protein-coupled receptors (GPCRs). Arrestins terminate receptor interactions with G proteins, redirect the signaling to a variety of alternative pathways, and orchestrate receptor internalization and subsequent intracellular trafficking. The elucidation of the structural basis and fine molecular mechanisms of the arrestin–receptor interaction paved the way to the targeted manipulation of this interaction from both sides to produce very stable or extremely transient complexes that helped to understand the regulation of many biologically important processes initiated by active GPCRs. The elucidation of the structural basis of arrestin interactions with numerous non-receptor-binding partners is long overdue. It will allow the construction of fully functional arrestins in which the ability to interact with individual partners is specifically disrupted or enhanced by targeted mutagenesis. These “custom-designed” arrestin mutants will be valuable tools in defining the role of various interactions in the intricate interplay of multiple signaling pathways in the living cell. The identification of arrestin-binding sites for various signaling molecules will also set the stage for designing molecular tools for therapeutic intervention that may prove useful in numerous disorders associated with congenital or acquired disregulation of GPCR signaling.
Keywords: Arrestin, G-protein-coupled receptors, Desensitization, Structure, Conformational change
1. Introduction
In the animal kingdom from Caenorhabditis elegans to humans, 3–4% of the genes encode various members of the largest and most diverse family of signaling proteins, G-protein-coupled receptors (GPCRs). The growing realization over the last 20 years of the amazing conservation of the core 7 transmembrane domain structure of these receptors, their signaling via heterotrimeric G proteins, and the regulation of their signaling and trafficking by G-protein-coupled receptor kinases (GRKs) and arrestins led to the formulation of the “classic” model of these processes (Fig. 1) (reviewed in Carman & Benovic, 1998; Claing et al., 2002; Marchese et al., 2003). In a nutshell, the model posits that the same active receptor conformation that preferentially interacts with G proteins is specifically phosphorylated by GRKs. Arrestin binds the active phosphoreceptor and shields its cytoplasmic surface, thereby precluding further G protein activation (desensitization). Receptor-bound arrestin also serves as an adaptor linking receptors to the internalization machinery of the coated pit, promoting receptor endocytosis. The internalized receptor can then be recycled back to the plasma membrane (resensitization) or directed to lysosomes and destroyed (down-regulation). Although it is not clear how the cell decides the fate of the internalized receptor, overall the model is beautiful and logical. The accumulating mechanistic evidence fit this model perfectly. The major activation-dependent conformational change in the receptor was demonstrated in a series of elegant experiments (reviewed in Hubbell et al., 2003). GRKs were found to specifically phosphorylate active GPCRs simply because the active receptor itself activates the kinase (Palczewski et al., 1991a). An ingenious mechanism involving activation and phosphorylation sensors in arrestin, and its transition into its active receptor-binding conformation when both sensors are engaged simultaneously, was found to ensure arrestin selectivity for the active phosphorylated receptor (Gurevich & Benovic, 1993). The arrestin sensor for receptor-attached phosphates was first identified by mutagenesis (Gurevich & Benovic, 1995) and then nicely confirmed by the crystal structure (Hirsch et al., 1999; Vishnivetskiy et al., 1999). A simple competition between G protein and arrestin was shown to underlie receptor desensitization (Krupnick et al., 1997b). The absence of arrestin (Xu et al., 1997a), receptor kinase (Chen et al., 1999), or receptor sites for GRK phosphorylation (Mendez et al., 2000) produced essentially the same expected phenotype: a severe deficit in receptor desensitization. Finally, direct interaction of receptor-bound arrestin with clathrin (Goodman et al., 1996) and clathrin adaptor complex AP-2 (Laporte et al., 1999) logically explained arrestin’s role in receptor endocytosis, and receptor sequestration was found to be deficient in cells lacking non-visual arrestins (Kohout et al., 2001).
Fig. 1.

The “classical” model of arrestin-mediated GPCR desensitization. The agonist-activated receptor activates cognate heterotrimeric G proteins that subsequently stimulate various signaling cascades increasing the activity of protein kinases PKA, PKC, etc. Active receptor is specifically phosphorylated by GRKs. Arrestin binds the active phosphoreceptor with high affinity, precluding further G protein activation. Arrestin serves as an adaptor linking the receptor to the internalization machinery of the coated pit (clathrin, adaptor complex AP-2), facilitating receptor internalization. Low pH in the endosome promotes agonist dissociation, which facilitates the release of arrestin, whereupon the receptor can be dephosphorylated and recycled back to the plasma membrane (resensitization). Alternatively, the receptor can be transported to lysosomes and destroyed (down-regulation).
However, the great majority of these experiments were performed with just two model GPCRs, rhodopsin and the β2-adrenergic receptor (b2AR). Although most GPCRs studied are phosphorylated and interact with arrestins, with other receptors things do not seem so simple and straightforward. Arrestins were shown to bind a number of unphosphorylated receptors (Mukherjee et al., 1999a; Min & Ascoli, 2000; Min et al., 2002; Mukherjee et al., 2002; Galliera et al., 2004; Jala et al., 2005). Every imaginable mechanism of internalization of different GPCRs has been described: arrestin- and clathrin-dependent; arrestin- and clathrin-independent; arrestin-independent and clathrin-dependent; as well as the most puzzling arrestin-dependent dynamin- and clathrin-independent (reviewed in Marchese et al., 2003; Prossnitz, 2004). In some cases the same receptor apparently uses different internalization pathways under different circumstances (Pals-Rylaarsdam et al., 1997; Lee et al., 2000). Arrestin was found to be necessary for desensitization but not for the internalization of some receptors (Pals-Rylaarsdam et al., 1997), and even the active receptor conformation recognized by G proteins and GRKs/arrestins was reported to be different in some cases (Qian et al., 2001; Vilardaga et al., 2001; Whistler et al., 2002b; Kohout et al., 2004; Hunton et al., 2005; Ponimaskin et al., 2005). Thus, one is left wondering whether these data can be reconciled with the “central dogma” of GRK and arrestin function in GPCR regulation.
Here we attempt to understand the mechanistic basis of these disparate results and provide the conceptual framework for their interpretation based on the available structural and functional information about receptors and arrestins. Because more than 40 review articles covering various aspects of arrestin function have been published since 2000, to avoid unnecessary repetition we did not attempt to make this review comprehensive. We also focus on vertebrate arrestins due to the relative paucity of the structural data on invertebrate homologues.
2. The molecular mechanism of the arrestin–receptor interaction
The first (and undisputed) arrestin function in life is to stop (arrest) receptor signaling via G proteins. This is how the first member of the family, visual arrestin, was discovered (Kuhn, 1978; Kuhn et al., 1984; Pfister et al., 1985) and how it got its name. The fact that rhodopsin activation and phosphorylation both enhance the binding of arrestin (known at the time as 48 kDa protein) was established before the functional role of this phenomenon was understood (Kuhn, 1978; Kuhn et al., 1984). The search for non-visual arrestin homologues begun after the unexpected discovery that the apparent ability of β-adrenergic receptor kinase (now known as GRK2) to reduce the coupling of purified β2-adrenergic receptor (b2AR) to G protein is greatly diminished by progressive kinase purification (Benovic et al., 1987). Because visual arrestin partially restored b2AR desensitization by purified kinase in reconstituted system, it became clear that there must be arrestin analogues that work with non-visual GPCRs (Benovic et al., 1987). Soon after the cloning of rod arrestin (Shinohara et al., 1987), further efforts yielded 2 ubiquitously expressed non-visual arrestins (each having at least 2 splice variants) that desensitize b2AR more effectively than visual (Lohse et al., 1990; Attramadal et al., 1992; Sterne-Marr et al., 1993).
Obviously, in any GPCR-driven signaling system, the exact timing and affinity of arrestin binding determine the magnitude of the signal that the active receptor generates. Most GPCRs are “reusable”, hence arrestin must dissociate at some point to allow receptor recycling back into the active pool. Thus, the rate of arrestin release regulates the reuse of the receptor, making the timing of this event equally important. Because arrestin has to dissociate before the receptor-attached phosphates are removed (Palczewski et al., 1989), arrestin binding cannot be regulated solely by the phosphorylation status of the receptor.
2.1. Arrestin activation: a multi-step process
The molecular mechanism that governs the arrestin–receptor interaction was first elucidated on the visual arrestin–rhodopsin model (Gurevich & Benovic, 1993). Visual arrestin binds phosphorylated light-activated rhodopsin (P-Rh*) with remarkable selectivity: its binding to an equal amount of dark (inactive) phosphorhodopsin (P-Rh) or active unphosphorylated rhodopsin (light-activated rhodopsin; Rh*) is 10–20 times lower, whereas its binding to inactive unphosphorylated rhodopsin [dark (inactive) rhodopsin; Rh] is barely detectable (Fig. 2). Thus, receptor activation or phosphorylation alone promotes relatively weak arrestin interaction. However, arrestin binding to P-Rh* is many times greater than the sum of dark P-Rh and Rh* levels, suggesting that the binding mechanism is more sophisticated than a simple cooperative 2-site interaction. Two other observations strongly support this idea. First, arrestin was found to undergo a significant conformational change in the process of its binding, as evidenced by an unusually high activation energy (Schleicher et al., 1989). Second, arrestin binding to dark P-Rh and to Rh* is very sensitive to high salt inhibition, indicating that it is mediated primarily by ionic interactions, whereas its binding to P-Rh* is actually enhanced by 150–200 mM NaCl and quite resistant to very high salt concentrations, suggesting the involvement of hydrophobic interactions that do not participate in arrestin binding to either P-Rh or Rh* (Gurevich & Benovic, 1993). The model of sequential multi-site arrestin–receptor interaction (Gurevich & Benovic, 1993), based on these and other data, explains arrestin selectivity (Fig. 2). This model posits that arrestin has 2 “sensor” sites: an “activation sensor” that binds receptor elements that change conformation upon activation, and a “phosphate sensor” that binds receptor-attached phosphates. These sensors by themselves mediate low affinity binding to Rh* and inactive P-Rh, respectively. The sensors allow arrestin to “probe” the functional state of the receptor molecule it encounters, and rapidly dissociate after the “exploration” of inactive Rh, P-Rh, or unphosphorylated Rh*. When the receptor is phosphorylated and active at the same time, both sensors bind. Simultaneous engagement of the 2 sensor sites allows arrestin transition into the active high-affinity receptor-binding state. The transition involves a major conformational change in arrestin, bringing into play additional receptor-binding sites that ensure tight semi-irreversible interaction. In this model receptor deactivation serves as a signal for arrestin to get off, whereupon the receptor can be dephosphorylated and reused.
Fig. 2.

The sequential multisite binding mechanism ensures high arrestin selectivity for the phosphorylated active receptor. (A) Direct arrestin binding to the 4 functional forms of rhodopsin (inactive phosphorylated, P-Rh; light-activated phosphorylated, P-Rh*; inactive, Rh; light-activated, Rh*). P-Rh* is the preferred arrestin target. (B) Model of the arrestin–receptor interaction. First, arrestin binds via its activation sensor to receptor elements that change conformation upon activation or via the phosphate sensor to receptor-attached phosphates, respectively. If the receptor is active and phosphorylated, simultaneous engagement of both sensors promotes arrestin transition into the active state with concomitant engagement of additional binding sites, stabilizing the arrestin–receptor complex. Eventual loss of the active receptor conformation reverses this sequence of events and induces arrestin dissociation (adapted from Gurevich & Benovic, 1993).
Several lines of evidence suggest that in its basal state arrestin has “repressed” binding ability that the receptor must “unleash”. One comes from deletion mutagenesis. The elimination of the last 37 residues yields arrestin(1–367) with virtually normal affinity for P-Rh* and dramatically reduced selectivity: it demonstrates comparable binding to P-Rh*, inactive P-Rh, and Rh* (but very little binding to inactive Rh, indicating that the other interactions are quite specific) (Gurevich & Benovic, 1992) (Fig. 3). The binding of this mutant to all functional forms of rhodopsin is similarly resistant to high salt inhibition, suggesting that truncated arrestin always uses its additional hydrophobic binding site that wild-type (WT) arrestin only engages for P-Rh* binding (Gurevich & Benovic, 1993). This phenotype suggests that the arrestin C-terminus is a regulatory region that does not directly participate in rhodopsin binding but suppresses high-affinity interactions with the “wrong” targets, phosphorylated inactive and active unphosphorylated rhodopsin. Interestingly, a structurally and functionally similar splice variant of visual arrestin, p44, was found a few years later in some species (Smith et al., 1994). Arrestin(1–191), lacking the whole C-terminal half of the molecule, is another very instructive deletion mutant. This “mini-arrestin” also binds best to the proper target, but its binding to P-Rh* is virtually equal to the sum of its binding to inactive P-Rh and Rh* (Fig. 3), demonstrating a typical cooperative 2-site interaction. Importantly, its binding to all forms of rhodopsin is as sensitive to salt inhibition as the binding of WT arrestin to inactive P-Rh and Rh* (Gurevich & Benovic, 1993). Thus, the additional binding site that full-length arrestin uses only for P-Rh* interaction is localized in the C-terminal half of the molecule, and the complex mechanism ensuring arrestin selectivity for P-Rh* involves the mobilization of this site.
Fig. 3.

The function of the arrestin C-domain and C-tail. The deletion of the arrestin C-tail yields truncated arrestin(1–367) that binds P-Rh* essentially as well as full-length (WT) arrestin. This deletion dramatically decreases arrestin selectivity for P-Rh*, enhancing the binding to dark P-Rh and unpho-sphorylated Rh*, suggesting that the C-tail is a regulatory element “suppressing” the interactions with non-preferred forms of rhodopsin. The deletion of the whole C-terminal half of the molecule yields arrestin(1–191). This “mini-arrestin” demonstrates essentially the same binding to dark P-Rh and unphosphorylated Rh* as WT arrestin, but its binding to P-Rh* is many times lower. In the case of this mutant (in sharp contrast to WT arrestin), P-Rh* binding roughly equals the sum of the binding to dark P-Rh and Rh*. Thus, arrestin(1–191) does not have an additional binding site that can be mobilized to ensure arrestin selectivity (Gurevich & Benovic, 1992).
2.2. The identification of the arrestin phosphate sensor
The testable prediction of the sequential multisite interaction model is that arrestin has two “buttons” which the receptor must “push” simultaneously to activate it and induce high-affinity binding. According to the model, one “button” has to be pushed by receptor-attached phosphates, the other by the part of the GPCR that changes conformation as a result of receptor activation. Two other classes of receptor-binding partners, G proteins and GRKs, also preferentially interact with the active receptor conformation, making it likely that all 3 classes of proteins use essentially the same structural clues to discriminate between active and inactive receptor (R). The obvious corollary of the model is that these “buttons” on arrestin can be constitutively “pushed” by appropriately targeted mutations, yielding arrestin proteins that would not require either receptor phosphorylation or activation. The mutants with the phosphate sensor turned “on” permanently would bind any active receptor, phosphorylated or not, whereas arrestins with the activation sensor turned on by mutagenesis would bind phosphoreceptor regardless of its activation. Thus, the most direct way to test the model experimentally is to identify these sensors in the arrestin molecule, figure out how they work, and turn them on by the appropriate mutations.
In most GPCRs, GRK phosphorylation sites come in clusters (3–4 serines or threonines within 5–6 residues), and it has been established in several models that multi-phosphor-ylation of the receptor is necessary for arrestin binding (Gurevich & Benovic, 1993; Gurevich et al., 1995; Mendez et al., 2000). Thus, even before the arrestin crystal structure was solved, it was pretty clear that the arrestin phosphate sensor is likely to contain a cluster of 3 or more positive charges within a relatively short sequence. One would also expect the relevant charges to be conserved in the arrestin family and localized in the N-terminal half of the molecule (Gurevich & Benovic, 1993). This identifies the stretch of residues 166–176 as the “prime suspect” (residue numbers here and below are given for bovine visual arrestin unless otherwise specified) (Fig. 4A). Neutralization of several positive charges in this element (K167S, R171Q, and K176S) yields mutants with reduced binding to P-Rh* and inactive P-Rh, and normal binding to Rh*, consistent with direct participation of these residues in phosphate interaction (Fig. 4B and C) (Gurevich & Benovic, 1995). However, the mutant that turned out to be the most interesting in this series, R175N, binds inactive P-Rh and P-Rh* better than WT arrestin. Most importantly, this mutant binds Rh* essentially as well as WT arrestin binds P-Rh* (Fig. 4B and C). In the context of the model, this phosphorylation-independent binding suggests that Arg175 is the phosphate sensor, and that the neutralization of its charge by mutagenesis turns it “on”. There seemed to be one inconsistency, though: the model posits that the sensor directly binds phosphates, hence one would expect a decrease (rather than the observed increase) of the mutant binding to dark P-Rh and P-Rh*. However, in the context of full-length arrestin, the interpretation of the effect of a mutation of this type is far from straightforward: the expected reduction of the direct binding of this site to the receptor-attached phosphates can be counterbalanced by its “activating” effect. Luckily, the impact of any mutation on phosphate binding per se can be determined directly in the context of arrestin(1–191), which does not have any sophisticated regulatory mechanisms. In this context, the R175N mutation behaves just like the neutralization of the other phosphate-binding positive charges; it decreases P-Rh* binding without affecting Rh* interaction (Fig. 4D and E).
Fig. 4.

Identification of the main phosphate-binding element of visual arrestin. (A) The N-domain element 161–179 contains a cluster of positively charged residues that are conserved in arrestin family (shown in bold). (B) The neutralization of several of these charges reduces the binding to P-Rh*, identifying these residues as phosphate-binding elements. In contrast, the R175N mutation enhances the binding to P-Rh* and dramatically increases the binding to unphosphorylated Rh* (C). (D) The same mutation in the context of the “mini-arrestin” (1–191) reduces P-Rh* binding without affecting arrestin interaction with Rh* (E). These data identify K166, R171, R175, and K176 as phosphate-binding elements. R175 also serves as the main “phosphate sensor”. Apparently, the neutralization of its charge by receptor-attached phosphates (or by the R175N mutation) is necessary to make the high-affinity arrestin binding possible (Gurevich & Benovic, 1995).
Thus, Arg175 fits the description of the phosphate sensor: it binds phosphates, and its neutralization by mutation makes arrestin phosphorylation-independent. Actually, charge reversal in this position (R175E) is even more effective than neutralization in promoting arrestin binding to Rh*, whereas the conservative R175K mutation that preserves the charge also preserves arrestin selectivity for P-Rh* (Gurevich & Benovic, 1997). Based on these data, we proposed the simplest conceivable mechanism of the function of the phosphate sensor (Gurevich & Benovic, 1995). In the basal (inactive) state, Arg175 interacts with a negatively charged residue within the arrestin molecule itself. The binding of receptor-attached phosphate neutralizes its charge (like the mutation), breaking this intra-molecular interaction. The disruption of this salt bridge lets the rest of the arrestin molecule “know” that the phosphate is in place. When the salt bridge is pre-disrupted by an appropriate mutation, the phosphates’ “job” is already done, hence the mutant does not need receptor-attached phosphates for high-affinity binding. Obviously, this hypothesis calls for the identification of the intra-molecular interaction partner of Arg175. The arrestin crystal structure revealed that Arg175 is right in the middle of the “polar core”, an unusual (for a soluble protein) arrangement of 5 shielded, essentially solvent-excluded, interacting charged residues in the center of the arrestin molecule (Hirsch et al., 1999) (Fig. 5). Exhaustive mutagenesis of all 5 residues corroborated the salt bridge idea and identified Asp296 as the most important negatively charged partner of Arg175 (Vishnivetskiy et al., 1999). Breaking this salt bridge by charge reversal mutations on either side (R175E or D296R) yields phosphorylation-independent arrestin species, whereas its reconstruction by the combination of these mutations (R175E+D296R) restores arrestin selectivity for P-Rh* (Fig. 5). These data clearly demonstrate that this salt bridge is the phosphate sensor, and that receptor-attached phosphates simply need to break it to make possible arrestin transition into the high-affinity-binding state.
Fig. 5.

Polar core is the phosphate sensor in arrestin. (A) Five solvent-excluded interacting charged residues are localized at the center of the arrestin molecule (hence the term “polar core”). These include Asp30 and Arg175 of the N-domain, Asp296 and Asp303 of the C-domain, and Arg382 of the C-tail. (B) The disruption of the salt bridge between Arg175 and Asp296 from either side by R175E or D296R charge reversal mutations dramatically increases arrestin binding to Rh*. Simultaneous reversal of both charges restores the salt bridge. Functionally the double-reversal mutation restores high arrestin selectivity for P-Rh*. These data identify the Arg175–Asp296 salt bridge as the phosphate sensor and demonstrate that receptor-attached phosphates simply break it by neutralizing the positive charge of Arg175.
To accomplish this, the phosphates must reach Arg175. However, this residue is buried at the very bottom of the N-domain “bowl” that has plenty of other more exposed positive charges (Fig. 6). Judging by a dramatic reduction of P-Rh* and especially inactive P-Rh binding by charge neutralization and reversal mutations (Fig. 6B and C), two of these residues, Lys14 and Lys15, directly interact with phosphates. In fact, the K15E substitution is the most detrimental point mutation for P-Rh* binding ever described in visual arrestin (Vishnivetskiy et al., 2000). Interestingly, in the context of arrestins with the polar core destabilized by other mutations, substitutions of Lys14 and Lys15 do not reduce the binding to P-Rh* much (Fig. 6C), suggesting that the function of these lysines is only obligatory when the polar core is intact and must be destabilized by phosphates. The simplest explanation of these data is that in wild-type arrestin the highly exposed lysines 14 and 15 “meet” the receptor-attached phosphates first, and then “guide” or “deliver” them to the polar core. This ingenious two-step mechanism explains the strict requirement for receptor phosphorylation for high-affinity arrestin binding.
Fig. 6.

How do the phosphates get to the shielded Arg175? (A) In the basal conformation arrestin’s main phosphate sensor Arg175 (highlighted with the atoms shown) in the polar core is shielded, whereas numerous other positively charged residues in the N-domain (highlighted) are highly exposed. (B) Lysines 14 and15 (highlighted with the atoms shown in panel A) in β-strand I interact with receptor-attached phosphates, as evidenced by the progressive decrease of P-Rh* binding with neutralization and reversal of their charges. (C) The K15A mutation dramatically reduces WT arrestin binding to P-Rh*. However, in the context of arrestin mutants in which the polar core is already disrupted (R175E or D296R), the effects of the same K15A mutation on P-Rh* binding are mild (K15A+R175E and K15A+D296R). Importantly, the K15A mutation suppresses arrestin binding to dark P-Rh (which is mediated solely by phosphate interactions) in any context, supporting the identification of Lys15 as one of the residues directly binding phosphates. Thus, the presence of Lys15 is required for arrestin binding to P-Rh* only when the polar core is intact, suggesting that its function is to “meet” the phosphates first and then “guide” them to the polar core (adapted from Vishnivetskiy et al., 2000).
2.3. The molecular mechanism of arrestin activation
So far we only have crystal structures of the basal (inactive) conformation of free arrestins (Hirsch et al., 1999; Han et al., 2001; Sutton et al., 2005). Therefore, the inferences regarding the conformation of active receptor-bound arrestin have to be made based on the conformation of free arrestin and the changes that occur in the process of its binding to the receptor. In its basal conformation arrestin is an elongated two-domain molecule. The relative orientation of the two domains is supported by three groups of intra-molecular interactions (Fig. 7). One of these is a relatively extensive interaction between the bodies of the two domains mediated by numerous hydrophobic residues that so far has not been studied in detail. The second is the polar core (Fig. 5) that apparently must be destabilized to allow high-affinity arrestin binding to the receptor. The third is an interaction between β-strand I and α-helix I in the N-domain, and the arrestin C-tail, which folds back from the C-domain and makes a strong contact with these two elements. This 3-element interaction is mediated by bulky hydrophobic residues (Fig. 8). Its disruption by mutagenesis from any side yields “constitutively active” arrestins with dramatically reduced selectivity: high binding to P-Rh* and significantly enhanced binding to the non-preferred forms of rhodopsin, inactive P-Rh and unphosphorylated Rh* (Fig. 8) (Gurevich, 1998; Vishnivetskiy et al., 2000). These effects are reminiscent of the consequences of mutations destabilizing the polar core, suggesting that the 3-element interaction is also disrupted in the process of arrestin binding. According to the current model, this interaction is also destabilized by receptor-attached phosphates (Vishnivetskiy et al., 2000). Phosphate-binding lysines 14 and 15 (Fig. 6) are localized on the short β-strand I, right next to a group of 3 hydrophobic residues (Val11–Ile12–Phe13) interacting with α-helix I and the C-tail. Because these lysines are two adjacent residues in a β-strand, their side chains point in opposite directions (Fig. 8). To enable both lysines to “meet” the same cluster of phosphorylated receptor residues simultaneously, one of them must flip over. This would likely “melt” the short β-strand I, moving its hydrophobic residues out of a position favorable for interaction with their partners, thereby disrupting the 3-element interaction and releasing both α-helix I and the C-tail. Several pieces of indirect evidence are consistent with this model. The release of the visual arrestin C-tail in response to its binding to P-Rh* or even a poly-anion like heparin has been documented, based on a dramatic increase of arrestin susceptibility to limited proteolysis (Palczewski et al., 1991b; Vishnivetskiy et al., 2002; Raman et al., 2003). α-Helix I participates in receptor binding and trafficking (Han et al., 2001; Dinh et al., 2005), suggesting that it also must get free of its intra-molecular partners.
Fig. 7.

Three sets of intramolecular interactions hold arrestin in its basal (inactive) conformation. Side chains of participating residues are shown in CPK: (1) TE, the residues participating in the 3-element interaction; (2) polar core; (3) DI, hydrophobic residues participating in the extensive interaction between the bodies of the two domains are shown in lighter (N-domain residues) or darker (C-domain residues) pattern. The inter-domain hinge is also highlighted.
Fig. 8.

The 3-element interaction: another “clasp” holding arrestin in its basal state. (A) The 3-element interaction between β-strand I and α-helix I of the N-domain and β-strand XX of the C-tail involves triplets of bulky hydrophobic residues in each element (Val11+Ile12+Phe13, Leu103+Leu107+Leu111, and Phe375+Val376+Phe377, respectively). Disrupting the 3-element interaction by replacing the hydrophobic residues with alanines in β-strand XX (3A), β-strand I (N3A), or α-helix I (h3A) yields constitutively active mutants, suggesting that it is disrupted in WT arrestin by P-Rh*. Two highly conserved lysines (Lys14 and Lys15) are present immediately downstream of the participating hydrophobic residues in β-strand I. The movements accompanying phosphate binding to Lys15 and Lys14 likely melt the short β-strand I, disrupting the hydrophobic interaction of adjacent residues with the arrestin C-tail and α-helix (adapted from Vishnivetskiy et al., 2000).
Thus, structurally both “hot spots” in the arrestin molecule where mutations facilitate the binding to the preferred and non-preferred forms of the receptor are the intra-molecular interactions that serve as “clasps” holding it in its basal conformation. Notably, both support the relative orientation of the 2 domains in free (inactive) arrestin (Fig. 7). This strongly suggests that in the process of its binding to the receptor arrestin undergoes a global conformational rearrangement, likely involving the movement of the 2 domains relative to each other. As will be discussed in the following sections, this conclusion has profound implications for various aspects of arrestin function, yet so far it is supported only by indirect evidence. The idea that a major conformational change in arrestin accompanies its binding to the receptor was originally proposed on the basis of the unusually high energy barrier of this process (Schleicher et al., 1989). The finding that arrestin sites that do not participate in its binding to inactive P-Rh or Rh* are mobilized for the interaction with P-Rh* lent further support to this model (Gurevich & Benovic, 1993). The data that the C-tail of visual arrestin is released upon receptor binding (Palczewski et al., 1991b; Vishnivetskiy et al., 2002; Raman et al., 2003) and that the overall pattern of arrestin3 proteolysis changes in the presence of heparin or phosphopep-tide mimicking the phosphorylated vasopressin type II receptor C-terminus (Xiao et al., 2004) are also consistent with this notion. However, the absence of the C-tail in visual arrestin splice variant p44 (Smith et al., 1994) reduces the activation energy by 50% but does not completely eliminate the barrier (Pulvermuller et al., 1997), suggesting that there is more to this conformational rearrangement than just the release of the C-tail. Structurally, any global rearrangement of the arrestin molecule must involve domain movement, which would be limited by the length of the inter-domain connector termed the “hinge region” (Fig. 7) (Vishnivetskiy et al., 2002). The hinge has substantial “slack” in all 3 arrestin types with known crystal structure: it is 12 residues long, whereas just 5 residues in a fully extended conformation can cover the distance between the domains (Granzin et al., 1998; Hirsch et al., 1999; Han et al., 2001; Milano et al., 2002; Sutton et al., 2005). In its sequence, the only conserved residues are prolines, glycines, and alanines, indicating the importance of its conformational flexibility rather than anything else. If arrestin–receptor interaction requires domain movement, and if the main function of the extended hinge is to make this movement possible, one can predict that an increase in hinge length or the scrambling of its sequence should not be detrimental for receptor binding, whereas increasing deletions in the hinge region should progressively reduce arrestin’s ability to bind the receptor. The binding should be completely abolished in mutants where the hinge is just long enough to cover the distance between the 2 domains. These predictions were rigorously tested and found to be correct (Vishnivetskiy et al., 2002), adding the most convincing piece of indirect evidence supporting the idea that domain movement is necessary for arrestin binding to the receptor.
Collectively, these data along with the localization of receptor-binding arrestin elements on the concave sides of both domains (Kieselbach et al., 1994; Ohguro et al., 1994; Pulvermuller et al., 2000; Dinculescu et al., 2002; Vishnivetskiy et al., 2004) (Fig. 7) lead to the following model of arrestin binding to the phosphorylated ligand-activated receptor (P-R*) (Gurevich & Gurevich, 2004) (Fig. 9). Arrestin “activation sensor” binds to receptor elements that change conformation upon activation. This binding likely weakens the interaction between the bodies of the 2 domains. Although this interaction has not been rigorously probed, this seems a reasonable hypothesis: the N- and C-domain receptor-binding elements (Vishnivetskiy et al., 2004) meet at the inter-domain contact point (Figs. 8 and 14), and it is the only stabilizing intra-molecular interaction in arrestin that is not affected by the phosphates. Meanwhile, receptor-attached phosphates bind lysines 14 and 15, forcing one of them to flip over, thereby destabilizing the 3-element interaction. The disruption of this interaction releases the arrestin C-tail, removing Arg382 from the polar core in the process. The lysines “guide” the phosphates to the polar core, where they neutralize the charge of Arg175, breaking the Arg175-Asp296 salt bridge. Thus, the polar core “loses” both positive charges. With all 3 interactions that held arrestin in its basal state now disrupted, the mutual repulsion of the 3 remaining negative charges in the polar core (Asp30, Asp296, and Asp303) helps to drive the movement of the 2 arrestin domains. With their concave sides facing the receptor, the 2 domains likely grip its cytoplasmic tip (Fig. 9). This multi-step mechanism ensures high arrestin preference for P-R* over other functional forms of the receptor and makes the binding semi-irreversible. Bound arrestin apparently shields receptor-attached phosphates because arrestin dissociation must precede receptor dephosphorylation (Palczewski et al., 1989). That leaves receptor deactivation due to the loss of bound agonist the only conceivable signal for arrestin release. This makes perfect sense biologically, because to ensure high fidelity of regulation arrestin is supposed to stay bound and preclude receptor interactions with G proteins as long as it takes to fully deactivate the receptor.
Fig. 9.

Current model of arrestin–receptor interaction. Receptor-attached phosphates (blue circles with yellow stripe) bind Lys14 and Lys15 (blue), forcing one of them to flip over. The resulting distortion of β-strand I disrupts the 3-element interaction, allowing Lys14 and Lys15 with bound phosphates to move towards phosphate-binding residues in β-strand X (Lys166, Arg171, Arg175, and Lys176; blue). Ultimately the phosphates neutralize the charge on Arg175, thereby breaking its salt bridge with Asp296 and destabilizing the polar core. The breakup of the 3-element interaction also releases the arrestin C-tail (green), removing the remaining positive charge (Arg382; orange) from the polar core. Having lost both interaction partners, the amphipathic α-helix I (magenta) swings out and participates in receptor binding. After the constraints holding the 2 arrestin domains in their basal orientation are released, the N-domain and C-domain move relative to each other. This movement brings all receptor-binding elements into contact with the receptor, so that arrestin encloses its cytoplasmic tip. In the bound form the “patch” of the phosphate-binding residues is in contact with receptor-attached phosphates and an unidentified arrestin elements (possibly one of the flexible loops; red) occupies the inter-helical cavity that opens upon receptor transition into its active state.
Fig. 14.

Receptor-binding elements are localized on the concave sides of both arrestin domains. Upper panel: “side” view of the arrestin molecule. Lower panel: view down the cavities of both domains on the receptor-binding surface. Receptor-binding elements are pattern-coded, as indicated. Element identification is based on truncation (Gurevich & Benovic, 1992, 1993) and site-directed mutagenesis (Gurevich & Benovic, 1995, 1997; Sutton et al., 2005; Vishnivetskiy et al., 2000), chimera construction (Gurevich et al., 1995; Vishnivetskiy et al., 2004), chemical modification and H/D-exchange (Ohguro et al., 1994), peptide inhibition (Pulvermuller et al., 2000), and epitope insertion (Dinculescu et al., 2002).
2.4. Are all arrestins created equal?
The great majority of mechanistic structure–function studies were performed on the visual arrestin–rhodopsin model. Biologically, visual arrestin is unique: it is expressed almost exclusively in rod photoreceptors in the retina and it is the only member of the family designed to bind just one receptor, rhodopsin. Both non-visual arrestins are ubiquitously expressed and apparently interact with hundreds of different GPCRs, and even the other specialized arrestin expressed in cone photoreceptors regulates 2–4 different cone opsins (depending on the species). So it is necessary to test whether arrestins with broad receptor specificity use the same activation mechanism and have the same selectivity for just one functional form of their cognate receptors. It is important to keep in mind that due to very limited availability of purified GPCRs necessary for the direct binding assay, quantitative biochemical studies of other arrestins under strictly controlled experimental conditions were performed on a small set of model receptors: rhodopsin, the β2-adrenergic receptor (b2AR), and the m2 muscarinic cholinergic receptor (m2 mAChR).
Direct studies of the binding of different arrestin proteins to the 4 functional forms of these receptors reveal striking differences in their selectivity (Gurevich et al., 1993b, 1995; Kovoor et al., 1999; Celver et al., 2002; Sutton et al., 2005) (Fig. 10). First, non-visual arrestins do not demonstrate the remarkable preference for active phosphorylated receptor (P-R*) over inactive phosphoreceptor (P-R). Arrestins 2 and 3 show only a 2-fold difference in binding levels at best, as compared to about a 10-fold difference, which is the hallmark of visual arrestin. This appears to be to a large extent an intrinsic characteristic of these arrestins because their binding to inactive and active phosphorhodopsin is also similar (Fig. 10A). Second, the difference between non-visual arrestins binding to active phosphorylated (P-R*) and unphosphorylated (ligand-activated receptor; R*) forms is also less dramatic, no more than 5-fold (Fig. 10B and C).
Fig. 10.

The selectivity of visual arrestin is unrivaled in the family. Comparative binding of visual (rod) arrestin and its 2 non-visual “cousins” (arrestin2 and arrestin3) to the 4 functional forms of rhodopsin (A), m2 muscrarinic cholinergic receptor (B), and β2-adrenergic receptor (C). Visual arrestin binding to the active phosphorylated form (P-Rh*) of its cognate receptor, rhodopsin, is many times greater than its binding to inactive P-Rh or unphosphorylated Rh*. In contrast, the difference in binding of both non-visual arrestins to inactive and active phosphoreceptors is about 2-fold or less. The difference in their binding to phosphorylated and unphosphorylated forms of the receptors is also much less impressive. Visual arrestin also shows much stronger preference for its cognate receptor, rhodopsin, over the m2 mAChR and the b2AR, whereas non-visual arrestins bind all 3 receptors comparably. Both arrestin2 and 3 show a similar selectivity profile for rhodopsin binding, demonstrating that the difference lies in the functional characteristics of the arrestin rather than the receptor (adapted from Gurevich et al., 1995).
Thus, other arrestins show qualitatively the same binding pattern, but their receptor functional form selectivity is nowhere near that of rod arrestin. This raises the question whether all arrestins use the same activation mechanism, and if they do, what is the structural basis of their remarkably different behavior. Thus far, crystal structures of 3 out 4 types of vertebrate arrestin proteins have been solved: rod (Granzin et al., 1998; Hirsch et al., 1999), arrestin2 (Han et al., 2001; Milano et al., 2002), and cone (Sutton et al., 2005). All arrestins show high conservation of the overall fold. In fact, the differences between arrestins from bovine rod and arrestin2 to salamander cone do not exceed the differences between alternative crystal forms of the same protein (Han et al., 2001; Sutton et al., 2005). Moreover, the polar core and the 3-element interaction are invariably present and virtually identically configured, suggesting that the phosphate-sensing mechanism must be shared by all arrestins. Indeed, the introduction of various mutations homologous to known “activating” mutations in visual arrestin into arrestin2, arrestin3, and salamander or human cone arrestin has very similar functional consequences (Gurevich et al., 1997; Kovoor et al., 1999; Smith et al., 2000; Celver et al., 2002; Pan et al., 2003; Sutton et al., 2005) (Fig. 11). First, the binding of these mutants to the unphosphorylated active forms of their cognate receptors dramatically increases, often to the levels that corresponding WT arrestins show with P-R* (Fig. 11B). Second, these mutants become more promiscuous: their binding to the active phosphorylated non-cognate receptors also increases. Interestingly, the binding of these mutants to the active unphosphorylated forms of non-cognate receptors does not increase (Fig. 11), indicating that when the remaining “push” for the transition into the active receptor-binding state must be provided by the receptor via the arrestin “activation sensor”, proper fit is still required. The idea that “activating” mutations in the polar core and 3-element interaction promotes arrestin binding to non-preferred forms of their cognate receptor and to phosphorylated active non-cognate receptors by enhancing the conformational flexibility of the molecule was first proposed based on the studies of visual arrestin–rhodopsin model (Gurevich & Benovic, 1993). The positions of the residues affected by “activating” mutations in subsequently solved crystal structures of visual arrestin and arrestin2 are consistent with this idea. The results of a recent hydrogen/deuterium exchange study of arrestin2 yielded the first direct evidence supporting this hypothesis (Carter et al., 2005). Essentially the same increase in accessibility of several regions implicated in receptor binding was detected in 2 structurally distinct phosphorylation-independent mutant forms of arrestin2, R169E (polar core mutation homologous to R175E in visual) and 3A (triple alanine substitution of the bulky hydrophobic residues that anchor arrestin C-tail) (Carter et al., 2005).
Fig. 11.

Phosphate-dependent activation mechanism is conserved in the arrestin family. Comparative binding of WT and mutant forms of visual, arrestin2, and arrestin3 to phosphorylated and unphosphorylated light-activated rhodopsin (A) and b2AR (B). The mutations destabilizing the polar core and 3-element interaction produce a similar phenotype in all 3 arrestins: enhanced binding to the unphosphorylated active cognate and to the phosphorylated active non-cognate receptor, but not to the unphosphorylated active non-cognate receptor. The mutations in visual, arrestin2 and 3, respectively, are designated, as follows: RE: R175E, R169E, and R170E; 3A, triple alanine substitution in the C-tail: FVF→AAA (375–377), IVF→AAA (386–388), and IVF→AAA (386–388); T, C-terminal truncation yielding 1–378, 1–382, and 1–392 mutants.
Collectively, these data suggest that the phosphate sensing mechanism in all arrestin proteins works similarly, if not identically. Thus, for the structural basis of their functional differences one has to look elsewhere. Several more subtle structural characteristics that distinguish visual and non-visual arrestins have been described. Visual arrestin has an arginine (Arg18) in the loop following β-strand I, which in all arrestins contains 2 phosphate-binding lysines discussed above. Neutralization and reversal of its charge progressively reduce visual arrestin binding to P-Rh*. Most importantly, these mutations dramatically reduce the binding to inactive P-Rh, which is mediated solely by arrestin interactions with receptor-attached phosphates (Fig. 12). This identifies it as a phosphate-binding residue (Sutton et al., 2005). None of the other arrestins has any positive charge in this loop. Interestingly, both non-visual and cone arrestin in various species have at least one proline or glycine residue in this loop that rod arrestins do not have (Fig. 12). Considering that the 2 phosphate-bound lysines in β-strand I need to move in the direction of the polar core to “deliver” the phosphates, extra flexibility of the adjacent loop would likely facilitate arrestin activation, making it less “picky”, whereas the rigidity of this loop in rod arrestin would make it more selective. Thus, an extra phosphate-binding residue in the loop following β-strand I together with the reduced flexibility of this loop may account for the better discrimination by visual arrestin between the functional forms of its receptor target (Sutton et al., 2005).
Fig. 12.
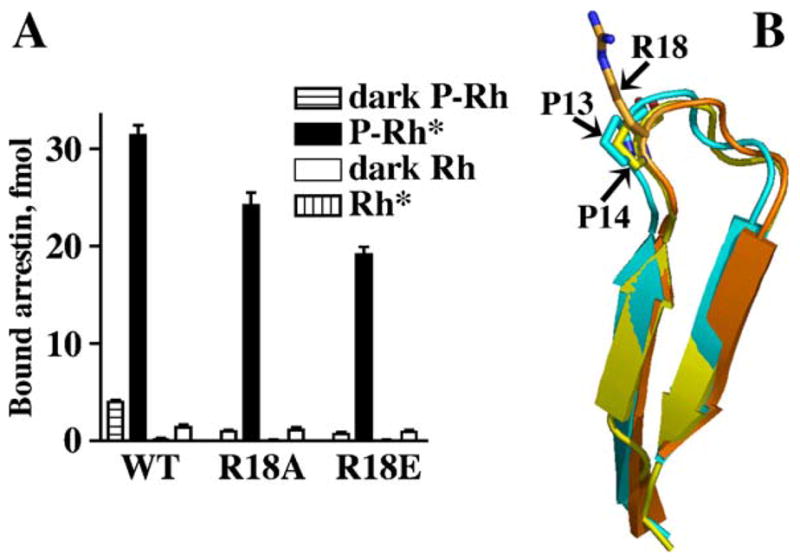
Arg18: an additional phosphate-binding residue in visual arrestin. (A) The neutralization (R18A) and reversal (R18E) of the charge in position 18 progressively reduces the binding of visual arrestin to P-Rh* and dramatically impedes its binding to dark P-Rh, which is mediated solely by phosphate interactions. These data identify Arg18 as yet another phosphate-binding residue. (B) Arg18 in visual arrestin is located in the loop between β-strands I and II, where no other arrestin has a positive charge. Instead, a proline is present in the equivalent position in non-visual arrestin2 (P14) and cone arrestin (P13) (adapted from Sutton et al., 2005). Non-visual and cone arrestins often have additional prolines and/or glycines in this loop. Higher rigidity of this loop and the presence of the additional phosphate-binding residue in visual arrestin likely contribute to its unparalleled selectivity for the active phosphorylated receptor.
Each arrestin domain is a β-strand “sandwich”, in which the two β-strand sheets are “glued” together via hydrophobic interactions between the side chains pointing inside the “sandwich”. In visual arrestin, Val90 is one of these residues, participating in multiple interactions (with Val45, Val57, Val59, and Phe118) (Hirsch et al., 1999). In both non-visual arrestins, this valine is conspicuously absent, being replaced with serine (arrestin2) or alanine (arrestin3). Even though all its potential “partners” are conserved in arrestin2 (Val41, Val53, Val55, and Phe115), the absence of this valine makes the N-domain more flexible. Visual arrestin shows very little binding to the phosphorylated active m2 muscarinic cholinergic receptor (P-m2 mAChR*), whereas arrestin2 binds this receptor well (Fig. 13). Importantly, the V90S mutation in visual arrestin, which “loosens up” its N-domain, dramatically reduces its receptor specificity, enhancing its binding to P-m2 mAChR* virtually to the level of arrestin2, while somewhat reducing binding to its cognate P-Rh* (Han et al., 2001) (Fig. 13). The magnitude of the effect of the mutation of a single residue (the side chain of which is not even exposed) strongly suggests that a relatively rigid N-domain stabilized by the interactions of Val90 with its partners is an important contributor to the high specificity of visual arrestin. The absence of these stabilizing interactions in non-visual arrestins makes their N-domains more flexible, thereby enhancing their ability to “mold” themselves to fit a wide variety of structurally diverse GPCRs. However, the introduction of the valine in the homologous position in arrestin2 (S86V) does not appreciably change its receptor preference (Fig. 13), suggesting that even with this valine its N-domain is still “loose” enough to bind its cognate receptor. Thus, there are likely other structural elements that make rod arrestin more rigid that its non-visual homologues.
Fig. 13.

Enhanced stability of the N-domain β-strand sandwich contributes to the receptor specificity of visual arrestin. (A) The substitution of Val90 with serine in visual arrestin greatly reduces its receptor specificity, allowing the mutant to bind the P-m2 mAChR* almost as well as arestin2. However, the introduction of valine in the homologous position in arrestin2 (S86V) does not appreciably change its binding selectivity, suggesting that there are other structural features in non-visual arrestins contributing to their broad receptor specificity. (B) The interactions between the 2 “layers” of the β-strands in visual arrestin are stabilized by the presence of Val90 (highlighted) that interacts with several hydrophobic partners (Val45, Val57, Val59, and Phe118; highlighted), whereas arrestins 2 and 3 have Ser86 and Ala87, respectively, in the equivalent positions (adapted from Han et al., 2001). Note that arrestin in panel B is shown with its N-domain on the right-hand side (in contrast to Figs. 6, 7, 9, and 14) to show the interactions of Val90 better.
To summarize, despite the remarkable conservation of the overall fold, identifiable structural differences underlie certain functional peculiarities of arrestin subtypes. We are far from a comprehensive understanding of the structural basis for many functional properties of arrestin proteins, but this goal seems achievable. The conservation of the key structural elements and regulatory mechanisms in the arrestin family suggests that, as far as receptor binding is concerned, the differences in their functional properties are still variations on the same theme. Therefore, the mechanistic insights gleaned from the extensive structure–function studies of visual arrestin help us understand the interactions of different arrestins with various receptors.
2.5. What is arrestin looking for in a GPCR?
All arrestins discriminate between active and inactive receptors, albeit the resulting differences in binding levels vary widely. All arrestins also recognize the phosphorylated state of the receptor. Thus, in order to bind the receptor with high affinity, any arrestin needs to find the “active” conformation of certain receptor elements (which must be different in the inactive state) and the part of the receptor that can activate the phosphate sensor.
The arrestin phosphate sensor is specifically designed to respond to at least 2 phosphates (Gurevich & Benovic, 1993; Gurevich et al., 1995; Mendez et al., 2000; Huttenrauch et al., 2002) in close proximity to each other largely independently of the sequence context in which the phosphorylated residues are found (Vishnivetskiy et al., 1999). This type of mechanism explains how just 2 non-visual arrestins in mammals are “primed” for binding to hundreds of different GPCRs. The requirement for a spatially concentrated charge explains why relevant serines and threonines are usually found in clusters, such as STSVSKTETS in rhodopsin (Wilden & Kuhn, 1982), STSVS and TVSTS in the m2 muscarinic cholinergic receptor (Lee et al., 2000), TTIST and TPSS in the substance P receptor, SSS and TSS in the vasopressin-2 receptor, STS in the neurotensin-1 receptor, two SSS clusters in the oxytocin receptor (Oakley et al., 2001), STQTS and TATNST in the N-formyl peptide receptor (FPR; Key et al., 2003), SS and TRTS in the D2 dopamine receptor (Namkung & Sibley, 2004), SSTS in the prostaglandin E2 receptor EP4 (Neuschafer-Rube et al., 2004), or STTT in all splice variants of the 5-HT4 serotonin receptor (Barthet et al., 2005). This mechanism also explains why arrestins do not care which particular kinase phosphorylates the receptor (as long as it adds 2 or more phosphates in close proximity); although in most cases relevant phosphates are added by GRKs (Carman & Benovic, 1998), phosphates introduced by PKC on the D2 dopamine receptor (Namkung & Sibley, 2004) or by casein kinase II on the thyrotropin-releasing hormone receptor (TRHR; Hanyaloglu et al., 2001) work for arrestins just as well.
This design of the phosphate sensor also suggests that the activating element does not necessarily have to contain receptor-attached phosphates: any spatially concentrated negative charge can disrupt the polar core. Indeed, poly-anions such as heparin or phosphopeptides were found to induce the release of the arrestin C-tail (Palczewski et al., 1991b; Gurevich et al., 1994; McDowell et al., 1999; Vishnivetskiy et al., 2002; Xiao et al., 2004), and even stimulate the binding of the most selective member of the family, visual arrestin, to unphosphorylated light-activated rhodopsin (Gurevich et al., 1994; Puig et al., 1995).
Many GPCRs must be phosphorylated to bind arrestins with high affinity. This was first shown directly for rhodopsin (Kuhn et al., 1984), and then demonstrated for the b2AR (Benovic et al., 1987; Lohse et al., 1990; Attramadal et al., 1992; Lohse et al., 1992; Gurevich et al., 1995; Krasel et al., 2005), m2 muscarinic cholinergic receptor (Gurevich et al., 1993a), type 1 angiotensin II receptor (Qian et al., 2001), chemokine receptor CCR5 (Kraft et al., 2001; Huttenrauch et al., 2002), α2-adrenergic receptor (Wang & Limbird, 2002), neuropeptide Y Y1 receptor (Holliday et al., 2005), and a few other GPCRs. However, for a surprising number of receptors phosphorylation is not required for arrestin binding. These include the lutropin receptor (Mukherjee et al., 1999a; Min & Ascoli, 2000; Min et al., 2002; Mukherjee et al., 2002), substance P receptor (Richardson et al., 2003), D6 non-signaling chemokine receptor (Galliera et al., 2004), orexin-1 receptor (Milasta et al., 2005), protease-activated receptor-2 (Stalheim et al., 2005), several splice variants of the serotonin 5-HT4 receptor (Barthet et al., 2005), and leukotriene B4 receptor-1 (Jala et al., 2005). Although in some of these cases phosphorylation enhances the stability of the arrestin–receptor complex (Richardson et al., 2003; Milasta et al., 2005; Stalheim et al., 2005), arrestin binding to unphosphorylated receptors requires an explanation consistent with the known mechanism of arrestin activation. Structure–function studies of several receptors provide interesting clues. It has been established that an acidic region in the C-terminus rather than putative phosphorylation sites in the “scavenger” chemokine receptor D6 (which constitutively internalizes via arrestin-dependent pathway) plays a key role in arrestin binding (Galliera et al., 2004). Apparently, the concentration of negative charges in this region is sufficient to activate the arrestin phosphate sensor and promote the interaction. Another example is the lutropin receptor, where a single residue, aspartic acid 564 in the third cytoplasmic loop is crucial for phosphorylation-independent arrestin binding (Mukherjee et al., 2002). Although it is unlikely that a single negative charge does all the work of arrestin activation (i.e., there are probably other negatively charged residues involved that remain unidentified), the presence or absence of this one residue apparently tips the scales (Mukherjee et al., 2002). Aspartates and glutamates often successfully serve as phosphate mimics (Lin et al., 1997; Lin et al., 2002), hence it is hardly surprising that several spatially close acidic amino acids (that do not necessarily form a contiguous stretch in a linear sequence) can “trip” the arrestin phosphate sensor. Visual arrestin is the only member of the family with which this trick does not work: the substitution of all 7 serines and threonines in the C-terminus with aspartates or glutamates does not yield the functional equivalent of phosphorylated rhodopsin (McDowell et al., 2001), even though the presence of 2–3 phosphates works fine (Gurevich & Benovic, 1993; Mendez et al., 2000). It is likely that the presence of one additional phosphate-binding residue (Arg18) in visual arrestin explains its stricter requirements for the spatial concentration of the negative charge: after all, side chains of acidic residues have a charge of <−1, whereas a serine- or threonine-attached phosphate has a charge of ~−1.5.
2.6. Receptor elements implicated in arrestin binding
Arrestin elements involved in receptor binding have been identified by a variety of methods, including mutagenesis (Gurevich & Benovic, 1992, 1993; Gurevich et al., 1994; Gurevich & Benovic, 1995, 1997; Gurevich, 1998; Vishnivetskiy et al., 1999, 2000), chimera construction (Gurevich et al., 1993a, 1995; Vishnivetskiy et al., 2004), peptide inhibition (Kieselbach et al., 1994; Pulvermuller et al., 2000), differential chemical modification and hydrogen/deuterium exchange (Ohguro et al., 1994), and epitope insertion (Dinculescu et al., 2002). Collectively, these data implicate a substantial portion of the arrestin surface in receptor interaction: most of the concave sides of both domains and a few additional elements (Fig. 14). These include 6 (7 in visual arrestin) phosphate-binding lysines and arginines and quite a few other residues in β-strands I, V, VI, X, XV, XVI, and in several loops (Fig. 14), suggesting that a similarly extensive multi-element receptor surface must participate in arrestin binding.
To activate the phosphate sensor of arrestins, receptors have to be phosphorylated (or contain negatively charged phosphate mimics). The first identified sites of receptor phosphorylation relevant for arrestin binding were localized in the C-terminus of rhodopsin (Sitaramayya & Liebman, 1983). Phosphorylation sites were also found to be localized in the C-terminus of another prototypical GPCR, the b2AR (Dohlman et al., 1987; Bouvier et al., 1988). Moreover, PKA phosphorylation of the 3rd cytoplasmic loop (i3) of the b2AR did not promote arrestin binding (Lohse et al., 1992). These data led to the belief that the GRCR C-terminus is the place of phosphorylation necessary for arrestin interaction, and that only GRKs can do it right. However, as soon as the studies moved beyond these two classic models, it became clear that relevant phosphorylation sites in different receptors could be localized on any cytoplasmic element (Table 1). In many receptors they are in the C-terminus, but a large group of GPCRs carry relevant phosphorylation sites in the 3rd cytoplasmic loop, and in some cases the phosphates promoting arrestin binding are found in the first (i1) or the second (i2) cytoplasmic loops (Table 1).
Table 1.
The localization of the phosphorylation sites relevant for arrestin binding on the cytoplasmic elements of different GPCRs
| Cytoplasmic element | Receptor | References |
|---|---|---|
| C-terminus | Chemokine receptors CCR5 | Huttenrauch et al., 2002; Kraft et al., 2001 |
| Chemokine receptor CCR7 | Kohout et al., 2004 | |
| Angiotensin II receptor type 1A | Qian et al., 2001 | |
| Thyrotropin-releasing | Hanyaloglu et al., 2001 | |
| hormone receptor | ||
| Parathyroid hormone receptor | Vilardaga et al., 2002 | |
| N-formyl peptide receptor | Key et al., 2003 | |
| β1-Adrenergic receptor | Liang et al., 2003a | |
| Substance P receptor | Richardson et al., 2003 | |
| Prostaglandin E2 receptor subtype EP4 | Neuschafer-Rube et al., 2004 | |
| Corticotropin-releasing hormone receptor type 1alpha | Teli et al., 2005 | |
| Orexin-1 receptor | Milasta et al., 2005 | |
| Protease-activated receptor-2 | Stalheim et al., 2005 | |
| Cysteinyl leukotriene type 1 receptor | Naik et al., 2005 | |
| Neuropeptide Y Y1 receptor | Holliday et al., 2005 | |
| Somatostatin receptor 2A m2 muscarinic cholinergic receptor | Tulipano et al., 2004 | |
| i3 (3rd loop) | Lee et al., 2000, Pals-Rylaarsdam et al., 1997 | |
| α2A-adrenoceptor | Wang & Limbird, 2002 | |
| Gonadotropin receptor | Bhaskaran et al., 2003, Kishi et al., 2002 | |
| Dopamine receptor D1 | Kim et al., 2004 | |
| Dopamine receptor D2 | Namkung & Sibley, 2004 | |
| i2 (2nd loop) | mu-opioid receptor | Celver et al., 2001, 2004, Lowe et al., 2002 |
| i1 (1st loop) | Follitropin receptor | Kishi et al., 2002 |
Thus, the phosphates (or negatively charged mimics) that activate the arrestin phosphate sensor could be localized almost anywhere on the intracellular surface of the receptor. Apparently, this is also true for other non-phosphorylated arrestin-binding receptor elements. Several experimental paradigms were used by a number of groups to identify arrestin-binding elements of >40 different GPCRs. The data are often fragmentary and both existing relatively comprehensive data sets were generated by systematic receptor mutagenesis. Dr. Weiss’ group performed alanine-scanning mutagenesis of rhodopsin cytoplasmic loops and identified L72 and N73 in i1 and P142+M143 in i2 as residues crucial for arrestin interaction (Raman et al., 1999). Simultaneous mutation of any 2 of these 3 residues virtually eliminates arrestin binding. These effects were not caused by the phosphorylation deficit of mutant rhodopsin, suggesting direct involvement of these residues in the interaction. One important caveat of this approach is that if a mutation in a particular element reduces rhodopsin phosphorylation (which was observed with several promising i3 mutants), the defects in arrestin binding cannot be unambiguously interpreted (Raman et al., 1999). Using the phosphorylation-independent arrestin, mutant R175E (Gray-Keller et al., 1997) and unphosphorylated rhodopsin circumvented this problem and confirmed the central role of these residues in arrestin binding (Raman et al., 2003). These i1 and i2 residues also proved to be important in inducing arrestin transition into its active state (Raman et al., 2003). Dr. Ascoli’s group took advantage of the fact that rat and human lutropin receptors are highly homologous, yet the human version internalizes 7 times faster than the rat receptor via an arrestin-dependent pathway (Nakamura et al., 2000). This allowed the authors to zero in on the very few amino acids that differ in the 2 receptors and elegantly identify 7 non-contiguous residues that determine arrestin interactions. The substitutions that dramatically improved arrestin binding to the rat lutropin receptor turned out to be VQ→IH in i2; R→K, Q→R, T→M, and P→T in the i3; and L→F in i4 (proximal C-terminus preceding the palmitoylation site). The relatively mild (often conservative) nature of these substitutions suggests that they are unlikely to change the global conformation of the respective receptor domains, increasing the probability that these are indeed the residues that directly interact with arrestin.
In contrast to rhodopsin, many GPCRs have very large cytoplasmic loops and/or C-tails (Probst et al., 1992), making comprehensive alanine-scanning mutagenesis impractical. Convenient pairs of highly homologous receptors with dramatically different internalization and arrestin binding are also an exception rather than the rule. Therefore, a variety of alternative approaches have been used to identify arrestin-binding receptor elements. Several groups used synthetic or, more often, overexpressed and purified (e.g., as GST fusions) peptides representing intracellular receptor domains. Rhodopsin (Krupnick et al., 1994) and lutropin receptor (Mukherjee et al., 1999b) loop peptides were tested for their ability to compete with the native receptor for arrestin. Rhodopsin i1 and i3 competed with P-Rh* with 30–1000 μM affinities, whereas lutropin receptor i3 had an IC50 of 10 μM (Mukherjee et al., 1999b). The studies of arrestin interactions with receptor elements using surface plasmon resonance also yielded micromolar affinities (Cen et al., 2001a; Liu et al., 2004). Wild-type arrestins bound to the immobilized phosphorylated C-terminal peptide of the N-formyl peptide receptor with micromolar affinities, although several constitutively active mutants demonstrated sub-micromolar dissociation constants (Potter et al., 2002). Interestingly, in a few studies where the affinity of arrestin proteins for the native phosphoreceptor has been measured, it was found to be nanomolar for rhodopsin (Schleicher et al., 1989; Pulvermuller et al., 1997; Osawa et al., 2000) and sub-nanomolar for the b2AR or m2 muscarinic cholinergic receptor (Gurevich et al., 1993a, 1995). As the binding affinity directly reflects the energy of the interaction (ΔG°=−RT lnKA), one can calculate that cooperative 3- to 4-point binding, where each individual interaction has micromolar affinity, would yield a sub-nanomolar KD. Thus, all reported affinity measurements are in good agreement with the rest of the data, clearly indicating the multi-site nature of the arrestin–receptor interaction. Qualitative studies of direct arrestin binding to various elements of different receptors have implicated i3 of the m2 and m3 muscarinic, the α2A-adrenergic receptors (Wu et al., 1997), and the 5-HT2A receptor (Gelber et al., 1999), i3 and the C-terminus of delta-opioid and the C-terminus of kappa-opioid receptors (Cen et al., 2001b), as well as i2, i3, and the C-terminus of dopamine D2 (Macey et al., 2004) and D1 receptors (Macey et al., 2005). Where the binding of a combination of elements was tested (e.g., i3+C-terminus of the delta-opioid receptor (Cen et al., 2001b), it was found to be additive, suggesting that these elements bind to different sites on arrestin.
The most popular (although less mechanistically rigorous) approach used to delineate arrestin-binding receptor elements is the use of changes in (presumably) arrestin-dependent trafficking of mutant and chimerical receptors as the readout. These experiments also identified the same “usual suspects”, singly or in combinations: various cytoplasmic loops in many GPCRs, as well as the C-termini of most receptors known to man (Table 2).
Table 2.
Receptor elements involved in arrestin binding identified using receptor trafficking assays in cells
| Receptor element | Receptor type | References |
|---|---|---|
| C-terminus | Thyrotropin-releasing hormone receptors1 and 2 | Groarke et al., 2001; Hanyaloglu et al., 2001, 2002 |
| Chemokine receptor CCR5 | Huttenrauch et al., 2002, Kraft et al., 2001 | |
| Chemokine receptor CCR7 | Kohout et al., 2004 | |
| Angiotensin II receptor type 1 | Qian et al., 2001 | |
| Platelet-activating factor receptor | Chen et al., 2002 | |
| N-formyl peptide receptor | Key et al., 2003 | |
| β1- and β2-adrenergic receptors | Liang et al., 2003a | |
| V2 vasopressin receptor | Charest & Bouvier, 2003 | |
| Metabotropic glutamate receptor 1A | Dale et al., 2001; Mundell et al., 2003 | |
| Substance P receptor | Richardson et al., 2003 | |
| Scavenger chemokine receptor D6 | Galliera et al., 2004 | |
| Prostaglandin receptor EP4 | Neuschafer-Rube et al., 2004 | |
| Corticotropin-releasing hormone receptor | Teli et al., 2005 | |
| Orexin-1 receptor | Milasta et al., 2005 | |
| Protease-activated receptor-2 | Stalheim et al., 2005 | |
| Cysteinyl leukotriene receptor | Naik et al., 2005 | |
| Neuropeptide Y1 receptor | Holliday et al., 2005 | |
| Somatostatin 2A receptor | Tulipano et al., 2004 | |
| Serotonin 5-HT4 receptor | Barthet et al., 2005 | |
| Follitropin receptor | Kishi et al., 2002 | |
| Neurokinin 1 and 3 receptors | Schmidlin et al., 2003 | |
| i3 (3rd loop) | m2 muscarinic cholinergic receptor | Lee et al., 2000 |
| Dopamine D2 and D3 receptors | Kim et al., 2001 | |
| Dopamine D1 receptor | Kim et al., 2004 | |
| Dopamine D2 receptor | Namkung & Sibley, 2004 | |
| α2A-adrenoreceptor | Wang & Limbird, 2002 | |
| α2B-Adrenoreceptor | DeGraff et al., 2002 | |
| α2C-Adrenoreceptor | DeGraff et al., 2002 | |
| Follitropin receptor | Kishi et al., 2002 | |
| Lutropin receptor | Bhaskaran et al., 2003 | |
| Neurokinin 1 and 3 receptors | Schmidlin et al., 2003 | |
| i2 (2nd loop) | Dopamine D2 and D3 receptors | Kim et al., 2001 |
| i1 (1st loop) | Follitropin receptor | Kishi et al., 2002 |
Thus, an extensive body of evidence indicates that the arrestin-binding surface of GPCRs is as extensive as the receptor-binding arrestin surface. It likely includes non-contiguous residues distributed through all intracellular elements of the receptor. Each individual interaction is relatively low-affinity, but simultaneous engagement of several elements yields the high-affinity complex. An obvious corollary of this model is that not all potential interaction sites on both partners need to be engaged to allow arrestin to perform its functions. The complexes held together by fewer elementary interactions would have reduced affinity and stability. This is likely the mechanistic basis of the functional differences between class B GPCRs that hold tight onto arrestins and travel with them all the way to late endosomes and beyond, and class A receptors that readily release bound arrestins upon internalization (Oakley et al., 2000). Another important corollary of this model is that the same arrestin–receptor combination can form a variety of complexes by engaging a subset of potential sites. The complexes held together by different sets of elementary interactions could have somewhat different shapes, with important functional consequences (see Section 3.3).
2.7. The many faces of receptor phosphorylation
The first clearly established role of rhodopsin phosphorylation was to promote arrestin binding (Kuhn et al., 1984). A similar functional effect of b2AR phosphorylation fully corroborated this notion (Benovic et al., 1989; Lohse et al., 1990), suggesting that direct arrestin binding to receptor-attached phosphates is an important contributor to the overall affinity of the interaction. The construction of the first visual arrestin mutants that bind P-Rh* as well or better than wild-type arrestin, and also interact with comparably high affinity with unphosphorylated Rh* and even light-activated truncated rhodopsin that has the C-terminus along with all phosphorylation sites proteolytically removed (Gurevich & Benovic, 1992, 1993; Gurevich et al., 1994; Gurevich & Benovic, 1997), challenged this idea, suggesting that as far as the affinity of the complex is concerned, direct arrestin–phosphate interaction is dispensable. An alternative idea in which the phosphates are mostly necessary to activate the phosphate sensor in arrestin to unleash its binding power was proposed (Gurevich & Benovic, 1993, 1995). The solved arrestin crystal structure (Hirsch et al., 1999) and follow-up mutagenesis (Vishnivetskiy et al., 1999) revealed an elegant mechanism of the function of the arrestin phosphate sensor providing very strong support for this model (Figs. 5–7).
However, a compelling set of studies of the m2 muscarinic receptor (Pals-Rylaarsdam et al., 1997; Lee et al., 2000) suggested that there is more to receptor phosphorylation than previously thought. This receptor is primarily phosphorylated on the 2 clusters in i3. The phosphorylation of the C cluster (TVSTS) is absolutely required for arrestin binding in vivo and in vitro (Pals-Rylaarsdam et al., 1997). Mutation of this sequence to AVAAA virtually eliminates the receptor’s ability to bind wild-type arrestins, similar to mutations or deletions of the phosphorylation sites in the b2AR or delta opioid receptor (Kovoor et al., 1999). Phosphorylation-independent arrestin2 mutants with the phosphate sensor “pre-activated” bind phosphorylation-deficient b2AR and delta opioid receptors (Kovoor et al., 1999; Celver et al., 2002), but the same “super-arrestins” still fail to bind the C-cluster mutant of m2 receptor (Lee et al., 2000). Unexpectedly, the deletion of 15 residues (including the C-cluster) yields an m2 receptor that binds even wild-type arrestins 2 and 3 normally. Thus, it appears that the C-cluster and surrounding sequence is an inhibitory element suppressing m2 interactions with arrestins. The deletion or, in the wild-type receptor, the phosphorylation of this element relieves the inhibition, likely moving it out of the way to allow arrestin binding to other m2 receptor elements (Pals-Rylaarsdam et al., 1997; Lee et al., 2000). This was the first description in a mammalian GPCR of a receptor element serving as a “brake” for arrestin binding that is released by receptor phosphorylation (although the term was coined a bit later; Whistler et al., 2001). Soon it became clear that similar mechanisms operate in other receptors, as well. Alanine substitution of serines and threonines in the distal C-terminus of the delta opioid receptor yielded a mutant that did not bind arrestins and failed to internalize, whereas the deletion of this element containing the regulatory phosphorylation sites was found to enable phosphorylation-independent recruitment of arrestin3 and normal receptor internalization (Whistler et al., 2001). Interestingly, the substitution of serines and threonines with aspartates also enables arrestin binding and receptor internalization, suggesting that the brake is removed from its inhibitory position by electrostatic interactions. Another example of a similar brake was described for the rat follitropin receptor (Kishi et al., 2002). Agonist activation leads to the phosphorylation of this receptor on i1 and i3 rather than on the C-tail. The deletion of 10 internal residues in the C-tail of this receptor that do not appear to be phosphorylated facilitated arrestin binding and arrestin-mediated internalization severalfold (Kishi et al., 2002). Although this issue has not been explored, it is tempting to speculate that in this case the phosphorylation of other receptor elements, rather than that of the brake itself, relieves the inhibition of arrestin binding. In yet another example, in i3 of the D1 dopamine receptor, alanine substitutions of all serines and threonines, or of the 3-serine cluster (256, 258, 259), yielded receptors with reduced phosphorylation and impaired arrestin binding and desensitization (Kim et al., 2004). C-terminal truncation of the D1 receptor also reduced phosphorylation, but even the most severely truncated mutant bound arrestin and desensitized normally. These data suggest a similar scenario; activation-induced phosphorylation of the D1 receptor moves its C-tail out of the way, enabling direct phosphorylation independent association of arrestin with the elements of the 3rd cytoplasmic loop (Kim et al., 2004).
Thus, receptor phosphorylation can play many roles. In some cases clustered phosphates prime arrestin for binding by turning its phosphate sensor “on”, in others they primarily facilitate the removal of an inhibitory receptor element and enhance the accessibility of other regions for arrestin. It is entirely possible that for many receptors phosphorylation fulfills both functions simultaneously, albeit with different relative importance for tight arrestin binding to various GPCRs.
2.8. Do G proteins and arrestins require the same “active” receptor conformation?
Until recently this question has not been asked because the general assumption was that since arrestins are designed to stop G-protein-mediated signaling, they should preferentially bind to the same active receptor state that G proteins prefer. The same logic also applies to GRKs that are supposed to prepare the receptor for arrestin binding. Therefore, an early finding that the extent of b2AR phosphorylation by GRK2 in the presence of full and partial agonists correlates remarkably well with their intrinsic activity, that is, their ability to activate adenylyl cyclase via Gs, was not surprising (Benovic et al., 1988). An impressively extensive body of evidence accumulated since the late 1970s solidly supports this idea (just a few representative references are Kuhn, 1978; Schleicher et al., 1989; Gurevich et al., 1995; Krasel et al., 2005). Numerous receptor mutagenesis studies indicate that signaling-impaired receptors demonstrate reduced GRK-mediated phosphoryla-tion, arrestin binding, and internalization, whereas constitutively active receptors are often constitutively phosphorylated, associated with arrestins, and internalized (Rim & Oprian, 1995; Min & Ascoli, 2000; Barak et al., 2001; Kim et al., 2001; Min et al., 2002; Ponimaskin et al., 2005). As discussed in Section 4, this unfortunate feature of naturally occurring constitutively active receptor mutants apparently underlies certain congenital disorders (Rim & Oprian, 1995; Barak et al., 2001; Wilbanks et al., 2002).
Another line of evidence pointing in the same direction is the ability of G proteins and arrestins to stabilize the same functional (and, by extension, conformational) state of the receptor. This was first described for rhodopsin, where different functional states are conveniently spectrally distinct. Visual arrestin stabilizes the same metarhodopsin II (with λmax 380 nm) as does the visual G protein transducin (Schleicher et al., 1989). Both non-visual arrestins were later shown to stabilize the high agonist affinity state of β2-adrenergic and m2 muscarinic cholinergic receptors (Gurevich et al., 1997), similar to the effect of their cognate G proteins (De Lean et al., 1980; Samama et al., 1993; Simons et al., 2004). Moreover, the fraction of receptor molecules in the high agonist affinity state in the presence of both G protein and arrestin was found to be directly proportional to the intrinsic activity of the agonist (Samama et al., 1993; Gurevich et al., 1997; Simons et al., 2004). Similar observations with the N-formyl peptide receptor (Bennett et al., 2001; Key et al., 2001, 2003) and glucagon-like peptide-1 receptor (Jorgensen et al., 2005) strongly indicate that this is a general rule for the GPCR family. The translocation of arrestin-GFP fusion proteins to the plasma membrane in response to receptor activation is a popular indicator of arrestin binding to the receptor in living cells (Barak et al., 1997). “Green” arrestin recruitment by a variety of receptors shows the same pharmacological profile as receptor activation (Zhang et al., 1999), also indicating that the active receptor state required for both is usually the same.
Several studies, however, suggest that the conformational requirements for G protein activation and arrestin binding may not always be the same. Most of these data were obtained with receptor mutants. One example is the 5-HT4A receptor, one unusual feature of which is the presence of 2 palmitoylation sites in its C-terminus: the “standard” double cysteine motif in the proximal part, and an extra cysteine in the distal segment (Ponimaskin et al., 2005). The elimination of the proximal site yields a robust phenotype suggesting close similarity, if not identity, of arrestin and G protein requirements: constitutive activity, constitutive phosphorylation, and enhanced arrestin-dependent desensitization and internalization. In other palmi-toylation-deficient mutants, activation, phosphorylation, and arrestin binding appeared to be affected differently (Ponimaskin et al., 2005). Another example comes from the complement factor 5a receptor (Whistler et al., 2002b). Two structurally different mutations, F251A and NQ, confer similar constitutive G protein activation, but only the NQ mutant undergoes constitutive internalization. A compensatory N296A on the NQ background eliminates constitutive activity, but not constitutive endocytosis (Whistler et al., 2002b). An engineered Zn2+-binding site in the parathyroid hormone receptor precludes (in the presence of Zn2+) the relative movement of helices 3 and 6, thereby blocking G protein activation, but does not prevent agonist-induced receptor phosphorylation and arrestin recruitment (Vilardaga et al., 2001). Two studies of the angiotensin II receptor type 1A using a signaling-deficient agonist analogue found that it promotes receptor phosphorylation and arrestin binding (Qian et al., 2001), as well as arrestin-mediated chemotaxis (Hunton et al., 2005). Finally, one study came to the same conclusion based exclusively on the properties of the wild-type chemokine receptor CCR7 and two of its native ligands, CCL19 and CCL21 (Kohout et al., 2004). While both ligands similarly induce G-protein-mediated Ca2+ mobilization, only CCL19 promotes robust desensitization of CCR7 and induces 4-fold greater arrestin-dependent extracellular signal-regulated kinase (ERK1/2) activation than CCL21, suggesting that the two ligands induce different active receptor conformations, equipotent in terms of G protein activation but distinct as far as arrestin binding and arrestin-mediated signaling is concerned (Kohout et al., 2004).
Thus, even though as a rule arrestins and G proteins preferentially bind to the same active receptor state, in some cases there may be subtle differences between the conformational requirements of these two families of regulatory proteins.
3. The arrestin–receptor complex: one size does not fit all
Structural diversity of the members of the GPCR superfamily defies imagination (Probst et al., 1992). Even though only their cytoplasmic surface matters from arrestins’ point of view, at least 2 elements on the intracellular side of the receptor vary wildly. The 3rd loop (i3) ranges from a modest 16-residue-long connector to a large domain comparable in size with the arrestin itself. Similarly, receptor C-termini range from a dozen to several hundred residues. Arrestins, on the other hand, are remarkably conserved: about 400 residues, give or take a dozen or two. Having several crystal structures of only one receptor, bovine rhodopsin in its dark (inactive) state (Palczewski et al., 2000; Okada et al., 2002; Li et al., 2004), we do not really know the extent of the structural conservation even of the 7 transmembrane domain core in the GPCR family, although there are good reasons to believe (and even better ones to hope) that at least these parts of different receptors are structurally similar. In contrast, there is no reason to believe that the conformations of large and small cytoplasmic loops and C-termini have much in common. Moreover, receptor-attached phosphates that arrestin binds are localized in different receptors on various intracellular elements: i1, i2, i3, or the C-tail. Yet the conformations of the members of 3 crystallized vertebrate arrestin subtypes in their free (inactive) state are essentially the same (Granzin et al., 1998; Hirsch et al., 1999; Han et al., 2001; Milano et al., 2002; Sutton et al., 2005). Common sense suggests that there is no way in which this standard arrestin “hat” can be uniformly “pulled” over the wildly varying cytoplasmic “heads” of hundreds of receptors. Yet functionally arrestin interactions with numerous GPCRs have a lot in common: they preferentially bind active, in most cases phosphorylated, receptors and arrestin binding invariably stops receptor signaling via G proteins, indicating that there must be a structural basis for this functional commonality.
3.1. What parts of the receptor must arrestin block to justify its name?
To “arrest” G-protein-mediated signaling, bound arrestin must occupy or shield at least one receptor element obligatory for G protein activation. In and of itself this does not help much because G proteins also interact with multiple receptor elements (reviewed in Bohm et al., 1997). In fact, structurally a somewhat larger family of heterotrimeric G proteins appears almost as conserved as the arrestin family (Noel et al., 1993; Coleman et al., 1994; Lambright et al., 1994; Sondek et al., 1994; Mixon et al., 1995; Wall et al., 1995; Lambright et al., 1996; Sondek et al., 1996; Sunahara et al., 1997), creating a similar problem of fitting a uniform “hat” onto a wide variety of “heads”. Nonetheless the receptor G protein interaction yields important clues. Receptor activation (as studied in great detail on the rhodopsin model) involves a substantial rigid body motion of the α-helical domains (reviewed in Hubbell et al., 2003). Helix 6 moves up and away by 8Å opening up a cavity between the helices on the cytoplasmic side (Farrens et al., 1996). The C-terminal peptide of the transducin α-subunit inserts itself into this cavity (Janz & Farrens, 2004). This peptide is the only part of transducin that can stabilize metarhodopsin II similar to holotransducin itself (Hamm et al., 1988). It also effectively competes with transducin for Rh* (Hamm et al., 1988), suggesting that this is one of the strongest interactions between the G protein and active receptor. Arrestin also stabilizes metarhodopsin II (Schleicher et al., 1989), hence we were not surprised to find that an as yet unidentified part of arrestin also occupies the cavity between the rearranged α-helices in Rh* (Kolobova et al., 2003). The overlap between the arrestin- and G-protein-binding surface on the receptor may be even more extensive: arrestin2 interaction with i2 of chemokine receptor CCR5 (Huttenrauch et al., 2002) and arrestin binding to the N-formyl peptide receptor (Bennett et al., 2000) requires an intact Asp-Arg-Tyr (DRY) motif, which is one of the crucial G protein interaction elements. Mutations D142A and D142T in the DRY motif of the α1B-adrenergic receptor impair arrestin binding (Mhaouty-Kodja et al., 1999). The interactions with both the cavity and DRY motif are possible only when the receptor is in its active state, hence either (or both) may be responsible for the relatively low-affinity arrestin binding to R* (Fig. 10).
The overlap of the binding sites is the structural basis of direct competition between arrestin and G protein for the active receptor (Krupnick et al., 1997b), explaining why their binding is mutually exclusive. This competition along with structural analysis of the binding of these 2 proteins to the receptor indicates that the inter-helical cavity is a likely “anchoring point” that arrestin must occupy or shield to do its job. We do not know yet which element of arrestin interacts with this cavity.
3.2. Arrestin-binding elements in GPCRs: obligatory and optional
The only other interaction between arrestin and the receptor that is likely to be obligatory in the great majority (if not all) cases is between the arrestin polar core (Fig. 5) and phosphorylated (or otherwise negatively charged) receptor elements. The situation here is the opposite: we know exactly the part of arrestin that must be engaged, but the relevant receptor elements do not appear to have any structurally fixed position in GPCRs; although phosphorylated residues are more often found in the C-terminus than anywhere else, they can also be in any of the intracellular loops, or even in more than one part of the same receptor. To complicate matters even further, i3 and/or the C-terminus in many receptors can be equipped with several similar-looking serine/threonine-rich clusters, some of which appear crucial for arrestin binding, while others turn out to be irrelevant (Lee et al., 2000; Oakley et al., 2001; Key et al., 2003; Namkung & Sibley, 2004; Neuschafer-Rube et al., 2004). Because, at least in some cases, these functionally distinct clusters were actually shown to be phosphorylated (Lee et al., 2000; Key et al., 2003; Neuschafer-Rube et al., 2004), they must be exposed. This raises an obvious question: how does arrestin know which cluster is the “right” one? The only logical explanation is that when arrestin is appropriately anchored at the inter-helical cavity, certain clusters sterically fit onto its phosphate-binding elements (Fig. 14) whereas others do not.
It is likely that the position of phosphorylated elements in the 3-dimensional structure of different GPCRs varies as much as it does in the linear sequence of these receptors. Then bound arrestin simultaneously anchored via an unknown element at the inter-helical cavity and via its phosphate-binding residues at the phosphorylated cluster may well be oriented quite differently relative to the “long” horizontal axis (helix I–helix V) of the receptor (Fig. 15).
Fig. 15.

The structural basis for the formation of different arrestin–receptor complexes. Arrestin binding to the receptor is mediated by multiple elements in both proteins. Different relative positioning of the arrestin-binding parts of the receptor results in the variation of arrestin orientation on the receptor, in some cases with profound functional consequences. For the sake of simplicity, the example shows only 2 elementary interactions. One of the flexible loops of arrestin (filled pink circle) moves into the inter-helical cavity that opens upon GPCR activation (red oval) and interacts with receptor elements that become accessible upon activation (red circle within the red oval). At the same time, phosphate-binding arrestin residues (2 yellow elements on β-strand X and one on β-strand I) interact with receptor-attached phosphates (filled red circles). The difference in the positions of receptor-attached phosphates forces arrestin to orient itself in different ways on the receptor to achieve the best fit. The views of the receptor from its cytoplasmic side and of the arrestin with its receptor-binding side facing the receptor (away from the reader) are shown.
Several lines of evidence suggest that arrestin binding to the receptor usually involves more than just these two obligatory interactions. In the most studied visual arrestin–rhodopsin model, the binding of wild-type arrestin to Rh* (likely engaging receptor elements that become exposed upon activation, such as the cavity and/or DRY motif) has relatively low affinity; it is also highly sensitive to salt inhibition (Gurevich & Benovic, 1993). The binding of structurally diverse phosphorylation-independent mutants to the same functional form of full-length or even truncated C-tail-less rhodopsin (which, by definition, cannot involve phosphate interactions because there are no phosphates in these preparations) is much stronger and has a different chemical nature: it includes hydrophobic interactions that are enhanced at physiological ionic strength (Gurevich & Benovic, 1993; Gurevich & Benovic, 1995; Gurevich & Benovic, 1997; Gurevich, 1998). Truncated arrestin(1–191) does not have the hydrophobic binding site (Gurevich & Benovic, 1993). These data clearly indicate that 2 binding sites in arrestin (one in the N-and the other in the C-domain) can engage a minimum of 2 distinct elements in unphosphorylated Rh*, hence with the receptor-attached phosphates the total comes to 3. High (sub-nanomolar; Gurevich et al., 1995) affinity of non-visual arrestins for their cognate receptors and the structural analysis of the elementary interactions involved also indicate that 3 or more sites are simultaneously engaged. The additional contact sites (some of which may be optional) likely include interactions with i1, i2, and parts of i3 and the C-terminus other than the phosphorylated elements in these receptor domains (Nakamura et al., 2000).
3.3. Different flavors of the complex
Arrestin interactions with the inter-helical cavity and phosphorylated receptor elements likely determine its orientation relative to the receptor in the complex. Possible differences in its position and the presence or absence of particular additional points of contact have the potential to yield arrestin–receptor complexes of different shapes, within certain limits (Fig. 15). In addition, even though receptor-bound arrestin is usually depicted with the concave sides of the 2 domains facing the membrane (Gurevich & Gurevich, 2003; Gurevich & Gurevich, 2004; Lefkowitz & Shenoy, 2005), as in Figs. 2, 9, and 15, this is not necessarily the case. In fact, the arrestin molecule can be turned on its long axis by up to 60–70° either way relative to the plane of the membrane with the cavities of the 2 domains facing the cytoplasmic tip of the receptor. It is entirely possible that the same arrestin can orient itself differently relative to the plane of the membrane when approaching and binding the receptor. Another source of structural variability of the complex is the extent of the movement of the two arrestin domains. The 3 groups of intra-molecular interactions described above (Fig. 7) fix the relative orientation of the two domains in the basal arrestin conformation. Arrestin activation involves the release of all of these “clasps” to allow rigid body-like domain movement (Vishnivetskiy et al., 2002). High stability of the β-strand sandwich in both domains is invariably supported by numerous hydrophobic interactions between inwardly pointing β-sheet residues (Hirsch et al., 1999; Han et al., 2001; Sutton et al., 2005). This structure along with the truly devastating effects of mutations that destabilize the sandwich, such as V53D (Zhang et al., 1997; Orsini & Benovic, 1998), makes it extremely unlikely that any significant domain meltdown takes place in the process of arrestin activation. On the other hand, there is no discernible structural feature that would rigidly fix the relative orientation of the two domains in the active state. The contact surface between the bodies of the two domains, including that between the receptor-binding elements of all arrestins, is mostly hydrophobic (Fig. 7) (Sutton et al., 2005). This makes it highly adaptable because in the absence of polar interactions there are very few geometrical constraints. Therefore, it seems unlikely that the domains move from one predetermined orientation to another one preset with similar strictness. Rather, the “released” domains are probably free to “sample” a variety of relative orientations, allowing activated arrestin to “mold” itself on the particular receptor to achieve the best possible fit (Gurevich & Gurevich, 2004). The ultimate shape of the complex would depend on the receptor elements engaged by arrestin and the distances between them.
One might ask whether these theoretical considerations have any relevance in real life. Several unexpected findings in the last 2 years suggest that they have. The first indication that the complex of the same arrestin with the same receptor can come in different “flavors” came from studies of the N-formyl peptide receptor (FPR), which has 2 main serine/threonine clusters in its C-terminus: residues 328–332 (STQTS) and 334–339 (TATNST) (Key et al., 2003). It was found that although phosphorylation-independent arrestin2(1–382) binds the phosphorylation-deficient receptor, the complex does not demonstrate high agonist affinity, in sharp contrast to the wild-type arrestin2-phospho-FPR complex (Key et al., 2001). The restoration of the STQTS cluster increased mutant arrestin affinity for the receptor but did not restore high agonist affinity of the complex. In contrast, the restoration of the TATNST cluster did not significantly affect arrestin affinity for the receptor, but it restored high agonist affinity of the complex (Key et al., 2003). This was the first demonstration that the functional properties of the arrestin complex with the receptor phosphorylated at alternative clusters are different. The simplest structural interpretation of these data is that depending on the position of the phospho-cluster that is in contact with the phosphate binding “patch” of positively charged arrestin residues (Fig. 15) an appropriate arrestin element can or cannot reach the inter-helical cavity (or some other part of the receptor) and therefore does or does not stabilize the high agonist affinity state of the receptor.
The results of two recent studies revealed unexpectedly distinct functional consequences of the phosphorylation of the angiotensin II receptor (Kim et al., 2005a) and V2 vasopres-sin receptor (Ren et al., 2005) by different GRKs. In both cases, the bulk of receptor phosphorylation, arrestin recruitment, desensitization of G-protein-mediated signaling, and receptor internalization was shown to be mediated by GRKs 2 and 3. In contrast, most of the arrestin-mediated ERK1/2 activation depends on receptor phosphorylation by GRKs 5 and 6. The inhibition of GRK5/6 expression abolishes arrestin-mediated ERK signaling, whereas the inhibition of GRK2/3 expression enhances it. Accordingly, arrestin-mediated ERK activation is enhanced by GRK5/6 overexpression and inhibited by the overexpression of GRK2/3. These data clearly establish different functional capabilities of the arrestin complex with receptor phosphorylated by different GRKs. If one accepts the only logical assumption that GRK2/3 and GRK5/6 phosphorylate different receptor elements, which subsequently “compete” for the phosphate binding “patch” on arrestin, these apparently mystifying results lend themselves to the same mechanistic explanation as the FPR data. Different positions of phosphorylated receptor elements differentially orient arrestin on the receptor, yielding structurally and functionally distinct arrestin–receptor complexes (which, however, include the same arrestin and the same receptor).
Another recent study suggests that the same arrestin bound to different receptors assumes different conformations (Shenoy & Lefkowitz, 2005). The authors demonstrate that in complex with the angiotensin II type 1A (AT1A) receptor, lysines 11 and 12 in arrestin3 become ubiquitinated. This ubiquitination is required for stable arrestin3 association with the receptor, their co-trafficking, robust ERK activation, and the retention of ERK in the cytoplasm. The arrestin3(K11,12R) mutant cannot perform these functions, whereas the fusion of a ubiquitin moiety at the C-terminus of this double arginine mutant restores them. In contrast, the binding of the same arrest-in3(K11,12R) mutant to vasopressin V2 and neurokinin 1 receptors is indistinguishable from wild-type arrestin3, and it mediates vasopressin-induced ERK activation normally. However, quintuple mutant arrestin3(K18,107,108,207,296R) is impaired in endosomal trafficking with V2 vasopressin, but not with the AT1A receptor (Shenoy & Lefkowitz, 2005). Because ubiquitin ligases are not particularly “picky” and often modify the most exposed lysines, these data suggest that these arrestin3 residues are differentially exposed depending on the receptor type it binds.
Collectively, these data support the idea that arrestins’ potential to assume a variety of active conformations yielding structurally different arrestin–receptor complexes is actually realized and indicate that arrestin “shape-shifting” has profound functional consequences. It appears that arrestin can undergo further conformational rearrangements after it is bound, further attesting to the flexibility of the molecule after its intra-molecular clasps are “unfastened” by the receptor. Experiments with arrestin sandwiched between luciferase and yellow fluorescent protein, in which intra-molecular bioluminescence resonance energy transfer (BRET) can be used to monitor the distance between its N- and C-termini in living cells, showed that the 2 ends of the molecule come close together after receptor stimulation (Charest et al., 2005). Kinetic analysis shows that the spike in BRET occurs substantially later than arrestin binding to the receptor (Charest et al., 2005). This “second wave” of conformational changes in bound arrestin possibly reflects the docking of its non-receptor-binding partners attracted to the complex as a result of the first wave of conformational rearrangements (Gurevich & Gurevich, 2003).
3.4. Where do receptor dimers fit in?
Homo- and heterodimerization of GPCRs is a fairly common phenomenon (reviewed in Angers et al., 2002), hence the first issue that needs to be addressed is whether arrestin binds the receptor monomer, the dimer, or both. Direct unambiguous experimental evidence pointing one way or the other does not exist. Free arrestins in their basal conformation are elongated 2-domain molecules with a “wingspan” of slightly more than 75 Å (Granzin et al., 1998; Hirsch et al., 1999; Han et al., 2001; Milano et al., 2002; Sutton et al., 2005). The cytoplasmic tip of the only receptor for which the crystal structure was determined, dark (inactive) bovine rhodopsin (Palczewski et al., 2000; Okada et al., 2002; Li et al., 2004) has a diameter of 35–40 Å (Fig. 15). Both arrestin domains have a more or less cup-like shape, with identified receptor-binding elements invariably localized on the concave sides of the two domains (Fig. 14) (Vishnivetskiy et al., 2004). This rather provocative geometry led to attempts to “dock” a single arrestin to the rhodopsin dimer (Liang et al., 2003b). Let us consider whether this is at least theoretically possible. First, this “geometric” model implies that receptor-bound arrestin has essentially the same shape as free arrestin. Although only the solution of the crystal structure of at least one arrestin–receptor complex can drive the final nail into the coffin of this notion, the evidence that arrestin binding involves a global conformational rearrangement of its domains is overwhelming (Schleicher et al., 1989; Palczewski et al., 1991b; Gurevich & Benovic, 1993; Gurevich et al., 1994; Ohguro et al., 1994; Gurevich & Benovic, 1995; Pulvermuller et al., 1997; Gurevich, 1998; Hirsch et al., 1999; Vishnivetskiy et al., 1999; Vishnivetskiy et al., 2000; Han et al., 2001; Vishnivetskiy et al., 2002; Raman et al., 2003; Xiao et al., 2004; Shenoy & Lefkowitz, 2005; Sutton et al., 2005). Second, this model does not take into account activation-induced conformational changes in rhodopsin; all of the crystal structures we have represent inactive unphosphorylated rhodopsin, that is, the only form that does not bind any arrestin, wild type or mutant, under any circumstances (Figs. 3, 5–7, 10–12). Even though the crystal structure of light-activated rhodopsin is not available, thanks to enormous efforts by many investigators, particularly comprehensive collaborative site-directed spin labeling studies of the laboratories of Drs. Hubbell and Khorana, we know that rhodopsin activation involves the movements of helices and intracellular loops the net result of which is a significant expansion of its cytoplasmic tip (Resek et al., 1993; Farahbakhsh et al., 1995; Farrens et al., 1996; Altenbach et al., 1999a, 1999b; Altenbach et al., 2001a, 2001b) (reviewed in Hubbell et al., 2003). Thus, in the functional state that actually binds arrestin the cytoplasmic tip of rhodopsin becomes too big to fit into one arrestin domain and is perfect in size to engage both arrestin domains simultaneously. Third, the docking of one rhodopsin to each domain of arrestin implies that these domains are more or less equivalent, which directly contradicts all available experimental evidence including every solved arrestin crystal structure. In fact, arrestin has just one copy of each identified functional element (Figs. 3–8), and the “similarity” between the two domains is largely superficial. Finally, rhodopsin has one of the smallest sets of cytoplasmic elements among all GPCRs (Probst et al., 1992). For many receptors that robustly bind arrestins the combined size of the cytoplasmic elements actually exceeds the size of arrestin, creating the opposite problem of fitting such a large cytoplasmic tip into two domains of just one arrestin molecule. Thus, a vast body of experimental data suggests that simultaneous binding of one arrestin molecule to both receptors in a dimer is virtually impossible. Importantly, not a single piece of experimental evidence contradicts this conclusion.
Nonetheless the arrestin aspect of the “dimer problem” actually has a scientifically relevant dimension. Even when one arrestin binds one receptor, we need to know whether arrestin prefers its target in the form of monomer or as a part of the dimer (or whether it cares one way or the other). This issue has never been directly addressed experimentally. However, a few recent studies suggest that at least non-visual arrestins can bind receptors within a dimer. One study took advantage of the difference between arrestin preference of the 2 types of thyrotropin-releasing hormone receptor (TRHR): TRHR1 interacts with both arrestin2 and arrestin3, whereas TRHR2 interacts only with arrestin3, as evidenced by the enhanced internalization of TRHR1 by overexpression of either non-visual arrestin; TRHR2 endocytosis was enhanced only by arrestin3 (Hanyaloglu et al., 2002). Accordingly, the trafficking of TRHR2 was impaired in mouse embryonic fibroblasts lacking either arrestin3 or both arrestins 2 and 3, whereas the trafficking of TRHR1 was only impaired in fibroblasts lacking both non-visual arrestins. The formation of TRHR1/2 heterodimers increased the interaction of TRHR2 with arrestin2. Most importantly, even heterodimerization between TRHR2 and truncated TRHR1(C335Stop), which by itself does not interact with non-visual arrestins, enhanced TRHR2–arrestin2 interaction (Hanyaloglu et al., 2002). The simplest mechanistic interpretation of these findings is that in a heterodimer with TRHR1 (but not in the TRHR2 homo-dimer) the conformation of the TRHR2 cytoplasmic elements changes in such a way as to permit arrestin2 binding tight enough to promote arrestin2-dependent internalization. This interpretation implies that arrestin2 binds a receptor molecule that is part of a dimer.
Another study used two GPCRs with distinct modes of arrestin interaction and different trafficking patterns: the mu-opioid receptor (MOR) and neurokinin1 receptor (NK1) (Pfeiffer et al., 2003). Arrestin binds MOR expressed alone and brings it to the coated pit but does not internalize with the receptor, whereas arrestin internalizes in complex with NK1. Co-expressed MOR and NK1 form heterodimers, as evidenced by BRET and co-immunoprecipitation. Although heterodimerization does not appreciably change the ligand binding and signaling properties of these receptors, it gives rise to several interesting phenomena (Pfeiffer et al., 2003). First, exposure of the heterodimer to either MOR- or NK1-selective agonists promotes cross-phosphorylation and co-internalization of the other receptor. Second, regardless of which receptor is stimulated, the heterodimer co-internalizes with arrestin to endosomes. Consequently, resensitization of MOR is severely delayed in cells co-expressing both receptors, as compared to cells expressing MOR alone (Pfeiffer et al., 2003). Cross-phosphorylation is easily explained by the fact that GRK is activated by the active receptor itself, whereupon it phosphorylates any substrate that happens to be around (Palczewski et al., 1991a), in this case the other receptor in a dimer. Cross-internalization and especially co-trafficking of the whole dimer along with arrestin suggest not only that arrestin interacts with the receptor in a dimer, but also that this interaction is tight enough to bring the whole dimer inside. Moreover, it appears that in the pair the receptor with higher affinity for arrestin (in this case NK1) “wins”, dragging its partner (MOR) through the long recycling pathway, thereby slowing down its resensitization.
Thus, arrestins can apparently bind to a receptor monomer that is part of a dimer. Judging by robust virtually stoichiometric arrestin binding to receptors that were solubilized and reconstituted into liposomes at a very high lipid-to-protein ratio (so that in the final preparations there is 1 receptor molecule per 10–15 lipid vesicles, making the existence of dimers highly improbable) (Gurevich et al., 1993a; Gurevich et al., 1995; Kovoor et al., 1999; Celver et al., 2002; Pan et al., 2003), arrestins can also bind monomeric GPCRs. The important question whether there is any difference in arrestin binding to the same receptor in monomeric or dimeric form still remains unanswered.
3.5. What about other classes of receptors that bind arrestins?
Several recent studies show that arrestins also bind cell surface receptors that do not belong to the GPCR superfamily. The list of these unconventional arrestin partners includes the insulin-like growth factor I (IGF1) receptor (Lin et al., 1998), the type III transforming growth factor (TGF)-beta receptor (Chen et al., 2003), and the low density lipoprotein (LDL) receptor (Wu et al., 2003). Structurally, these receptors have no similarities with GPCRs. However, as far as arrestin interaction is concerned, certain functional commonalities emerge. Arrest-ins prefer the active forms of IGF1 receptor and type III TGF-beta receptor, and the constitutively cycling scavenger-type LDL receptor can be considered “constitutively active”. Naturally, arrestin binds to the cytoplasmic tail of these receptors because they have no intracellular loops. Similarly to GPCRs, the phosphorylation of TGF-beta type III receptor by its partner TGF-beta type II receptor promotes arrestin binding and endocytosis of both receptors along with arrestin (Chen et al., 2003). Notably, Tyr807Ala mutation abolishes, and Ser833-Asp mutation enhances arrestin binding and endocytosis of the LDL receptor (Wu et al., 2003). Arrestin also enhances MAP kinase activation by the IGF1 receptor (Lin et al., 1998).
Thus, even though these receptors are very different from GPCRs, arrestins still prefer them active and phosphorylated (or otherwise equipped with extra negative charges). Considering that phosphopeptides and other polyanions promote the release of the arrestin C-tail (Palczewski et al., 1991b; Gurevich et al., 1994; Puig et al., 1995; Vishnivetskiy et al., 2002; Xiao et al., 2004), which carries both clathrin (Goodman et al., 1996) and AP-2 (Laporte et al., 1999) interaction sites, the mechanisms whereby arrestins promote the internalization of these receptors and GPCRs via coated pits may be quite similar (Gurevich & Gurevich, 2003). However, we know too little about the interactions of arrestins with these receptors to glean any useful clues regarding the conformation of bound arrestin in this case.
4. The functional consequences of complexity
It seems hard to believe now that less than 10 years ago the commonly accepted paradigm was that as far as signaling is concerned arrestin binding to the receptor was the end of the story. The first indication that arrestins do more than simply “arrest” G-protein-mediated signaling was reported in 1996 (Ferguson et al., 1996). Arrestin and GRK2 overexpression was able to rescue the internalization of a mutant b2AR that did not internalize otherwise, indicating that arrestin binding plays a role in receptor trafficking (Ferguson et al., 1996). The mechanism whereby arrestin accomplishes this feat was soon revealed: both non-visual arrestins directly bind clathrin, apparently serving as adaptors tethering the bound receptor to the internalization machinery of the coated pit (Goodman et al., 1996). Later it was discovered that receptor-bound arrestins also interact with the clathrin adaptor complex AP-2 (Laporte et al., 1999). Both interactions are necessary for efficient endocytosis, but to a certain extent they also serve as backups for each other, because disabling or deletion of either binding site in arrestin does not completely block receptor internalization (Kim & Benovic, 2002). For example, truncated arrestin2(1–383), which binds receptors in phosphorylation-independent fashion (Kovoor et al., 1999) and lacks AP-2 binding site (Laporte et al., 1999), supports the internalization of unphosphorylated receptors and traffics into endocytic vesicles with receptors that normally release wild-type arrestin at the plasma membrane (Oakley et al., 2001). This additional function of non-visual arrestins was still viewed as an extension of its role in desensitization, as internalization was considered a simple “mopping up”, that is, the removal of the used desensitized receptor. However, soon it became clear that internalization does not just remove “unwanted” receptor from the cell surface but is a necessary step in receptor resensitization. Dominant-negative mutants of arrestin and dynamin, both of which block the internalization of the b2AR, were found to impair dephosphorylation and resensitization of this receptor (Zhang et al., 1997). Accordingly, impaired b2AR sequestration, dephosphorylation, and resensitization in COS-7 cells (expressing very low levels of endogenous arrestins) were completely rescued by the overexpression of arrestin3, suggesting that the levels of arrestin expression regulate the ability of the receptor to resensitize (Zhang et al., 1997).
Continuing discoveries of additional non-receptor arrestin-binding partners, many of which were bona fide signaling molecules, completely changed the overall perception of arrestin’s role in the cell (reviewed in Lefkowitz & Whalen, 2004; Lefkowitz & Shenoy, 2005). The ever-growing list of proteins that bind the arrestin–receptor complex (and in some cases free arrestin as well) now includes ADP ribosylation factor 6 (ARF6) and ARF nucleotide-binding site opener (ARNO) (Claing et al., 2001; Hunzicker-Dunn et al., 2002), cAMP phosphodiesterase 4 (PDE4) (Perry et al., 2002), N-ethylmaleimide-sensitive fusion protein (NSF) (McDonald et al., 1999), E3 ubiquitin ligase Mdm2 (Shenoy et al., 2001), IκBα, an inhibitor of NFκB (Gao et al., 2004), and a variety of protein kinases, such as cSrc (Luttrell et al., 1999) and the related kinase Yes (Imamura et al., 2001), extracellular signal-regulated kinases (ERK1 and 2) and cRaf1 (Luttrell et al., 2001), and c-Jun N-terminal kinase 3 (JNK3) and ASK1 (McDonald et al., 2000). This overabundance of binding partners raises many important questions. Which of these proteins preferentially interact with active receptor-bound arrestin, and how is this accomplished mechanistically? Do the partners that bind free arrestins stay bound through the process of its docking to the receptor? Are there proteins (other than GPCRs) that only bind free arrestins? Which partners bind arrestin simultaneously and which compete for overlapping binding sites? And last but not least, what are the functional consequences of all these interactions?
4.1. Does arrestin binding predetermine the internalization pathway?
Arrestin binding to the receptor induces the release of the arrestin C-tail, where both clathrin and AP-2 sites are localized (Goodman et al., 1996; Krupnick et al., 1997a; Orsini & Benovic, 1998; Laporte et al., 1999; Kim & Benovic, 2002). In addition, several positively charged residues in the C-domain of non-visual arrestins constitute a high-affinity phosphoinositide-binding site (Gaidarov et al., 1999). Phosphoinositide binding is an additional mechanism bringing arrestin to the coated pit: adaptin2 also binds phosphoinositides apparently for the same purpose (Gaidarov & Keen, 1999). Thus, arrestin binding creates so many reasons for the complex to end up in the coated pit that it is hard to imagine how any arrestin-binding receptor can escape this fate. Yet early on it became clear that some receptors do just that. One of the first examples was m2 muscarinic cholinergic receptor (m2 mAChR). This was one of the first receptors shown to bind arrestins (Gurevich et al., 1993a). The main GRK phosphorylation sites in m2 mAChR are localized in 2 clusters in i3, termed the N- and C-cluster for convenience (Pals-Rylaarsdam et al., 1997). Alanine substitution of serines and threonines in the N-cluster does not affect receptor desensitization in living cells or direct arrestin binding in vitro, whereas the elimination of phosphorylatable residues from the C-cluster or from both the N- and C-clusters impairs both desensitization and arrestin binding, indicating that the desensitization of this receptor is strictly arrestin dependent (Pals-Rylaarsdam et al., 1997). Yet even the receptor with both clusters mutated internalizes in HEK293 cells normally. Moreover, the internalization of the m2 mAChR and its mutant forms is not affected by the dominant-negative dynamin(K44A) mutant that blocks clathrin- and arrestin-dependent internalization of the b2AR in the same cells (Pals-Rylaarsdam et al., 1997). Thus, the m2 mAChR even with arrestin bound apparently chooses a different internalization pathway. Interestingly, overexpression of either non-visual arrestin enhances its internalization, and this additional receptor trafficking is suppressed by dynamin(K44A) (Pals-Rylaarsdam et al., 1997). So bound arrestin actually can direct the m2 mAChR to the coated pit, although usually this receptor uses an alternative route. What is the structural basis of this curious phenomenon?
The cytoplasmic elements of many GPCRs contain a variety of internalization and sorting motifs. One example is the dileucine motif recognized by several clathrin adaptors participating in the trafficking of other membrane proteins, which was identified in the C-termini of several receptors: the b2AR (Gabilondo et al., 1997), V2 vasopressin receptor (Schulein et al., 1998), lutropin receptor (Nakamura & Ascoli, 1999), leukotriene B4 receptor (Gaudreau et al., 2004), chemokine receptor CCR5 (Kraft et al., 2001), and a few others. In many cases the dileucine motif was shown to regulate receptor internalization, sorting, or membrane targeting. Other examples are interaction motifs for G-protein-coupled receptor-associated sorting protein (GASP), which has not been precisely structurally defined (Whistler et al., 2002a), N-ethylmaleimide-sensitive fusion protein (NSF) (Cong et al., 2001), and PDZ domain interaction motifs (Parker et al., 2003). GASP binding was documented for the C-termini of delta opioid, D4 dopamine, as well as β2- and α2B-adrenergic receptors (although the effect of this interaction on lysosomal targeting of only one receptor, delta opioid, was demonstrated) (Whistler et al., 2002a), the NSF interaction motif is localized to the last 3 amino acids of the b2AR (Cong et al., 2001), whereas PDZ domain interaction was shown for the 5-HT2C receptor (Parker et al., 2003). It is likely that other trafficking-related motifs in GPCRs remain to be discovered.
As discussed above, arrestin is not big enough to cover all of the cytoplasmic elements of the great majority of GPCRs. Therefore, even in the complex other structural motifs regulating receptor trafficking are apparently exposed and interact with their targets. In the ensuing competition between different trafficking motifs in the receptor itself and in bound arrestin, “pulling” the complex in different directions, arrestin does not always win. Therefore, arrestin binding does not predetermine the internalization route that the complex would follow.
The m2 muscarinic receptor is not the only member of the family that can internalize via different pathways. Chemokine receptor CCR5 can internalize in a phosphorylation- and arrestin-dependent, as well as independent, fashion (Kraft et al., 2001). Full-length A2B adenosine receptor internalizes via an arrestin-dependent pathway, but deep truncation of its C-terminus redirects it to an arrestin-independent route (Matharu et al., 2001). The cysteinyl leukotriene type 1 receptor internalizes normally in mouse embryonic fibroblasts in which both non-visual arrestins are knocked out, yet arrestin overexpression enhances its internalization (Naik et al., 2005), apparently linking it to an additional arrestin-dependent pathway, similar to the situation with the m2 mAChR (Pals-Rylaarsdam et al., 1997). The metabotropic glutamate receptor mGluR1a undergoes extensive constitutive agonist-independent internalization that does not involve arrestins, yet its internalization in response to agonist is selectively mediated by arrestin2 (Dale et al., 2001). In contrast, spontaneous internalization of the α1A-adrenoreceptor is arrestin dependent, and this receptor travels with bound arrestin all the way to the recycling endosomes (Pediani et al., 2005). WT serotonin 5-HT4 receptor internalizes via an arrestin-dependent pathway in colliculi neurons naturally expressing it, as well as in COS-7 and HEK293 cells, but the deletion of the main S/T cluster phosphorylated by GRK2 re-directs the receptor to an arrestin-independent pathway and actually increases the rate of its internalization (Barthet et al., 2005).
Not all receptors bind arrestins under usual circumstances, even though they appear to be structurally equipped for the interaction. In human airway smooth muscle, prostaglandin receptor EP2 does not recruit arrestins upon stimulation, does not internalize, and does not demonstrate any effects of arrestins on signaling (Penn et al., 2001). Nonetheless, the phosphorylation-independent arrestin2 mutant R169E induced marked desensitization of EP2 signaling and enabled substantial receptor internalization (Penn et al., 2001). The same arrestin2(R169E) mutant was found to induce agonist-independent internalization of the serotonin 5-HT2A receptor, which was previously considered arrestin insensitive (Gray et al., 2003).
Thus, arrestin binding functionally “equips” the receptor to interact with the internalization machinery of the coated pit but does not necessarily direct it to this pathway. The complex interplay of trafficking signals localized on bound arrestin and the receptor itself along with the availability of various internalization pathways in a particular cell type determine the actual route the receptor takes. Apparently, most (if not all) GPCRs have the structural potential to interact with arrestins as well as to internalize via more than one pathway, arrestin-dependent and -independent.
4.2. The properties of the arrestin–receptor complex and the fate of the internalized receptor
The internalized receptor can be deactivated, dephosphorylated, and recycled back to the plasma membrane, or it can be transported to lysosomes for degradation. Some receptors, like protease-activated receptors where the tethered ligand exposed by proteolysis serves as an agonist, cannot be deactivated and therefore they are “disposable” by design. The great majority of GPCRs can actually be either recycled or destroyed, and it is far from clear how the cells “decide” what to do with a particular receptor molecule. However, there is a growing body of evidence suggesting that the properties of the receptor–arrestin complex play a significant role in determining the route the receptor travels inside the cell and its ultimate fate.
The stability of different arrestin–receptor complexes varies. Extensive use of arrestin-GFP fusions revealed that the activation of most GPCRs induces rapid arrestin translocation to the plasma membrane, indicative of its binding to the receptor (Barak et al., 1997). After that, the scenario can be quite different and largely depends on the receptor studied. Some GPCRs, such as the b2AR, D1 dopamine, and endothelin type A receptors release bound arrestin near the plasma membrane, apparently soon after internalization, whereas others, including angiotensin II type 1A (AT1A) or neurotensin receptors, travel with bound arrestins inside the cell (Zhang et al., 1999). The exchange of the C-termini between the b2AR and AT1A reverses this pattern, suggesting that this receptor element largely determines the stability of the complex (Zhang et al., 1999). The difference in complex stability determines the rate of receptor resensitization: the exchange of the C-tail between the b2AR that recycles and resensitizes rapidly and the vasopressin V2 receptor that keeps bound arrestin longer and recycles and resensitizes slowly, switches the pattern of their trafficking and the rate of their dephosphorylation, recycling, and resensitization (Oakley et al., 1999). Similarly, the exchange of the C-termini between 2 vasopressin receptors, V1a that recycles rapidly via peripheral endosomes, and V2 that moves slowly via the perinuclear recycling compartment reversed their trafficking pattern and the rate of their recycling (Innamorati et al., 2001).
Based on the relative affinity for different arrestins and the stability of the arrestin–receptor complex, GPCRs were divided into 2 classes (Oakley et al., 2000). Class A receptors (b2AR, mu-opioid, endothelin type A, dopamine D1, and α1B-adrenergic) bind arrestin3 with higher affinity than arrestin2 and do not interact with visual arrestin. These receptors also tend to release arrestin soon after internalization and recycle rapidly. Class B receptors (AT1A, neurotensin receptor 1, vasopressin V2, TRHR1, and NK1) bind both non-visual arrestins equally well, interact with visual arrestin, and tend to internalize with bound arrestin and recycle more slowly (Oakley et al., 1999; Zhang et al., 1999; Oakley et al., 2000). Obviously, a number of GPCRs do not fit into this classification. For example, the metabotropic glutamate receptor 1a interacts selectively with arrestin2, but not with arrestin3 (Dale et al., 2001), somatostatin receptor 2A internalizes along with arrestin, yet is recycled and resensitized rapidly, whereas somatostatin receptor 3 releases arrestin near the plasma membrane, yet does not recycle rapidly and is largely degraded after internalization (Tulipano et al., 2004). Nonetheless it is clear that the stability of the arrestin–receptor complex significantly affects the trafficking pattern of different GPCRs.
These experiments do not explain how the choice between the degradation and recycling is made for the same receptor in the same cell. Even the b2AR that is rapidly internalized and recycled even in the presence of agonist (Morrison et al., 1996) becomes progressively degraded upon stimulation for many hours (Pan et al., 2003). Several studies using various arrestin mutants suggest that the stability of the complex may play a role in this choice, as well as in arrestin-dependent receptor trafficking beyond internalization. Arrestin release must precede receptor dephosphorylation, leaving the loss of active receptor conformation due to agonist dissociation in acidic endosomes as the only logical signal for arrestin to get off. Then the receptor has to undergo several cycles of dephosphorylation (because receptor multi-phosphorylation is required for arrestin binding) before it can finally emerge in a recycling-competent fully dephosphorylated form (Hsieh et al., 1999). However, both non-visual arrestins poorly discriminate between phosphorylated active and inactive forms of the receptor (Fig. 10), suggesting that arrestin release may be rate limiting in this process. On the other hand, the binding of phosphorylation-independent mutants to the unphosphorylated receptor is strictly activation dependent (Fig. 16A), so that in this case receptor deactivation would be immediately followed by arrestin release. Moreover, because both phosphorylation-independent arrestins and GRKs compete for the same functional form of the receptor, R*, these mutants directly inhibit receptor phosphorylation (Fig. 16B) (Pan et al., 2003). Thus, if the expression of the mutant arrestin is high enough to out-compete endogenous wild-type arrestins and GRKs, the receptor primarily internalizes in an unphosphorylated form in complex with the constitutively active arrestin. As could be expected, the b2AR internalizes in these complexes at a normal rate but recycles much faster than the same receptor internalized in “normal” phosphoreceptor–arrestin complexes (Pan et al., 2003). It turned out that this accelerated cycling prevents receptor down-regulation even after very long (up to 24 h) agonist exposure (Fig. 16C). Phosphorylation-independent arrestin mutants bind phosphorylated receptor at least as well as wild-type arrestin, and in this situation their ability to discriminate between active and inactive phosphoreceptor is, if anything, even worse than that of wild-type arrestin (Fig. 16A). Thus, one can predict that overexpression of a GRK along with the mutant would ensure that most receptors get phosphorylated before they have a chance to encounter the “super-arrestin” mutant. Indeed, overexpression of GRK2 along with mutant arrestin returns the situation back to normal, “rescuing” receptor down-regulation (Fig. 16C) (Pan et al., 2003). Thus, the formation of a less stable complex with exactly the same receptor tips the balance towards recycling, whereas the stabilization of the complex favors receptor degradation. This conclusion is in remarkable agreement with the correlation observed for a variety of different GPCRs that belong to classes A or B (Oakley et al., 1999; Zhang et al., 1999).
Fig. 16.

The formation of a transient arrestin–receptor complex prevents receptor down-regulation. In contrast to WT arrestin2, R169E mutant binds with high affinity to unphosphorylated b2AR* (A), thereby competing with GRK2 and inhibiting receptor phosphorylation (B). The expression of the R169E mutant in HEK cells at levels sufficiently high to out-compete GRKs and endogenous arrestins and to form complexes with unphosphorylated b2AR* prevents the loss of the receptor even after very long agonist exposure, in contrast to the overexpression of WT arrestin2 (C). However, concomitant overexpression of GRK2 (that ensures that the receptor is phosphorylated faster, so that both WT and R169E arrestins bind phosphorylated receptor) “rescues” receptor down-regulation (C). The R169E mutant facilitates receptor cycling in two ways. First, deactivation of P-b2AR (due to agonist loss in the endosome) does not dramatically reduce arrestin binding, whereas deactivation of unphosphorylated b2AR sharply reduces arrestin binding, so that upon internalization the mutant dissociates from the receptor faster. Second, after the release of WT arrestin the emerging multi-phosphorylated receptor must be fully dephosphorylated before it can be recycled, whereas the dissociation of R169E immediately releases the unphosphorylated recycling-competent receptor. Thus, the decrease in stability of the arrestin–receptor complex facilitates receptor cycling, and rapid recycling, in its turn, prevents receptor down-regulation (Pan et al., 2003).
This conclusion is also supported by studies where a different type of arrestin mutant was used to make the arrestin–receptor complex unusually stable. Rapid internalization of the b2AR requires transient ubiquitination of receptor-bound arrestin3, which is accomplished by E3 ubiquitin ligase Mdm2, that directly interacts with arrestin (Shenoy et al., 2001). The time course of arrestin3 ubiquitination and de-ubiquitination is consistent with the idea that arrestin carries a ubiquitin moiety as long as it is bound to the receptor (Shenoy et al., 2001). Indeed, the activation of the V2 vasopressin receptor, which stays in complex with arrestin longer than b2AR, results in more prolonged arrestin ubiquitination (Shenoy & Lefkowitz, 2003). Switching the b2AR and V2 C-termini, which to a large extent determine the stability of the complex (Oakley et al., 1999), also reverses the kinetics of arrestin ubiquitination induced by the activation of these receptors (Shenoy & Lefkowitz, 2003). An arrestin3–ubiquitin fusion protein that cannot be de-ubiquitinated in the cell stays in complex with the b2AR just as long as wild-type arrestin remains bound to class B receptors. Most importantly, the expression of the arrestin–ubiquitin fusion enhances the degradation of the b2AR (Shenoy & Lefkowitz, 2003). Thus, arrestin mutations that make b2AR–arrestin complex more transient prevent receptor degradation (Pan et al., 2003), whereas mutants forming unusually stable complexes shift the equilibrium towards degradation (Shenoy & Lefkowitz, 2003), suggesting that the stability of the complex plays a role in the determination of the fate of internalized receptor. One of the mechanisms whereby this can be accomplished appears to be arrestin-mediated recruitment of enzymes that ubiquitinate the receptor itself, as has been demonstrated for the b2AR (Shenoy et al., 2001) and the V2 vasopressin receptor (Martin et al., 2003).
The properties of the arrestin–receptor complex also affect post-endocytic receptor trafficking in other ways. An example of this is the N-formyl peptide receptor, which internalizes in an arrestin-independent fashion but requires arrestins for recycling (Vines et al., 2003). In normal cells this receptor recycles relatively fast, whereas in arrestin-deficient cells the receptor accumulates in the perinuclear recycling compartment and fails to recycle. The recycling can be rescued by the overexpression of arrestin2 or 3 (Vines et al., 2003). The recycling of this receptor is inhibited by the expression of a constitutively active arrestin2(I386A, V387A, F388A) mutant that binds the phosphorylated receptor with higher affinity than wild-type arrestin (Key et al., 2005). Importantly, the recycling was restored when receptor was only partially phosphorylated (Key et al., 2005), which reduces arrestin affinity for the receptor. Thus, excessively stable arrestin association stops the receptor in its tracks, inhibiting recycling of the N-formyl peptide receptor.
Thus, the stability of the arrestin–receptor complex is an important factor, which determines the receptor trafficking pattern and the ultimate fate of the internalized receptor. However, most of the data leading to this conclusion were generated using various receptor and arrestin mutants and chimeras, that is, the tools that the cell does not have at its disposal, raising the question whether the cell can actually regulate the properties of the arrestin–receptor complex by some normal, natural means. Biochemical studies of the mechanisms of the arrestin–receptor interaction suggest at least one relatively simple method the cell can employ to change the stability of the complex. The properties of the complex and the biological consequences of its formation depend to a large extent on the nature of the phosphorylatable receptor elements (Oakley et al., 1999; Zhang et al., 1999; Innamorati et al., 2001) and the actual level of their phosphorylation (Oakley et al., 2001; Pan et al., 2003; Key et al., 2005). Virtually all receptors have many more GRK phosphorylation sites than the minimum of 2 required for the activation of the arrestin phosphate sensor to induce arrestin transition into its high-affinity receptor-binding state. When arrestin binds, it physically covers the phosphorylation sites, whether they are “hit” by GRK or not, preventing further receptor phosphorylation (Fig. 16B) (Pan et al., 2003). Because active receptor encounters with GRK and arrestin are stochastic events governed by the law of mass action, in a cell that has relatively little GRK activity and high levels of arrestins most receptors will meet and bind arrestin as soon as they have the minimum requisite number of phosphates attached. Conversely, in cells expressing a lot of GRKs and relatively little arrestin, most receptors will be phosphorylated many times before they encounter and bind an arrestin molecule. The first situation would mostly generate arrestin–receptor complexes with the lowest possible stability, whereas in the latter case more stable complexes would emerge in abundance. The functionally relevant differences would persist even beyond the eventual dissociation of arrestin because the receptor that emerges would have varying numbers of attached phosphates requiring different time for full dephosphorylation.
This type of possible regulation remains inexplicably underappreciated, even though emerging evidence suggests that it may actually be used in vivo, especially by neurons, that is, the cells that constantly regulate and integrate complex networks of signaling pathways. As a rule, the total GRK/total arrestin ratio in neurons is fairly high, favoring receptor multi-phosphorylation, but there are intriguing variations between different brain regions (Gurevich et al., 2004). Complex changes in the expression levels of both non-visual arrestins and GRKs 2, 3, 5, and 6 during neural development have been documented (Penela et al., 2000; Sefton et al., 2000; Gurevich et al., 2002; Gurevich et al., 2004). A dramatic 7- to 10-fold increase in arrestin2 expression during neuronal maturation in the rat brain that is faithfully recapitulated in the process of the differentiation of neuronal precursors in vitro is particularly striking (Gurevich et al., 2004). Profound changes in arrestin2 and GRK6 expression levels in the brain have also been reported in the monkey model of Parkinson’s disease; importantly, these changes were reversed by L-DOPA treatment (Bezard et al., 2005). The changes in the relative levels of GRK2/3 and GRK5/6 groups yield arrestin–receptor complexes with dramatically different functions (Kim et al., 2005a; Ren et al., 2005). Relatively low levels of GRK2 expression are sufficient to promote arrestin-dependent internalization of the serotonin 5-HT4 receptor, but substantially higher GRK2 expression (similar to that in colliculi neurons that express it naturally) is required for its rapid desensitization (in the latter process GRK2 seems to function independently of its kinase activity because its K220R kinase-dead mutant works just as well as WT GRK2) (Barthet et al., 2005). All these data indicate that changes in the expression of arrestins and GRKs are important and are probably widespread regulatory mechanisms. Our mechanistic analysis suggests that these changes (among other things) may regulate the properties of the arrestin–receptor complex and the functional consequences of its formation.
4.3. Constitutively desensitized receptor mutants in congenital disorders
Rhodopsin, with its covalently bound inverse agonist, 11-cis-retinal, is unique among GPCRs in that its noise level (constitutive activity) is virtually nil: based on the rod photoreceptor noise level, it has been calculated that one rhodopsin molecule undergoes spontaneous activation once in 2000 years (Burns & Baylor, 2001). Most GPCRs have detectable levels of constitutive (agonist-independent) activity (Seifert & Wenzel-Seifert, 2002). Certain naturally occurring mutations in different receptors enhancing their constitutive activity have been shown to underlie a variety of human congenital disorders, ranging from stationary night blindness to various forms of cancer (recently reviewed in Seifert & Wenzel-Seifert, 2002). Because receptor conformations preferred by G proteins, GRKs, and arrestins are in most cases the same (or very similar), on purely theoretical grounds one would expect these constitutively active mutants to be subject to GRK- and arrestin-dependent desensitization. The first experimental proof that this is the case was found in the visual system: several rhodopsin mutants that constitutively activate transducin in biochemical assays and cause night blindness or retinal degeneration in humans were shown to be constitutively phosphorylated by rhodopsin kinase and bind visual arrestin, suggesting that the disease phenotype may be the result of either their uncontrolled signaling or persistent desensitization (Rim & Oprian, 1995). On the same lines, two forms of constitutively active luteinizing hormone receptor were found to internalize faster than the wild-type receptor via an arrestin-dependent pathway (Bradbury & Menon, 1999). The most intriguing discovery was the finding that the naturally occurring vasopressin receptor mutation R137H associated with familial nephrogenic diabetes insipidus that was originally described as loss-of-function actually induces constitutive arrestin-mediated desensitization. In contrast to the wild-type vasopressin receptor, the “non-signaling” R137H receptor is phosphorylated and sequestered in arrestin-associated intracellular vesicles even in the absence of agonist. Eliminating molecular determinants on the receptor that promote high-affinity arrestin–receptor interaction reestablishes plasma membrane localization and the ability of the mutated receptors to signal (Barak et al., 2001). Thus, in the case of constitutively active GPCRs, persistent desensitization can overwhelm persistent signaling and directly contribute to the etiology of a mutation-induced disease. This finding suggests that some of the other disease-associated GPCR mutants that were classified as “non-signaling” in cell-based assays need to be re-examined biochemically for possible constitutive activity and consequent persistent desensitization in cells. It also indicates that pharmacological targeting of the desensitization machinery, GRKs and/or arrestins, may be a viable therapeutic strategy.
4.4. Non-receptor-binding partners of arrestin proteins
As if the binding to hundreds of different GPCRs and a few unrelated receptors were not enough, arrestins also interact with a fairly diverse group of non-receptor binding partners (recently reviewed in Lefkowitz & Shenoy, 2005). The ever-growing list of proteins that bind arrestins includes trafficking-related partners (clathrin, adaptin2, NSF) and a variety of signaling proteins: small G proteins and guanine nucleotide exchange factors, protein kinases, ubiquitin ligase, etc. Obviously, arrestin-mediated signaling can only be receptor activation-dependent if receptor-bound arrestin interacts with the downstream partner with higher affinity than free arrestin, which is always present in the cell (Gurevich & Gurevich, 2003). Thus, the most important question from a functional viewpoint is which of these partners bind free arrestins in the cytoplasm, which preferentially interact with receptor-bound arrestins in the active conformation, and which bind to both functional states of arrestin with comparable affinity. With the exception of clathrin (Goodman et al., 1996; Kim & Benovic, 2002; Xiao et al., 2004), we do not know the answer to these questions because in most cases the interactions were demonstrated by co-immunoprecipitation and other inherently non-quantitative methods. Careful biochemical experiments with purified proteins reconstructing putative arrestin-containing complexes necessary to measure the affinity of any interaction partner for free arrestin in the basal conformation and for active receptor-bound arrestin have yet to be performed. However, several lines of evidence suggest that virtually any arrestin partner binds both functional forms of arrestin, albeit likely with different affinities. Even the arrestin–clathrin interaction was first discovered using free arrestin (Goodman et al., 1996) and its facilitation due to the destabilization of the basal arrestin conformation by various means was demonstrated only later (Kim & Benovic, 2002; Xiao et al., 2004). However, it is clear that the affinity of receptor-bound arrestin for clathrin must be much higher than that of free arrestin, because otherwise overexpressed non-visual arrestins would compete with the receptor–arrestin complexes for clathrin, effectively acting as inhibitors of receptor internalization. In fact, this mechanism is perfectly feasible because the overexpressed arrestin2 C-terminus, which carries clathrin and AP-2 binding sites, effectively acts as a “dominant-negative” arrestin as far as receptor internalization is concerned (Orsini & Benovic, 1998), but WT arrestin does not.
Sometimes the reports that free or receptor-bound arrestin bind a particular partner are perceived as conflicting, as is the case with ARF6 and ARNO (Mukherjee et al., 2000; Claing et al., 2001; Hunzicker-Dunn et al., 2002), whereas it is likely that these partners simply bind both functional forms of arrestin. Another example is JNK3. Arrestin3 was reported to serve as a scaffold for receptor activation-dependent JNK3 phosphoryla-tion by the ASK1-MKK4-JNK3 activation cascade (McDonald et al., 2000). Yet arrestin3 that has a nuclear export signal (NES) in its C-terminus also redistributes JNK3 from the nucleus to the cytoplasm (Scott et al., 2002). Because membrane-bound GPCRs obviously cannot move between the nucleus and the cytoplasm via aqueous nuclear pore where NES-dependent transport operates, this must be the function of free arrestin, suggesting that it also binds JNK3. Similarly, bound non-visual arrestins mobilize ubiquitin ligase Mdm2 to GPCRs where it ubiquitinates the receptor (Shenoy et al., 2001), whereas free arrestins also redistribute Mdm2 from the nucleus to the cytoplasm (Wang et al., 2003).
Mechanistically, the change in arrestin conformation upon receptor binding likely explains an enhanced affinity of receptor-bound arrestin for many non-receptor-binding partners (Gurevich & Gurevich, 2003). However, free arrestin (like any normal protein) is not as rigid as the crystal structure may lead one to believe: it “breathes”, likely “sampling” a number of conformational variations, although for the sake of simplicity we call this whole group of conformational states “basal”. In the process, it may occasionally expose the same sites for its binding partners that become fully exposed in the receptor-bound state (which again is likely a whole family of conformations that we classify as “active”). The destabilization of the basal conformation of free arrestin by mutagenesis (Kim & Benovic, 2002), by the phosphopeptide mimicking the phosphorylated receptor C-terminus, or even by heparin (Xiao et al., 2004), enhances clathrin binding apparently because “loose” arrestin assumes the conformation somewhere in between the basal and active-like more often. It is likely that this would also be true for other binding partners that prefer receptor-bound arrestin, but so far this has never been tested experimentally.
Arrestin elements involved in receptor binding have been fairly well mapped (Fig. 14) (Gurevich & Benovic, 1995; Gurevich et al., 1995; Vishnivetskiy et al., 2000; Vishnivetskiy et al., 2004). In sharp contrast, the binding sites for the non-receptor partners, with the exception of clathrin and AP-2 (Krupnick et al., 1997a; Laporte et al., 1999; Kim & Benovic, 2002), were either identified imprecisely or simply remain unknown. For example, proline-rich motifs in arrestin2 were shown to participate in its interaction with the SH3 domain of c-Src (Luttrell et al., 1999), yet both non-visual arrestins bind Src comparably, apparently via its catalytic domain (Miller et al., 2000). The binding site for JNK3 was tentatively localized to the RRSLHL motif in rat arrestin3 (Miller et al., 2001), yet this sequence is not conserved in arrestin3 in other mammals. To achieve a mechanistic understanding of arrestin interaction with various signaling proteins and develop tools affecting these interactions, this gap needs to be filled. The most straightforward approach to achieving this goal is the use of direct biochemical and biophysical methods to study the complexes reconstructed under carefully controlled conditions with purified proteins, both wild-type and targeted mutants.
4.5. ERK activation and beyond
Cells have an enormous number of pathways leading to ERK activation. Even the subset of these pathways initiated by active GPCRs is relatively large: ERK activation can be mediated by several heterotrimeric G proteins (Gs via cAMP and PKA, Go via PLCβ and PKC, Gi via its βγ-subunit and Src), by trans-activation of receptor tyrosine kinases, etc. (recently reviewed in Luttrell, 2003). The arrestin-mediated pathway is unique in one important respect: active ERK does not go to the nucleus to phosphorylate its usual transcription factor substrates and induce proliferation but stays in the cytoplasm instead (Tohgo et al., 2002; Luttrell, 2003; Tohgo et al., 2003). Interestingly, one of the known cytoplasmic ERK substrates is arrestin2 itself, where the phosphorylation of Ser412 in its C-terminus reduces its affinity for the internalization machinery (Lin et al., 1997). Free arrestin2 is constitutively phosphorylated, and the phosphate is apparently removed upon its binding to the receptor. Its subsequent phosphorylation by ERK2 facilitates the dissociation of the arrestin–receptor complex from clathrin, but the phosphorylation status of Ser412 does not affect receptor interaction (Lin et al., 1997). Another interesting ERK substrate is GRK2 (Pitcher et al., 1999; Elorza et al., 2000). ERK phosphorylates the C-terminal Ser670 in GRK2, thereby attenuating its Gβγ-mediated mobilization to the membrane and reducing its activity towards the receptor (Pitcher et al., 1999; Elorza et al., 2000). In essence, ERK stops the events that lead to its arrestin-dependent activation in the first place, making this a typical negative feedback loop. ERK also phosphorylates Gα-interacting protein (GAIP), which is a regulator of G protein signaling (RGS) (Ogier-Denis et al., 2000). Its phosphorylation at Ser151 in the RGS domain enhances GAIP-dependent acceleration of the rate of GTP hydrolysis by the α-subunit of the G protein Gi3 (Ogier-Denis et al., 2000). Interestingly, all these ERK substrates are regulators of GPCR signaling. Another cytoplasmic ERK substrate, tyrosine hydroxylase (Salvatore et al., 2001), is the rate-limiting enzyme in the synthesis of catecholamines, which are endogenous agonists of several GPCRs. Still, the relative paucity of known cytoplasmic ERK substrates makes it very likely that quite a few more will be discovered before we can fully appreciate the functional significance of the retention of active ERK in the cytoplasm.
Theoretically, one can envision two mechanisms responsible for the cytoplasmic retention of active ERK. Arrestin3 with its native nuclear export signal (NES) was shown to bring some of its interaction partners, such as JNK3 (Scott et al., 2002) and Mdm2 (Wang et al., 2003), out of the nucleus, so that (assuming that in its free state it also binds ERK) it could redistribute active ERK to the cytoplasm. However, this seems unlikely, as arrestin overexpression is necessary to see this effect, and upon receptor activation the concentration of free arrestin3 in the cytoplasm actually decreases (Barak et al., 1997). Besides, this mechanism does not explain why it matters how ERK was activated; free arrestin could redistribute ERK activated by any pathway. The formation of stable receptor–arrestin complexes facilitates ERK activation and the retention of active ERK in the cytoplasm (Tohgo et al., 2003). ERK has been repeatedly detected by co-immunoprecipitation in complexes containing arrestin and receptor (DeFea et al., 2000a, 2000b; Luttrell et al., 2001). Thus, the most likely mechanism of retention is that ERK activated on the scaffold of the arrestin–receptor complex simply stays bound. One important corollary of such a mechanism is that the c-Raf1-MEK1-ERK1/2 cascade assembled with the help of receptor-bound arrestin does not work as a catalyst sequentially activating and releasing multiple ERK molecules. If the complex activates only 1 molecule of ERK, then during arrestin-assisted ERK activation there cannot be any amplification of the signal. Another implication is that for the system to work this way, the affinity of receptor-bound arrestin for active ERK must be at least as high as that for the inactive ERK it presumably binds first. This raises the question of how the bound ERK (active or inactive) eventually leaves arrestin, and when this happens.
4.6. Scaffolding and its limitations
Arrestins are relatively small proteins (about 80×35×40 Å) with a total exposed surface of less than 10,000 Å2 (Hirsch et al., 1999; Han et al., 2001; Sutton et al., 2005), a significant proportion of which is occupied by the GPCR in the arrestin–receptor complex (Figs. 9 and 14) (Gurevich & Gurevich, 2004; Vishnivetskiy et al., 2004), with which Src, c-Raf1, MEK1, ERK2, ASK1, JNK3, and many other arrestin-binding partners also interact (reviewed in Gurevich & Gurevich, 2003; Lefkowitz & Shenoy, 2005). Most protein–protein interaction surfaces are 500–2000 Å2, hence it is unlikely that more than 4–6 proteins can be bound to a single arrestin–receptor complex simultaneously. Therefore, the sheer number of arrestin-binding partners and their size relative to arrestin suggests that some of them must compete with each other for the limited number of “parking spaces” available on the arrestin molecule (Fig. 17). This suggests that there has to be a certain sequence of arrestin interactions with various non-receptor partners following its binding to the GPCR. Because receptor internalization is required for ERK activation in some cases (Daaka et al., 1998; Ignatova et al., 1999; DeFea et al., 2000a; Luttrell et al., 2001) but is not in many others (Budd et al., 1999; Schramm & Limbird, 1999; Kramer & Simon, 2000), the binding of clathrin+AP-2 and that of other arrestin partners is likely mutually independent. The detachment of the fairly long arrestin C-tail carrying both clathrin and AP-2 interaction sites upon arrestin binding to the receptor is perfectly suited to move these sites far enough from the rest of the arrestin molecule and the binding sites for the other partners to achieve this independence. c-Src likely binds early on, because it has to phosphorylate dynamin to promote receptor internalization (Perry & Lefkowitz, 2002), but it is not known how long it stays in the complex. Even though Src and ERK have been found associated with immunoprecipitated arrestin–receptor complexes simultaneously (DeFea et al., 2000a), this does not mean that both proteins are present in the same complex. Because free arrestin in its basal conformation obviously interacts with some of the non-receptor partners, such as ubiquitin ligase Mdm2 and JNK3 (Scott et al., 2002; Wang et al., 2003), arrestin might come to the receptor already “loaded” by one or more other proteins. It is unclear whether this is the case and whether the “company” arrestin brings to the receptor affects the functional consequences of its binding.
Fig. 17.

Relative sizes of the receptor, arrestin, and its non-receptor-binding partners. Crystal structures of rhodopsin (Okada et al., 2004), arrestin (Hirsch et al., 1999) (shown from the “back”, that is, from the side that does not interact with receptors), the arrestin-binding N-terminal domain of clathrin heavy chain (ter Haar et al., 1998), the arrestin-binding appendage of β2-subunit of the AP-2 clathrin adaptor complex (Owen et al., 2000; Traub, 2003), ARF6 (Shiba et al., 2003), c-Src (Xu et al., 1997b), JNK3 (Xie et al., 1998), MEK1 (Ohren et al., 2004), Mdm2 (Grasberger et al., 2005), ERK2 (Canagarajah et al., 1997), the kinase domain of Raf (Wan et al., 2004), the catalytic domain of phosphodiesterase PDE4 (Huai et al., 2003), and the sec7 (exchange activity) domain of ARNO (Cherfils et al., 1998). The structures are shown to scale. The relative sizes of arrestin and its partners suggest that only a few of them can be bound to a single receptor-associated arrestin simultaneously.
Unless a single arrestin molecule can simultaneously scaffold the c-Raf1-MEK1-ERK1/2 and ASK1-MKK4-JNK3 cascades (which, given the relative sizes of arrestin and these kinases, seems virtually impossible; Fig. 17), there also must be inter-dependence of the binding of certain partners. For example, if arrestin with bound JNK3 or ERK1/2 were equally likely to bind c-Raf1 and ASK1, a lot of resulting complexes would be unproductive. Therefore, it would make sense biologically if the binding of JNK3 made the binding of ASK1 more likely (and/or vice versa), whereas the binding of ERK1/2 should facilitate the binding of c-Raf1 at the expense of ASK1. The mechanistic information regarding these interactions is sadly missing.
Another important and unclear issue is when these partners ultimately dissociate from arrestin and what makes them do so. The release of the partners that preferentially interact with the arrestin–receptor complex is likely prompted by arrestin dissociation from the receptor and its return to its basal conformation (Gurevich & Gurevich, 2003). However, in the case of JNK3, which clearly binds free arrestin with high enough affinity to be removed by it from the nucleus (Scott et al., 2002), and some other proteins, the situation is not that simple. Just as receptor activation and phosphorylation make arrestin bind and receptor deactivation prompts arrestin to leave, there must be regulatory mechanisms that “tell” other arrestin partners when to bind and when to go away. Experimental studies of the regulation of arrestin interactions with various signaling proteins are long overdue.
4.7. Competition and cooperation
Every cell type that has been carefully studied has multiple GPCR subtypes (human airway smooth muscle is one well-characterized example; Penn et al., 2001). Yet the pool of GRKs and arrestins in the cell appears to be common and shared by all. Thus, it is not surprising that a receptor that forms tight complexes with arrestin can deplete this pool, leaving other receptors with little arrestin to bind. This competition between receptors for arrestin was first demonstrated in a heterologous system expressing the b2AR and vasopressin V2 receptors at similar levels (Klein et al., 2001). Activation of the V2 receptor dramatically inhibited isoproter-enol-induced internalization of the b2AR. In contrast, the activation of the b2AR, which releases arrestin soon after internalization, did not affect agonist-induced trafficking of V2. Overexpression of either non-visual arrestin, expanding the pool, abolished vasopressin-induced inhibition of b2AR internalization, as did the mutations in the V2 receptor that prevented the formation of its stable complexes with arrestin (Klein et al., 2001). Thus, the 2 receptors actually compete for the limited pool of arrestin, and the one that forms more stable complexes with it “wins”, thereby affecting the trafficking (and presumably desensitization and phosphorylation level) of the other. Similar observations were later reported with neurokinin receptors NK1 and NK3; active NK1 forms stable complexes with arrestin, inhibiting internalization and desensitization of the co-expressed NK3, whereas NK3, which does not traffic with arrestin, does not take enough of it out of circulation to affect NK1 regulation (Schmidlin et al., 2002). Most importantly, the same phenomenon was observed in enteric neurons that naturally co-express NK1 and NK3 (Schmidlin et al., 2002). Thus, the size of the arrestin pool accessible to one receptor can be regulated not only by arrestin expression, but also by the rate of its utilization by other “competing” GPCRs. The importance of the size of the arrestin pool for receptor regulation may be one of the reasons for the dramatic increase in arrestin2 expression during neuronal maturation (Gurevich et al., 2002; Gurevich et al., 2004), that is, at the time when the expression of various GPCRs also increases.
A number of proteins other than the “usual suspects” (GRKs, arrestins, and G proteins) bind to the cytoplasmic side of many GPCRs, suggesting that if arrestins cover a substantial part of a receptor’s cytoplasmic tip they should compete with other receptor-binding partners as well. The third cytoplasmic loop of the α2A-adrenoceptor interacts with spinophilin (Richman et al., 2001) and 14-3-3ζ (Prezeau et al., 1999). Arrestin has higher affinity for the unphosphorylated i3 loop than 14-3-3ζ (Wang & Limbird, 2002). Agonist binding enhances α2A-adrenoceptor association with spinophilin, and arrestin actually competes with it. The elimination of GRK phosphorylation sites from the i3 loop tips the balance in favor of spinophilin (Wang & Limbird, 2002). This competition was shown to be functionally relevant in vivo (Wang et al., 2004). Spinophilin antagonizes arrestin effects on receptor signaling and trafficking, reducing arrestin-stabilized receptor phosphorylation, receptor endocytosis, and the stimulation of mitogen-activated protein kinases. Spinophilin knockout mice are more sensitive than wild-type animals to sedation elicited by stimulation of α2A-adrenoceptors, whereas arrestin3 knockout mice are more resistant (Wang et al., 2004). Sometimes arrestin competition with other receptor-binding proteins depends on the functional state of the receptor. The D3 dopamine receptor in its resting state was found to form complexes containing both arrestin3 and filamin (Kim et al., 2005b). The reduction of GRK2/3 activity enhances receptor association with both proteins, whereas an increase of receptor phosphorylation due to agonist activation or higher GRK2/3 expression enhances arrestin3 association with the D3 receptor at the expense of filamin (Kim et al., 2005b).
Certain biological responses require the cooperation of receptor signaling via both G proteins and arrestins. Angio-tensin II receptor type 1A (AT1A) stimulation results in the activation of the small G protein RhoA, which leads to the reorganization of stress fibers (Barnes et al., 2005). Whereas neither arrestin2 mobilization to the active receptor nor Gq/11 activation alone is sufficient to activate RhoA, the concurrent recruitment of arrestin2 and activation of Gq/11 leads to full activation of RhoA and to the subsequent stress fiber formation (Barnes et al., 2005). In the case of chemotaxis induced by the AT1A receptor, only arrestin-mediated signaling is required, whereas the same process induced via the LPA receptor requires simultaneous activation of Gi- and arrestin-mediated signaling pathways (Hunton et al., 2005). Thus, even though arrestin binding to the receptor terminates coupling to the G protein, subsequent arrestin-mediated signaling can cooperate with the G protein signaling, dramatically changing the biological outcome of the latter.
5. Conclusions
In the 20 years since the discovery that arrestin binding to rhodopsin is enhanced by rhodopsin phosphorylation (Kuhn et al., 1984), our understanding of arrestin structure and function has improved enormously. In the first 10 years after this seminal discovery, it became increasingly clear that receptor phosphorylation followed by arrestin binding is a common mechanism regulating the signaling of the great majority of GPCRs. Subsequent identification of arrestin and receptor elements involved in the interaction and the solution of the arrestin crystal structure led to a fairly good understanding of the simple and ingenious sequential multi-site binding mechanism that governs arrestin binding to its cognate receptors and subsequent dissociation (which is equally important from a functional viewpoint). Deep understanding of the “inner workings” of arrestin led to the targeted construction of a number of mutants with interesting and useful functional characteristics, in particular phosphorylation-independent “constitutively active” versions of visual and both non-visual arrestins that bind active phosphorylated and unphosphorylated receptors with comparable affinity and dramatically change the trafficking patterns of various receptors. All of these developments consolidated the perception that arrestin is just what its name implies: a terminator of G-protein-mediated signaling.
In a parallel development, since clathrin was identified as the first non-receptor interaction partner of arrestins in 1996 (Goodman et al., 1996), additional arrestin-binding proteins have been identified at a rapid pace, such that in less than 10 years more than 20 other proteins were reported to bind arrestins. In this direction, the description of the phenomenology far outpaces the mechanistic understanding of these interactions, hence we do not really know where many of these partners bind and what makes them bind or dissociate at a particular phase of the arrestin functional cycle. This lack of mechanistic information hampers our understanding of the regulation of these interactions, including possible cooperativity of the binding and/or competition between various proteins for arrestin. The reconstruction of the putative signaling complexes that include arrestin with and without bound receptor in vitro from purified components is necessary to get definitive answers regarding the actual sequence of events in arrestin-mediated signaling cascades and to fully appreciate the biological significance of arrestin’s ability to interact with so many different proteins.
It is entirely possible that arrestins emerged in evolution as signaling adaptors (rather than terminators), directing receptor signaling to particular pathways, essentially functioning as analogues of the Ste5 scaffolding protein in yeast that couples the α-mating factor receptor to the MAP kinase cascade (notably, yeast have a GPCR but do not have arrestin homologues). Because proteins that bind active GPCRs inevitably compete with each other due to the limitations of available space on the cytoplasmic surface of the receptor, arrestins may have started “moonlighting” as inhibitors of G-protein-mediated signaling. This latter function of arrestin appears to be the main one only in the visual system, where the selectivity of the highly specialized rod arrestin for the active phosphorylated receptor was ultimately perfected to levels unattainable for the other members of the family, which needed to preserve their other important functions. Because arrestin and its functional role was first discovered in rod photoreceptors, which are arguably the most unusually designed and highly specialized cells in the body, we may have read the evolution of arrestin backwards, from its “arresting” capability to its “roots” as an ubiquitous adaptor and facilitator of signaling.
It is clear that arrestins represent an important intersection in the intricate network of signaling pathways in the cell, which makes them an inviting target for experimental and therapeutic manipulation. The modular structure of the arrestin molecule, where receptor-binding elements are collected on one side, and the binding sites for the non-receptor partners are apparently localized on the other side, as well as on the conveniently detachable C-tail, is perfect for this kind of manipulation. The identification of the interaction sites used by various arrestin-binding partners will allow the construction of arrestins in which the ability to bind a particular partner can be selectively disrupted or enhanced by targeted mutagenesis, leaving all other functional properties virtually intact. There is no doubt that these “custom-designed” arrestins will be extremely useful in unraveling the biological significance of arrestin-mediated signaling and the mechanisms of its regulation. The identification of arrestin-binding sites for various non-receptor partners will also set the stage for designing peptides and/or small molecule mimics to selectively disrupt individual interactions without affecting the others. These novel tools may be useful for therapeutic intervention in a growing number of disorders associated with congenital or acquired disregulation of GPCR signaling.
Abbreviations
- ARF6
ADP ribosylation factor 6
- ARNO
ARF nucleotide-binding site opener
- AT1A
angiotensin II type 1A
- b2AR
β2-adrenergic receptor
- BRET
bioluminescence resonance energy transfer
- ERK
extracellular signal-regulated kinase
- FPR
N-formyl peptide receptor
- GPCR
G-protein-coupled receptor
- GRK
G-protein-coupled receptor kinase
- m2 mAChR
m2 muscarinic cholinergic receptor
- MOR
mu-opioid receptor
- NK1
neurokinin1 receptor (a.k.a. substance P receptor)
- P-R
inactive phosphoreceptor
- P-R*
phosphorylated ligand-activated receptor
- P-Rh*
phosphorylated light-activated rhodopsin
- P-Rh
dark (inactive) phosphorhodopsin
- R
inactive receptor
- R*
ligand-activated receptor
- Rh
dark (inactive) rhodopsin
- Rh*
light-activated rhodopsin
- TRHR
thyrotropin-releasing hormone receptor
- WT
wild type
References
- Altenbach C, Cai K, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 306–322 extending from helix VII to the palmitoylation sites in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999a;38:7931–7937. doi: 10.1021/bi9900121. [DOI] [PubMed] [Google Scholar]
- Altenbach C, Klein-Seetharaman J, Hwa J, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 59–75 connecting helices I and II in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999b;38:7945–7949. doi: 10.1021/bi990014l. [DOI] [PubMed] [Google Scholar]
- Altenbach C, Cai K, Klein-Seetharaman J, Khorana HG, Hubbell WL. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 65 in helix TM1 and residues in the sequence 306–319 at the cytoplasmic end of helix TM7 and in helix H8. Biochemistry. 2001a;40:15483–15492. doi: 10.1021/bi011546g. [DOI] [PubMed] [Google Scholar]
- Altenbach C, Klein-Seetharaman J, Cai K, Khorana HG, Hubbell WL. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 316 in helix 8 and residues in the sequence 60–75, covering the cytoplasmic end of helices TM1 and TM2 and their connection loop CL1. Biochemistry. 2001b;40:15493–15500. doi: 10.1021/bi011545o. [DOI] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, Caron MG. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- Barak LS, Oakley RH, Laporte SA, Caron MG. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc Nat Acad Sci U S A. 2001;98:93–98. doi: 10.1073/pnas.011303698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes WG, Reiter E, Violin JD, Ren XR, Milligan G, Lefkowitz RJ. beta-Arrestin 1 and Galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem. 2005;280:8041–8050. doi: 10.1074/jbc.M412924200. [DOI] [PubMed] [Google Scholar]
- Barthet G, Gaven F, Framery B, Shinjo K, Nakamura T, Claeysen S, Bockaert J, Dumuis A. Uncoupling and endocytosis of 5-hydroxytryptamine 4 receptors. Distinct molecular events with different GRK2 requirements. J Biol Chem. 2005;280:27924–27934. doi: 10.1074/jbc.M502272200. [DOI] [PubMed] [Google Scholar]
- Bennett TA, Maestas DC, Prossnitz ER. Arrestin binding to the G protein-coupled N-formyl peptide receptor is regulated by the conserved “DRY” sequence. J Biol Chem. 2000;275:24590–24594. doi: 10.1074/jbc.C000314200. [DOI] [PubMed] [Google Scholar]
- Bennett TA, Key TA, Gurevich VV, Neubig R, Prossnitz ER, Sklar LA. Real-time analysis of G protein-coupled receptor reconstitution in a solubilized system. J Biol Chem. 2001;276:22453–22460. doi: 10.1074/jbc.M009679200. [DOI] [PubMed] [Google Scholar]
- Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science. 1989;246:235–240. doi: 10.1126/science.2552582. [DOI] [PubMed] [Google Scholar]
- Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc Nat Acad Sci U S A. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL, Staniszewski C, Mayor FJ, Caron MG, Lefkowitz RJ. beta-Adrenergic receptor kinase. Activity of partial agonists for stimulation of adenylate cyclase correlates with ability to promote receptor phosphorylation. J Biol Chem. 1988;263:3893–3897. [PubMed] [Google Scholar]
- Bezard E, Gross CE, Qin L, Gurevich VV, Benovic JL, Gurevich EV. L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis. 2005;18:323–335. doi: 10.1016/j.nbd.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Bhaskaran RS, Min L, Krishnamurthy H, Ascoli M. Studies with chimeras of the gonadotropin receptors reveal the importance of third intracellular loop threonines on the formation of the receptor/nonvisual arrestin complex. Biochemistry. 2003;42:13950–13959. doi: 10.1021/bi034907w. [DOI] [PubMed] [Google Scholar]
- Bohm A, Gaudet R, Sigler PB. Structural aspects of heterotrimeric G-protein signaling. Curr Opin Biotechnol. 1997;8:480–487. doi: 10.1016/s0958-1669(97)80072-9. [DOI] [PubMed] [Google Scholar]
- Bouvier M, Hausdorff WP, De Blasi A, O’Dowd BF, Kobilka BK, Caron MG, et al. Removal of phosphorylation sites from the beta 2-adrenergic receptor delays onset of agonist-promoted desensitization. Nature. 1988;333:370–373. doi: 10.1038/333370a0. [DOI] [PubMed] [Google Scholar]
- Bradbury FA, Menon KMJ. Evidence that constitutively active luteinizing hormone/human chorionic gonadotropin receptors are rapidly internalized. Biochemistry. 1999;38:8703–8712. doi: 10.1021/bi990169t. [DOI] [PubMed] [Google Scholar]
- Budd Dc, Rae A, Tobin AB. Activation of the mitogen-activated protein kinase pathway by a Gq/11-coupled muscarinic receptor is independent of receptor internalization. J Biol Chem. 1999;274:12355–12360. doi: 10.1074/jbc.274.18.12355. [DOI] [PubMed] [Google Scholar]
- Burns ME, Baylor DA. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu Rev Neurosci. 2001;24:779–805. doi: 10.1146/annurev.neuro.24.1.779. [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- Carter JM, Gurevich VV, Prossnitz ER, Engen JR. Conformational differences between arrestin2 and pre-activated mutants as revealed by hydrogen exchange mass spectrometry. J Mol Biol. 2005;351:865–878. doi: 10.1016/j.jmb.2005.06.048. [DOI] [PubMed] [Google Scholar]
- Celver J, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonine 180 is requred for G protein-coupled receptor kinase 3 and b-arrestin mediated desensitization of the m-opioid receptor in Xenopus oocytes. J Biol Chem. 2001;276:4894–4900. doi: 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- Cen B, Xiong Y, Ma L, Pei G. Direct and differential interaction of beta-arrestins with the intracellular domains of different opioid receptors. Mol Pharmacol. 2001a;59:758–764. doi: 10.1124/mol.59.4.758. [DOI] [PubMed] [Google Scholar]
- Cen B, Yu Q, Guo J, Wu Y, Limng K, Cheng Z, et al. Direct binding of beta-arrestins to two distinct intracellular domains of the delta opioid receptor. J Neurochem. 2001b;76:1887–1894. doi: 10.1046/j.1471-4159.2001.00204.x. [DOI] [PubMed] [Google Scholar]
- Charest PG, Bouvier M. Palmitoylation of the V2 vasopressin receptor carboxyl tail enhances beta-arrestin recruitment leading to efficient receptor endocytosis and ERK1/2 activation. J Biol Chem. 2003;278:41541–41551. doi: 10.1074/jbc.M306589200. [DOI] [PubMed] [Google Scholar]
- Charest PG, Terrillon S, Bouvier M. Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO Rep. 2005;6:334–340. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, et al. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Nat Acad Sci U S A. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Dupre DJ, Le Gouill C, Rola-Pleszczynski M, Stankova J. Agonist-induced internalization of the platelet-activating factor receptor is dependent on arrestins but independent of G protein activation. Role of the C-terminus and the (D/N)PXXY motif. J Biol Chem. 2002;277:7356–7362. doi: 10.1074/jbc.M110058200. [DOI] [PubMed] [Google Scholar]
- Chen W, Kirkbride KC, How T, Nelson CD, Mo J, Frederick JP, et al. Beta-arrestin 2 mediates endocytosis of type III TGF-beta receptor and down-regulation of its signaling. Science. 2003;301:1394–1397. doi: 10.1126/science.1083195. [DOI] [PubMed] [Google Scholar]
- Cherfils J, Menetrey J, Mathieu M, Le Bras G, Robineau S, Beraud-Dufour S, et al. Structure of the Sec7 domain of the Arf exchange factor ARNO. Nature. 1998;392:101–105. doi: 10.1038/32210. [DOI] [PubMed] [Google Scholar]
- Claing A, Chen W, Miller WE, Vitale N, Moss J, Premont RT, et al. beta-Arrestin-mediated ADP-ribosylation factor 6 activation and beta 2-adrenergic receptor endocytosis. J Biol Chem. 2001;276:42509–42513. doi: 10.1074/jbc.M108399200. [DOI] [PubMed] [Google Scholar]
- Claing A, Laporte SA, Caron MG, Lefkowitz RJ. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog Neurobiol. 2002;66:61–79. doi: 10.1016/s0301-0082(01)00023-5. [DOI] [PubMed] [Google Scholar]
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- Cong M, Perry SJ, Hu LA, Hanson PI, Claing A, Lefkowitz RJ. Binding of the beta2 adrenergic receptor to N-ethylmalei-mide-sensitive factor regulates receptor recycling. J Biol Chem. 2001;276:45145–45152. doi: 10.1074/jbc.M106087200. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, Caron MG, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- Dale LB, Bhattacharya M, Seachrist JL, Anborgh PH, Ferguson sSG. Agonist-stimulated and tonic internalization of metabotropic glutamate receptor 1A in human embryonic kidney 293 cells: agonist-stimulated endocytosis is beta-arrestin1 isoform-specific. Mol Pharmacol. 2001;60:1243–1253. doi: 10.1124/mol.60.6.1243. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- DeFea KA, Vaughn ZD, O’Bryan EM, Nishijima D, Dery O, Bunnett NW. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta-arrestin-dependent scaffolding complex. Proc Nat Acad Sci U S A. 2000;97:11086–11091. doi: 10.1073/pnas.190276697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGraff JL, Gurevich VV, Benovic JL. The third intracellular loop of alpha2-adrenergic receptors determines subtype specificity of arrestin interaction. J Biol Chem. 2002;277:43247–43252. doi: 10.1074/jbc.M207495200. [DOI] [PubMed] [Google Scholar]
- Dinculescu A, McDowell JH, Amici SA, Dugger DR, Richards N, Hargrave PA, et al. Insertional mutagenesis and immunochemical analysis of visual arrestin interaction with rhodopsin. J Biol Chem. 2002;277:11703–11708. doi: 10.1074/jbc.M111833200. [DOI] [PubMed] [Google Scholar]
- Dinh DT, Qian H, Seeber R, Lim E, Pfleger K, Eidne KA, et al. Helix I of beta-arrestin is involved in postendocytic trafficking but is not required for membrane translocation, receptor binding, and internalization. Mol Pharmacol. 2005;67:375–382. doi: 10.1124/mol.104.004721. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Bouvier M, Benovic JL, Caron MG, Lefkowitz RJ. The multiple membrane spanning topography of the beta 2-adrenergic receptor. Localization of the sites of binding, glycosylation, and regulatory phosphorylation by limited proteolysis. J Biol Chem. 1987;262:14282–14288. [PubMed] [Google Scholar]
- Elorza A, Sarnago S, Mayor FJ. Agonist-dependent modulation of G protein-coupled receptor kinase 2 by mitogen-activated protein kinases. Mol Pharmacol. 2000;57:778–783. doi: 10.1124/mol.57.4.778. [DOI] [PubMed] [Google Scholar]
- Farahbakhsh ZT, Ridge KD, Khorana HG, Hubbell WL. Mapping light-dependent structural changes in the cytoplasmic loop connecting helices C and D in rhodopsin: a site-directed spin labeling study. Biochemistry. 1995;34:8812–8819. doi: 10.1021/bi00027a033. [DOI] [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Downey WE, Colapietro AM, Barak LS, Menard L, Caron MG. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- Gabilondo AM, Hegler J, Krasel C, Boivin-Jahns V, Hein L, Lohse MJ. A dileucine motif in the C terminus of the beta2-adrenergic receptor is involved in receptor internalization. Proc Nat Acad Sci U S A. 1997;94:12285–12290. doi: 10.1073/pnas.94.23.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov I, Keen JH. Phosphoinositide-AP-2 interactions required for targeting to plasma membrane clathrin-coated pits. J Cell Biol. 1999;146:755–764. doi: 10.1083/jcb.146.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 1999;18:871–881. doi: 10.1093/emboj/18.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliera E, Jala VR, Trent JO, Bonecchi R, Signorelli P, Lefkowitz RJ, et al. beta-Arrestin-dependent constitutive internalization of the human chemokine decoy receptor D6. J Biol Chem. 2004;279:25590–25597. doi: 10.1074/jbc.M400363200. [DOI] [PubMed] [Google Scholar]
- Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, et al. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell. 2004;14:303–317. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- Gaudreau R, Beaulieu ME, Chen Z, Le Gouill C, Lavigne P, Stankova J, et al. Structural determinants regulating expression of the high affinity leukotriene B4 receptor: involvement of dileucine motifs and alpha-helix VIII. J Biol Chem. 2004;279:10338–10345. doi: 10.1074/jbc.M309207200. [DOI] [PubMed] [Google Scholar]
- Gelber EI, Kroeze WK, Willins DL, Gray JA, Sinar CA, Hyde EG, et al. Structure and function of the third intracellular loop of the 5-hydroxytriptamine-2A receptor: the third intracellular loop is a-helical and binds purified arrestins. J Neurochem. 1999;72:2206–2214. doi: 10.1046/j.1471-4159.1999.0722206.x. [DOI] [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, Buldt G. X-ray crystal structure of arrestin from bovine rod outer segments. Nature. 1998;391:918–921. doi: 10.1038/36147. [DOI] [PubMed] [Google Scholar]
- Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK, et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem. 2005;48:909–912. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- Gray JA, Bhatnagar A, Gurevich VV, Roth BL. The interaction of a constitutively active arrestin with the arrestin-insensitive 5-HT2A receptor induces agonist-independent internalization. Mol Pharma-col. 2003;63:961–972. doi: 10.1124/mol.63.5.961. [DOI] [PubMed] [Google Scholar]
- Gray-Keller MP, Detwiler PB, Benovic JL, Gurevich VV. Arrestin with a single amino acid substitution quenches light-activated rhodopsin in a phosphorylation0independent fasion. Biochemistry. 1997;36:7058–7063. doi: 10.1021/bi963110k. [DOI] [PubMed] [Google Scholar]
- Groarke DA, Drmota T, Bahia DS, Evans NA, Wilson S, Milligan G. Analysis of the C-terminal tail of the rat thyrotropin-releasing hormone receptor-1 in interactions and co-internalization with beta-arrestin 1-green fluorescent protein. Mol Pharmacol. 2001;59:375–385. doi: 10.1124/mol.59.2.375. [DOI] [PubMed] [Google Scholar]
- Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation–recignition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Mechanism of phosphorylation–recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol Pharmacol. 1997;51:161–169. doi: 10.1124/mol.51.1.161. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. TIPS. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem. 1993a;268:16879–16882. [PubMed] [Google Scholar]
- Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem. 1993b;268:16879–16882. [PubMed] [Google Scholar]
- Gurevich VV, Chen C-Y, Kim CM, Benovic JL. Visual arrestin binding to rhodopsin: intramolecular interaction between the basic N-terminus and acidic C-terminus of arrestin may regulate binding selectivity. J Biol Chem. 1994;269:8721–8727. [PubMed] [Google Scholar]
- Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, et al. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109:421–436. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 expression selectively increases during neural differentiation. J Neurochem. 2004;91:1404–1416. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science. 1988;241:832–835. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, Vrecl M, Kroeger KM, Miles LE, Qian H, Thomas WG, et al. Casein kinase II sites in the intracellular C-terminal domain of the thyrotropin-releasing hormone receptor and chimeric gonadotropin-releasing hormone receptors contribute to beta-arrestin-dependent internalization. J Biol Chem. 2001;276:18066–18074. doi: 10.1074/jbc.M009275200. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, Seeber RM, Kohout TA, Lefkowitz RJ, Eidne KA. Homo- and hetero-oligomerization of thyrotropin-releasing hormone (TRH) receptor subtypes. Differential regulation of beta-arrestins 1 and 2. J Biol Chem. 2002;277:50422–50430. doi: 10.1074/jbc.M209340200. [DOI] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Holliday ND, Lam CW, Tough IR, Cox HM. Role of the C terminus in neuropeptide Y Y1 receptor desensitization and internalization. Mol Pharmacol. 2005;67:655–664. doi: 10.1124/mol.104.006114. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Huai Q, Colicelli J, Ke H. The crystal structure of AMP-bound PDE4 suggests a mechanism for phosphodiesterase catalysis. Biochemistry. 2003;42:13220–13226. doi: 10.1021/bi034653e. [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- Hunton DL, Barnes WG, Kim J, Ren XR, Violin JD, Reiter E, et al. bete-arrestin 2 dependent AT1A receptor mediated pathway of chemotaxis. Mol Pharmacol. 2005;67:1229–1236. doi: 10.1124/mol.104.006270. [DOI] [PubMed] [Google Scholar]
- Hunzicker-Dunn M, Gurevich VV, Casanova JE, Mukherjee S. ARF6: a newly appreciated player in G protein-coupled receptor desensitization. FEBS Lett. 2002;521:3–8. doi: 10.1016/s0014-5793(02)02822-3. [DOI] [PubMed] [Google Scholar]
- Huttenrauch F, Nitzki A, Lin FT, Honing S, Oppermann M. Beta-arrestin binding to CC chemokine receptor 5 requires multiple C-terminal receptor phosphorylation sites and involves a conserved Asp-Arg-Tyr sequence motif. J Biol Chem. 2002;277:30769–30777. doi: 10.1074/jbc.M204033200. [DOI] [PubMed] [Google Scholar]
- Ignatova EG, Belcheva MM, Bohn LM, Neuman MC, Coscia CJ. Requirement of receptor internalization for opioid stimulation of mitogen-activated protein kinase: biochemical and immunofluorescence confocal microscopic evidence. J Neurosci. 1999;19:56–63. doi: 10.1523/JNEUROSCI.19-01-00056.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, et al. beta-Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J Biol Chem. 2001;276:43663–43667. doi: 10.1074/jbc.M105364200. [DOI] [PubMed] [Google Scholar]
- Innamorati G, Le Gouill C, Balamotis M, Birnbaumer M. The long and the short cycle. Alternative intracellular routes for trafficking of G-protein-coupled receptors. J Biol Chem. 2001;276:13096–13103. doi: 10.1074/jbc.M009780200. [DOI] [PubMed] [Google Scholar]
- Jala VR, Shao WH, Haribabu B. Phosphorylation-independent beta-arrestin translocation and internalization of leukotriene B4 receptors. J Biol Chem. 2005;280:4880–4887. doi: 10.1074/jbc.M409821200. [DOI] [PubMed] [Google Scholar]
- Janz JM, Farrens DL. Rhodopsin activation exposes a key hydrophobic binding site for the transducin alpha-subunit C terminus. J Biol Chem. 2004;279:29767–29773. doi: 10.1074/jbc.M402567200. [DOI] [PubMed] [Google Scholar]
- Jorgensen R, Martini L, Schwartz TW, Elling CE. Characterization of glucagon-like peptide-1 receptor beta-arrestin2 interaction: a high-affinity receptor phenotype. Mol Endocrinol. 2005;19:812–823. doi: 10.1210/me.2004-0312. [DOI] [PubMed] [Google Scholar]
- Key TA, Bennett TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. Regulation of formyl peptide receptor agonist affinity by reconstitution with arrestins and heterotrimeric G proteins. J Biol Chem. 2001;276:49204–49212. doi: 10.1074/jbc.M109475200. [DOI] [PubMed] [Google Scholar]
- Key TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. N-formyl peptide receptor phosphorylation domains differentially regulate arrestin and agonist affinity. J Biol Chem. 2003;278:4041–4047. doi: 10.1074/jbc.M204687200. [DOI] [PubMed] [Google Scholar]
- Key TA, Vines CM, Wagener BM, Gurevich VV, Sklar LA, Prossnitz ER. Inhibition of chemoattractant N-formyl peptide receptor trafficking by active arrestins. Traffic. 2005;6:87–99. doi: 10.1111/j.1600-0854.2004.00248.x. [DOI] [PubMed] [Google Scholar]
- Kieselbach T, Irrgang KD, Ruppel H. A segment corresponding to amino acids Val170-Arg182 of bovine arrestin is capable of binding to phosphorylated rhodopsin. Eur J Biochem. 1994;226:87–97. doi: 10.1111/j.1432-1033.1994.tb20029.x. [DOI] [PubMed] [Google Scholar]
- Kim YM, Benovic JL. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J Biol Chem. 2002;277:30760–30768. doi: 10.1074/jbc.M204528200. [DOI] [PubMed] [Google Scholar]
- Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem. 2001;276:37409–37414. doi: 10.1074/jbc.M106728200. [DOI] [PubMed] [Google Scholar]
- Kim OJ, Gardner BR, Williams DB, Marinec PS, Cabrera DM, Peters JD, et al. The role of phosphorylation in D1 dopamine receptor desensitization: evidence for a novel mechanism of arrestin association. J Biol Chem. 2004;279:7999–8010. doi: 10.1074/jbc.M308281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, et al. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Nat Acad Sci U S A. 2005a;102:1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Gainetdinov RR, Laporte SA, Caron MG, Barak LS. G protein-coupled receptor kinase regulates dopamine D3 receptor signaling by modulating the stability of a receptor-filamin-beta-arrestin complex. A case of autoreceptor regulation. J Biol Chem. 2005b;280:12774–12780. doi: 10.1074/jbc.M408901200. [DOI] [PubMed] [Google Scholar]
- Kishi H, Krishnamurthy H, Galet C, Bhaskaran RS, Ascoli M. Identification of a short linear sequence in the C-terminal tail of the rat follitropin receptor that modulates arrestin-3 binding in a phosphorylation-independent fashion. J Biol Chem. 2002;277:21939–21946. doi: 10.1074/jbc.M110894200. [DOI] [PubMed] [Google Scholar]
- Klein U, Muller C, Chu P, Birnbaumer M, von Zastrow M. Heterologous inhibition of G protein-coupled receptor endocytosis mediated by receptor-specific trafficking of beta-arrestins. J Biol Chem. 2001;276:17442–17447. doi: 10.1074/jbc.M009214200. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Nat Acad Sci U S A. 2001;98:1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- Kolobova EA, Klug CS, Buckheister SM, Kuznetzow AK, Hubbell WL, Gurevich VV. Identification of elements involved in arrestin-rhodopsin interaction by site-directed spin-labelling. Investig Ophthalmol Vis Sci. 2003;44:3176S. [Google Scholar]
- Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- Kraft K, Olbrich H, Majoul I, Mack M, Proudfoot A, Oppermann M. Characterization of sequence determinants within the carboxyl-terminal domain of chemokine receptor CCR5 that regulate signaling and receptor internalization. J Biol Chem. 2001;276:34408–34418. doi: 10.1074/jbc.M102782200. [DOI] [PubMed] [Google Scholar]
- Kramer HK, Simon EJ. mu and delta-opioid receptor agonists induce mitogen-activated protein kinase (MAPK) activation in the absence of receptor internalization. Neuropharmacology. 2000;39:1707–1719. doi: 10.1016/s0028-3908(99)00243-9. [DOI] [PubMed] [Google Scholar]
- Krasel C, Bunemann M, Lorenz K, Lohse MJ. Beta-arrestin binding to the beta2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J Biol Chem. 2005;280:9528–9535. doi: 10.1074/jbc.M413078200. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Gurevich VV, Schepers T, Hamm HE, Benovic JL. Arrestin-rhodopsin interaction. Multi-site binding delineated by peptide inhibition. J Biol Chem. 1994;269:3226–3232. [PubMed] [Google Scholar]
- Krupnick JG, Goodman OB, Jr, Keen JH, Benovic JL. Arrestin/clathrin interaction. Localization of the clathrin binding domain of nonvisual arrestins to the carboxy terminus. J Biol Chem. 1997a;272:15011–15016. doi: 10.1074/jbc.272.23.15011. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Gurevich VV, Benovic JL. Mechanism of Quenching of Phototransduction: binding competition between arrestin and transducin for phosphorhodopsin. J Biol Chem. 1997b;272:18125–18131. doi: 10.1074/jbc.272.29.18125. [DOI] [PubMed] [Google Scholar]
- Kuhn H. Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry. 1978;17:4389–4395. doi: 10.1021/bi00614a006. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Hall SW, Wilden U. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 1984;176:473–478. doi: 10.1016/0014-5793(84)81221-1. [DOI] [PubMed] [Google Scholar]
- Lambright DG, Noel JP, Hamm HE, Sigler PB. Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SSG, Caron MG, et al. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Nat Acad Sci U S A. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KB, Ptasienski JA, Pals-Rylaarsdam R, Gurevich VV, Hosey MM. Arrestin binding to the M2 muscarinic acetylcholine receptor is precluded by an inhibitory element in the third intracellular loop of the receptor. J Biol Chem. 2000;275:9284–9289. doi: 10.1074/jbc.275.13.9284. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Whalen EJ. beta-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162–168. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- Liang W, Austin S, Hoang Q, Fishman PH. Resistance of the human beta 1-adrenergic receptor to agonist-mediated down-regulation. Role of the C-terminus in determining beta-subtype degradation. J Biol Chem. 2003a;278:39773–39781. doi: 10.1074/jbc.M304482200. [DOI] [PubMed] [Google Scholar]
- Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem. 2003b;278:21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FT, Daaka Y, Lefkowitz RJ. beta-arrestins regulate mitogenic signaling and clathrin-mediated endocytosis of the insulin-like growth factor I receptor. J Biol Chem. 1998;273:31640–31643. doi: 10.1074/jbc.273.48.31640. [DOI] [PubMed] [Google Scholar]
- Lin FT, Krueger KM, Kendall HE, Daake Y, Fredericks ZL, Pitcher JA, et al. Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. J Biol Chem. 1997;272:31051–31057. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- Lin FT, Chen W, Shenoy S, Cong M, Exum ST, Lefkowitz RJ. Phosphorylation of beta-arrestin2 regulates its function in internalization of beta(2)-adrenergic receptors. Biochemistry. 2002;41:10692–10699. doi: 10.1021/bi025705n. [DOI] [PubMed] [Google Scholar]
- Liu P, Roush ED, Bruno J, Osawa S, Weiss ER. Direct binding of visual arrestin to a rhodopsin carboxyl terminal synthetic phosphopeptide. Mol Vis. 2004;10:712–719. [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Andexinger S, Pitcher J, Trukawinski S, Codina J, Faure JP, et al. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. J Biol Chem. 1992;267:8558–8564. [PubMed] [Google Scholar]
- Lowe J, Celver J, Gurevich VV, Chavkin C. mu-opioid receptors desensitize less rapidly than d-opioid receptors due to less efficient activation of arrestin. J Biol Chem. 2002;277:15729–15735. doi: 10.1074/jbc.M200612200. [DOI] [PubMed] [Google Scholar]
- Luttrell LM. ‘Location, location, location’: activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol. 2003;30:117–126. doi: 10.1677/jme.0.0300117. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Nat Acad Sci U S A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey TA, Gurevich VV, Neve KA. Preferential interaction between the dopamine D2 receptor and arrestin2 in neostriatal neurons. Mol Pharmacol. 2004;66:1635–1642. doi: 10.1124/mol.104.001495. [DOI] [PubMed] [Google Scholar]
- Macey TA, Liu Y, Gurevich VV, Neve KA. Dopamine D1 receptor interaction with arrestin3 in neostriatal neurons. J Neurochem. 2005;93:128–134. doi: 10.1111/j.1471-4159.2004.02998.x. [DOI] [PubMed] [Google Scholar]
- Marchese A, Chen C, Kim YM, Benovic JL. The ins and outs of G protein-coupled receptor trafficking. Trends Biochem Sci. 2003;28:369–376. doi: 10.1016/S0968-0004(03)00134-8. [DOI] [PubMed] [Google Scholar]
- Martin NP, Lefkowitz RJ, Shenoy SK. Regulation of V2 vasopressin receptor degradation by agonist-promoted ubiquitination. J Biol Chem. 2003;278:45954–45959. doi: 10.1074/jbc.M308285200. [DOI] [PubMed] [Google Scholar]
- Matharu AL, Mundell SJ, Benovic JL, Kelly E. Rapid agonist-induced desensitization and internalization of the A2B adenosine receptor is mediated by a serine residue close to the COOH-terminus. J Biol Chem. 2001;276:30199–30207. doi: 10.1074/jbc.M010650200. [DOI] [PubMed] [Google Scholar]
- McDonald PH, Cote NL, Lin FT, Premont RT, Pitcher JA, Lefkowitz RJ. Identification of NSF as a beta-arrestin1-binding protein. Implications for beta2-adrenergic receptor regulation. J Biol Chem. 1999;274:10677–10680. doi: 10.1074/jbc.274.16.10677. [DOI] [PubMed] [Google Scholar]
- McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, et al. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1515–1518. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- McDowell JH, Smith WC, Miller RL, Popp MP, Arendt A, Abdulaeva G, et al. Sulfhydryl reactivity demonstrates different conformational states for arrestin, arrestin activated by a synthetic phosphopeptide, and constitutively active arrestin. Biochemistry. 1999;38:6119–6125. doi: 10.1021/bi990175p. [DOI] [PubMed] [Google Scholar]
- McDowell JH, Robinson PR, Miller RL, Brannock MT, Arendt A, Smith WC, et al. Activation of arrestin: requirement of phosphorylation as the negative charge on residues in synthetic peptides from the carboxyl-terminal region of rhodopsin. Investig Ophthalmol Vis Sci. 2001;42:1439–1443. [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, et al. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Mhaouty-Kodja S, Barak LS, Scheer A, Abuin L, Diviani D, Caron MG, et al. Constitutively active alpha-1b adrenergic receptor mutants display different phosphorylation and internalization features. Mol Pharmacol. 1999;55:339–347. doi: 10.1124/mol.55.2.339. [DOI] [PubMed] [Google Scholar]
- Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41:3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- Milasta S, Evans NA, Ormiston L, Wilson S, Lefkowitz RJ, Milligan G. The sustainability of interactions between the orexin-1 receptor and beta-arrestin-2 is defined by a single C-terminal cluster of hydroxy amino acids and modulates the kinetics of ERK MAPK regulation. Biochem J. 2005;387(Pt 3):573–584. doi: 10.1042/BJ20041745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WE, Maudsley S, Ahn S, Khan KD, Luttrell LM, Lefkowitz RJ. beta-arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. Role of beta-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J Biol Chem. 2000;275:11312–11319. doi: 10.1074/jbc.275.15.11312. [DOI] [PubMed] [Google Scholar]
- Miller WE, McDonald PH, Cai SF, Field ME, Davis RJ, Lefkowitz RJ. Identification of a motif in the carboxyl terminus of beta -arrestin2 responsible for activation of JNK3. J Biol Chem. 2001;276:27770–27777. doi: 10.1074/jbc.M102264200. [DOI] [PubMed] [Google Scholar]
- Min L, Ascoli M. Effect of activating and inactivating mutations on the phosphorylation and trafficking of the human lutropin/choriogonado-tropin receptor. Mol Endocrinol. 2000;14:1797–1810. doi: 10.1210/mend.14.11.0555. [DOI] [PubMed] [Google Scholar]
- Min L, Galet C, Ascoli M. The association of arrestin-3 with the human lutropin/choriogonadotropin receptor depends mostly on receptor activation rather than on receptor phosphorylation. J Biol Chem. 2002;277:702–710. doi: 10.1074/jbc.M106082200. [DOI] [PubMed] [Google Scholar]
- Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- Morrison KJ, Moore RH, Carsrud ND, Trial J, Millman EE, Tuvim M, et al. Repetitive endocytosis and recycling of the beta 2-adrenergic receptor during agonist-induced steady state redistribution. Mol Pharmacol. 1996;50:692–699. [PubMed] [Google Scholar]
- Mukherjee S, Palczewski K, Gurevich VV, Benovic JL, Banga JP, Hunzicker-Dunn M. A direct role for arrestins in desensitization of luteinizing hormone/choriogonatropin receptor in porcine ovarian follicular membranes. Proc Nat Acad Sci U S A. 1999a;96:493–498. doi: 10.1073/pnas.96.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Palczewski K, Gurevich VV, Hunzicker-Dunn M. Beta-arrestin dependent desensitization of luteinizing hormone/-choriogonadotropin receptor is prevented by a synthetic peptide corresponding to the third intracellular loop of the receptor. J Biol Chem. 1999b;274:12984–12989. doi: 10.1074/jbc.274.19.12984. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Gurevich VV, Jones JCR, Casanova JE, Frank SR, Maizels ET, et al. The ADP ribosylation factor nucleotide exchange factor ARNO promotes beta-arrestin release necessary for luteinizing hormone/choriogonadotropin receptor desensitization. Proc Nat Acad Sci U S A. 2000;97:5901–5906. doi: 10.1073/pnas.100127097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Gurevich VV, Preninger A, Hamm HE, Bader M-F, Fazleabas AT, et al. Aspartic acid 564 in the third cytoplasmic loop of luteinizing hormone/choriogonadotropin receptor is crucial for phosphorylation-independent interaction with arrestin2. J Biol Chem. 2002;277:17916–17927. doi: 10.1074/jbc.M110479200. [DOI] [PubMed] [Google Scholar]
- Mundell SJ, Pula G, Carswell K, Roberts PJ, Kelly E. Agonist-induced internalization of metabotropic glutamate receptor 1A: structural determinants for protein kinase C- and G protein-coupled receptor kinase-mediated internalization. J Neurochem. 2003;84:294–304. doi: 10.1046/j.1471-4159.2003.01515.x. [DOI] [PubMed] [Google Scholar]
- Naik S, Billington CK, Pascual RM, Deshpande DA, Stefano FP, Kohout TA, et al. Regulation of cysteinyl leukotriene type 1 receptor internalization and signaling. J Biol Chem. 2005;280:8722–8732. doi: 10.1074/jbc.M413014200. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Ascoli M. A dileucine-based motif in the C-terminal tail of the lutropin/choriogonadotropin receptor inhibits endocytosis of the agonist-receptor complex. Mol Pharmacol. 1999;56:728–736. [PubMed] [Google Scholar]
- Nakamura K, Liu X, Ascoli M. Seven non-contiguous intracellular residues of the lutropin/choriogonadotropin receptor dictate the rate of agonist-induced internalization and its sensitivity to non-visual arrestins. J Biol Chem. 2000;275:241–247. doi: 10.1074/jbc.275.1.241. [DOI] [PubMed] [Google Scholar]
- Namkung Y, Sibley DR. Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J Biol Chem. 2004;279:49533–49541. doi: 10.1074/jbc.M408319200. [DOI] [PubMed] [Google Scholar]
- Neuschafer-Rube F, Hermosilla R, Rehwald M, Ronnstrand L, Schulein R, Wernstedt C, et al. Identification of a Ser/Thr cluster in the C-terminal domain of the human prostaglandin receptor EP4 that is essential for agonist-induced beta-arrestin1 recruitment but differs from the apparent principal phosphorylation site. Biochem J. 2004;379:573–585. doi: 10.1042/BJ20031820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel JP, Hamm HE, Sigler PB. The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, barrestin1, and barrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:30995–39090. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- Ohguro H, Palczewski K, Walsh KA, Johnson RS. Topographic study of arrestin using differential chemical modifications and hydrogen/deuterium exchange. Protein. 1994:2428–2434. doi: 10.1002/pro.5560031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Biol. 2004;11:1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Nat Acad Sci U S A. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- Orsini MJ, Benovic JL. Characterization of dominant negative arrestins that inhibit beta2-adrenergic receptor internalization by distinct mechanisms. J Biol Chem. 1998;273:34616–34622. doi: 10.1074/jbc.273.51.34616. [DOI] [PubMed] [Google Scholar]
- Osawa S, Raman D, Weiss ER. Heterologous expression and reconstitution of rhodopsin with rhodopsin kinase and arrestin. Methods Enzymol. 2000;315:411–422. doi: 10.1016/s0076-6879(00)15858-6. [DOI] [PubMed] [Google Scholar]
- Owen DJ, Vallis Y, Pearse BM, McMahon HT, Evans PR. The structure and function of the beta 2-adaptin appendage domain. EMBO J. 2000;19:4216–4227. doi: 10.1093/emboj/19.16.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, McDowell H, Jakes S, Ingebritsen TS, Hargrave PA. Regulation of rhodopsin dephosphorylation by arrestin. J Biol Chem. 1989;264:15770–15773. [PubMed] [Google Scholar]
- Palczewski K, Buczylko J, Kaplan MW, Polans AS, Crabb JW. Mechanism of rhodopsin kinase activation. J Biol Chem. 1991a;266:12949–12955. [PubMed] [Google Scholar]
- Palczewski K, Pulvermuller A, Buczylko J, Hofmann KP. Phosphorylated rhodopsin and heparin induce similar conformational changes in arrestin. J Biol Chem. 1991b;266:18649–18654. [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Pals-Rylaarsdam R, Gurevich VV, Lee KB, Ptasienski J, Benovic JL, Hosey MM. Internalization of the m2 muscarinic acetylcholine receptor: arrestin-independent and -dependent pathways. J Biol Chem. 1997;272:23682–23689. doi: 10.1074/jbc.272.38.23682. [DOI] [PubMed] [Google Scholar]
- Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin×receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- Parker LL, Backstrom JR, Sanders-Bush E, Shieh BH. Agonist-induced phosphorylation of the serotonin 5-HT2C receptor regulates its interaction with multiple PDZ protein 1. J Biol Chem. 2003;278:21576–21583. doi: 10.1074/jbc.M210973200. [DOI] [PubMed] [Google Scholar]
- Pediani JD, Colston JF, Caldwell D, Milligan G, Daly CJ, McGrath JC. Arrestin-dependent spontaneous alpha1a-adreno-ceptor endocytosis causes intracellular transportation of alpha-blockers via recycling compartments. Mol Pharmacol. 2005;67:992–1004. doi: 10.1124/mol.104.008417. [DOI] [PubMed] [Google Scholar]
- Penela P, Alvarez-Dolado M, Munoz A, Mayor FJ. Expression patterns of the regulatory proteins G protein-coupled receptor kinase 2 and beta-arrestin 1 during rat postnatal brain development: effect of hypothyroidism. Eur J Biochem. 2000;267:4390–4396. doi: 10.1046/j.1432-1327.2000.01484.x. [DOI] [PubMed] [Google Scholar]
- Penn RB, Pascual RM, Kim YM, Mundell SJ, Krymskaya VP, Panettieri RA, et al. Arrestin specificity for G protein-coupled receptors in human airway smooth muscle. J Biol Chem. 2001;276:32648–32656. doi: 10.1074/jbc.M104143200. [DOI] [PubMed] [Google Scholar]
- Perry SJ, Lefkowitz RJ. Arresting developments in heptahelical receptor signaling and regulation. Trends Cell Biol. 2002;12:130–138. doi: 10.1016/s0962-8924(01)02239-5. [DOI] [PubMed] [Google Scholar]
- Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002;298:834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Kirscht S, Stumm R, Koch T, Wu D, Laugsch M, et al. Heterodimerization of substance P and mu-opioid receptors regulates receptor trafficking and resensitization. J Biol Chem. 2003;278:51630–51637. doi: 10.1074/jbc.M307095200. [DOI] [PubMed] [Google Scholar]
- Pfister C, Chabre M, Plouet J, Tuyen VV, De Kozak Y, Faure JP, et al. Retinal S antigen identified as the 48K protein regulating light-dependent phosphodiesterase in rods. Science. 1985;228:891–893. doi: 10.1126/science.2988124. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Tesmer JJ, Freeman JL, Capel WD, Stone WC, Lefkowitz RJ. Feedback inhibition of G protein-coupled receptor kinase 2 (GRK2) activity by extracellular signal-regulated kinases. J Biol Chem. 1999;274:34531–34534. doi: 10.1074/jbc.274.49.34531. [DOI] [PubMed] [Google Scholar]
- Ponimaskin E, Dumuis A, Gaven F, Barthet G, Heine M, Glebov K, Richter DW, Oppermann M. Palmitoylation of the 5-hydroxytryptamine4a receptor regulates receptor phosphorylation, desensitization, and beta-arrestin-mediated endocytosis. Mol Pharmacol. 2005;67:1434–1443. doi: 10.1124/mol.104.008748. [DOI] [PubMed] [Google Scholar]
- Potter RM, Key TA, Gurevich VV, Sklar LA, Prossnitz ER. Arrestin variants display differential binding characteristics for the phosphorylated N-formyl peptide receptor carboxyl terminus. J Biol Chem. 2002;277:8970–8978. doi: 10.1074/jbc.M111086200. [DOI] [PubMed] [Google Scholar]
- Prezeau L, Richman JG, Edwards SW, Limbird LE. The zeta isoform of 14-3-3 proteins interacts with the third intracellular loop of different alpha2-adrenergic receptor subtypes. J Biol Chem. 1999;274:13462–13469. doi: 10.1074/jbc.274.19.13462. [DOI] [PubMed] [Google Scholar]
- Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon SC. Sequence alignment of the G protein coupled receptor superfamily. DNA Cell Biol. 1992;11:1–20. doi: 10.1089/dna.1992.11.1. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER. New roles from arrestins in the post-endocytic trafficking of G protein-coupled receptors. Life Sci. 2004;75:893–899. doi: 10.1016/j.lfs.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Puig J, Arendt A, Tomson FL, Abdulaeva G, Miller R, Hargrave PA, et al. Synthetic phosphopeptide from rhodopsin sequence induces retinal arrestin binding to photoactivated unphosphorylated rhodopsin. FEBS Lett. 1995;362:185–188. doi: 10.1016/0014-5793(95)00225-x. [DOI] [PubMed] [Google Scholar]
- Pulvermuller A, Maretzki D, Rudnicka-Nawrot M, Smith WC, Palczewski K, Hofmann KP. Functional differences in the interaction of arrestin and its splice variant, p44, with rhodopsin. Biochemistry. 1997;36:9253–9260. doi: 10.1021/bi970772g. [DOI] [PubMed] [Google Scholar]
- Pulvermuller A, Schroder K, Fischer T, Hofmann KP. Interactions of metarhodopsin II. Arrestin peptides compete with arrestin and transducin. J Biol Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- Qian H, Pipolo L, Thomas WG. Association of beta-Arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol Endocrinol. 2001;15:1706–1719. doi: 10.1210/mend.15.10.0714. [DOI] [PubMed] [Google Scholar]
- Raman D, Osawa S, Weiss ER. Binding of arrestin to cytoplasmic loop mutants of bovine rhodopsin. Biochemistry. 1999;38:5117–5123. doi: 10.1021/bi9824588. [DOI] [PubMed] [Google Scholar]
- Raman D, Osawa S, Gurevich VV, Weiss ER. The interaction with the cytoplasmic loops of rhodopsin plays a crucial role in arrestin activation and binding. J Neurochem. 2003;84:1040–1050. doi: 10.1046/j.1471-4159.2003.01598.x. [DOI] [PubMed] [Google Scholar]
- Ren XR, Reiter E, Ahn S, Kim J, Chen W, Lefkowitz RJ. Different G protein-coupled receptor kinases govern G protein and beta-arrestin mediated signaling of V2 vasopressin receptor. Proc Nat Acad Sci U S A. 2005;102:1448–1453. doi: 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resek JF, Farahbakhsh ZT, Hubbell WL, Khorana HG. Formation of the meta II photointermediate is accompanied by conformational changes in the cytoplasmic surface of rhodopsin. Biochemistry. 1993;32:12025–12032. doi: 10.1021/bi00096a012. [DOI] [PubMed] [Google Scholar]
- Richardson MD, Balius AM, Yamaguchi K, Freilich ER, Barak LS, Kwatra MM. Human substance P receptor lacking the C-terminal domain remains competent to desensitize and internalize. J Neurochem. 2003;84:854–863. doi: 10.1046/j.1471-4159.2003.01577.x. [DOI] [PubMed] [Google Scholar]
- Richman JG, Brady AE, Wang Q, Hensel JL, Colbran RJ, Limbird LE. Agonist-regulated Interaction between alpha2-adrenergic receptors and spinophilin. J Biol Chem. 2001;276:15003–15008. doi: 10.1074/jbc.M011679200. [DOI] [PubMed] [Google Scholar]
- Rim J, Oprian DD. Constitutive activation of opsin: interaction of mutants with rhodopsin kinase and arrestin. Biochemistry. 1995;34:11938–11945. doi: 10.1021/bi00037a035. [DOI] [PubMed] [Google Scholar]
- Salvatore MF, Waymire JC, Haycock JW. Depolarization-stimulated catecholamine biosynthesis: involvement of protein kinases and tyrosine hydroxylase phosphorylation sites in situ. J Neurochem. 2001;79:349–360. doi: 10.1046/j.1471-4159.2001.00593.x. [DOI] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Schleicher A, Kuhn H, Hofmann KP. Kinetics, binding constant, and activation energy of the 48-kDa protein–rhodopsin complex by extra-metarhodopsin II. Biochemistry. 1989;28:1770–1775. doi: 10.1021/bi00430a052. [DOI] [PubMed] [Google Scholar]
- Schmidlin F, Dery O, Bunnett NW, Grady EF. Heterologous regulation of trafficking and signaling of G protein-coupled receptors: beta-arrestin-dependent interactions between neurokinin receptors. Proc Nat Acad Sci U S A. 2002;99:3324–3329. doi: 10.1073/pnas.052161299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin F, Roosterman D, Bunnett NW. The third intracellular loop and carboxyl tail of neurokinin 1 and 3 receptors determine interactions with beta-arrestins. Am J Physiol Cell Physiol. 2003;285:945–958. doi: 10.1152/ajpcell.00541.2002. [DOI] [PubMed] [Google Scholar]
- Schramm NL, Limbird LE. Stimulation of mitogen-activated protein kinase by G protein-coupled alpha(2)-adrenergic receptors does not require agonist-elicited endocytosis. J Biol Chem. 1999;274:24935–24940. doi: 10.1074/jbc.274.35.24935. [DOI] [PubMed] [Google Scholar]
- Schulein R, Hermosilla R, Oksche A, Dehe M, Wiesner B, Krause G, et al. A dileucine sequence and an upstream glutamate residue in the intracellular carboxyl terminus of the vasopressin V2 receptor are essential for cell surface transport in COS, M6 cells. Mol Pharmacol. 1998;54:525–535. doi: 10.1124/mol.54.3.525. [DOI] [PubMed] [Google Scholar]
- Scott MG, Le Rouzic E, Perianin A, Pierotti V, Enslen H, Benichou S, et al. Differential nucleocytoplasmic shuttling of beta-arrestins. Characterization of a leucine-rich nuclear export signal in beta-arrestin2. J Biol Chem. 2002;277:37693–37701. doi: 10.1074/jbc.M207552200. [DOI] [PubMed] [Google Scholar]
- Sefton M, Blanco MJ, Penela P, Mayor FJ, Nieto MA. Expression of the G protein-coupled receptor kinase 2 during early mouse embryogenesis. Mech Dev. 2000;98:127–131. doi: 10.1016/s0925-4773(00)00441-x. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn-Schmiedeberg’s Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Receptor-specific ubiquitination of beta-arrestin directs assembly and targeting of 7TM receptor-signalosomes. J Biol Chem. 2005;280 doi: 10.1074/jbc.M412418200. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- Shiba T, Kawasaki M, Takatsu H, Nogi T, Matsugaki N, Igarashi N, et al. Molecular mechanism of membrane recruitment of GGA by ARF in lysosomal protein transport. Nat Struct Biol. 2003;10:386–393. doi: 10.1038/nsb920. [DOI] [PubMed] [Google Scholar]
- Shinohara T, Dietzschold B, Craft CM, Wistow G, Early JJ, Donoso LA, et al. Primary and secondary structure of bovine retinal S antigen (48-kDa protein) Proc Nat Acad Sci U S A. 1987;84:6975–6979. doi: 10.1073/pnas.84.20.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons PC, Biggs SM, Waller A, Foutz T, Cimino DF, Guo Q, et al. Real-time analysis of ternary complex on particles: direct evidence for partial agonism at the agonist-receptor-G protein complex assembly step of signal transduction. J Biol Chem. 2004;279:13514–13521. doi: 10.1074/jbc.M310306200. [DOI] [PubMed] [Google Scholar]
- Sitaramayya A, Liebman PA. Mechanism of ATP quench of phosphodiesterase activation in rod disc membranes. J Biol Chem. 1983;258:1205–1209. [PubMed] [Google Scholar]
- Smith WC, Milam AH, Dugger D, Arendt A, Hargrave PA, Palczewski K. A splice variant of arrestin. Molecular cloning and localization in bovine retina. J Biol Chem. 1994;269:15407–15410. [PubMed] [Google Scholar]
- Smith WC, Gurevich EV, Dugger DR, Shelamer CL, Vishnivetskiy SA, McDowell H, et al. Cloning and functional characterization of salamander rod and cone arrestins. Investig Ophthalmol Vis Sci. 2000;41:2445–2455. [PubMed] [Google Scholar]
- Sondek J, Lambright DG, Noel JP, Hamm HE, Sigler PB. GTPase mechanism of Gproteins from the 1.7-A crystal structure of transducin alpha-GDP-AIF-4. Nature. 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB. Crystal structure of a G-protein beta gamma dimer at 2.1A resolution. Nature. 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, et al. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharmacol. 2005;67:78–87. doi: 10.1124/mol.104.006072. [DOI] [PubMed] [Google Scholar]
- Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, et al. Polypeptide variants of beta-arrestin and arrestin3. J Biol Chem. 1993;268:15640–15648. [PubMed] [Google Scholar]
- Sunahara RK, Tesmer JJ, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gsalpha. Science. 1997;278:1943–1947. doi: 10.1126/science.278.5345.1943. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal structure of cone arrestin at 2.3A: Evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Teli T, Markovic D, Levine MA, Hillhouse EW, Grammatopoulos DK. Regulation of corticotropin-releasing hormone receptor type 1alpha signaling: structural determinants for G protein-coupled receptor kinase-mediated phosphorylation and agonist-mediated desensitization. Mol Endocrinol. 2005;19:474–490. doi: 10.1210/me.2004-0275. [DOI] [PubMed] [Google Scholar]
- ter Haar E, Musacchio A, Harrison SC, Kirchhausen T. Atomic structure of clathrin: a beta propeller terminal domain joins an alpha zigzag linker. Cell. 1998;95:563–573. doi: 10.1016/s0092-8674(00)81623-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, et al. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- Traub LM. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J Cell Biol. 2003;163:203–208. doi: 10.1083/jcb.200309175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulipano G, Stumm R, Pfeiffer M, Kreienkamp HJ, Hollt V, Schulz S. Differential beta-arrestin trafficking and endosomal sorting of somatostatin receptor subtypes. J Biol Chem. 2004;279:21374–21382. doi: 10.1074/jbc.M313522200. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Frank M, Krasel C, Dees C, Nissenson RA, Lohse MJ. Differential conformational requirements for activation of G proteins and the regulatory proteins arrestin and G protein-coupled receptor kinase in the G protein-coupled receptor for parathyroid hormone (PTH)/PTH-related protein. J Biol Chem. 2001;276:33435–33443. doi: 10.1074/jbc.M011495200. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Krasel C, Chauvin S, Bambino T, Lohse MJ, Nissenson RA. Internalization determinants of the parathyroid hormone receptor differentially regulate beta-arrestin/receptor association. J Biol Chem. 2002;277:8121–8129. doi: 10.1074/jbc.M110433200. [DOI] [PubMed] [Google Scholar]
- Vines CM, Revankar CM, Maestas DC, LaRusch LL, Cimino DF, Kohout TA, et al. N-formyl peptide receptors internalize but do not recycle in the absence of arrestins. J Biol Chem. 2003;278:41581–41584. doi: 10.1074/jbc.C300291200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez M-G, Gurevich VV. An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J Biol Chem. 2000;275:41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Hirsch JA, Velez M-G, Gurevich YV, Gurevich VV. Transition of arrestin in the active receptor-binding state requires an extended interdomain hinge. J Biol Chem. 2002;277:43961–43968. doi: 10.1074/jbc.M206951200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Wang Q, Limbird LE. Regulated interactions of the alpha 2A adrenergic receptor with spinophilin, 14-3-3zeta, and arrestin 3. J Biol Chem. 2002;277:50589–50596. doi: 10.1074/jbc.M208503200. [DOI] [PubMed] [Google Scholar]
- Wang P, Wu Y, Ge X, Ma L, Pei G. Subcellular localization of beta-arrestins is determined by their intact N domain and the nuclear export signal at the C terminus. J Biol Chem. 2003;278:11648–11653. doi: 10.1074/jbc.M208109200. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhao J, Brady AE, Feng J, Allen PB, Lefkowitz RJ, et al. Spinophilin blocks arrestin actions in vitro and in vivo at G protein-coupled receptors. Science. 2004;304:1940–1944. doi: 10.1126/science.1098274. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Tsao P, von Zastrow M. A phosphorylation-regulated brake mechanism controls the initial endocytosis of opioid receptors but is not required for post-endocytic sorting to lysosomes. J Biol Chem. 2001;276:34331–34338. doi: 10.1074/jbc.M104627200. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda P, et al. Modulation of postendocytic sorting of G protein-coupled receptors. Science. 2002a;297:615–620. doi: 10.1126/science.1073308. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Gerber BO, Meng EC, Baranski TJ, von Zastrow M, Bourne HR. Constitutive activation and endocytosis of the complement factor 5a receptor: evidence for multiple activated conformations of a G protein-coupled receptor. Traffic. 2002b;3:866–877. doi: 10.1034/j.1600-0854.2002.31203.x. [DOI] [PubMed] [Google Scholar]
- Wilbanks AM, Laporte SA, Bohn LM, Barak LS, Caron MG. Apparent loss-of-function mutant GPCRs revealed as constitutively desensitized receptors. Biochemistry. 2002;41:11981–11989. doi: 10.1021/bi020275m. [DOI] [PubMed] [Google Scholar]
- Wilden U, Kuhn H. Light-dependent phosphorylation of rhodopsin: number of phosphorylation sites. Biochemistry. 1982;21:3014–3022. doi: 10.1021/bi00541a032. [DOI] [PubMed] [Google Scholar]
- Wu G, Krupnick JG, Benovic JL, Lanier SM. Interaction of arrestins with intracellular domains of muscarinic and alpha2-adrenergic receptors. J Biol Chem. 1997;272:17836–17842. doi: 10.1074/jbc.272.28.17836. [DOI] [PubMed] [Google Scholar]
- Wu JH, Peppel K, Nelson CD, Lin FT, Kohout TA, Miller WE, et al. The adaptor protein b-arrestin2 enhances endocytosis of the low density lipoprotein receptor. J Biol Chem. 2003;278:44238–44245. doi: 10.1074/jbc.M309450200. [DOI] [PubMed] [Google Scholar]
- Xiao K, Shenoy SK, Nobles K, Lefkowitz RJ. Activation-dependent conformational changes in {beta}-arrestin 2. J Biol Chem. 2004;279:55744–55753. doi: 10.1074/jbc.M409785200. [DOI] [PubMed] [Google Scholar]
- Xie X, Gu Y, Fox T, Coll JT, Fleming MA, Markland W, et al. Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure. 1998;6:983–991. doi: 10.1016/s0969-2126(98)00100-2. [DOI] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997a;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997b;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Barak LS, Winkler KE, Caron MG, Ferguson SS. A central role for beta-arrestins and clathrin-coated vesicle-mediated endocytosis in beta2-adrenergic receptor resensitization. Differential regulation of receptor resensitization in two distinct cell types. J Biol Chem. 1997;272:27005–27014. doi: 10.1074/jbc.272.43.27005. [DOI] [PubMed] [Google Scholar]
- Zhang J, Barak LS, Anborgh PH, Laporte SA, Caron MG, Ferguson SS. Cellular trafficking of G protein-coupled receptor/-beta-arrestin endocytic complexes. J Biol Chem. 1999;274:10999–11006. doi: 10.1074/jbc.274.16.10999. [DOI] [PubMed] [Google Scholar]