

Abstract
A lithium-halogen exchange reaction occurs when the chloro[bis(diisopropylamino)]cyclopropenium tetrafluoroborate salt 1 (X = BF4) is treated with n-butyllithium. The resulting cyclopropenylidene-lithium adduct 3 has been isolated in 45% yield. In the solid state, this compound exists as a polymeric chain with an overall stoichiometry of two LiBF4 per carbene ligand. Addition of 12-crown-4-ether does not liberate the carbene from the lithium cation, but affords a monomeric tertiary complex (60% yield) that includes the crown ether. Moreover, complex 3 can also be synthesized by depro tonation of the bis(diisopropylamino)cyclopropenium tetrafluoroborate salt 2 (X = BF4) with n-butyllithium, whereas using potassium bis(trimethylsilyl)amide the free cyclopropenylidene was isolated in 53% yield. These results as whole seem to demonstrate that only certain counteranions allow for the isolation of cyclopropenylidene-lithium adducts, and only bases not containing lithium allow for the isolation of the free cyclopropenylidene. The former and the latter presumably prevented Weiss and Yoshida from isolating what would have been the first example of a stable carbene-lithium adduct and a free carbene, respectively.
Keywords: Carbenes, cyclopropenylidene, Li, Complex
In the 1950s, Breslow[1] and Wanzlick[2] realized that the stability of a carbene could be dramatically enhanced by the presence of amino substituents, but they were unable to isolate a “monomeric” carbene.[3] It was only in 1991, three years after the isolation of a (phosphino)(silyl)carbene,[4] that a bottle-able diamino carbene, namely an imidazol-2-ylidene, was prepared.[5] Even more strikingly, in a paper entitled “1,2,3,4-Tetraphenylimidazol-2-ylidene: The Realization of Wanzlick's Dream”, Arduengo[6] reported that a modification of the experimental procedure published by Wanzlick's group makes it possible to isolate one of the exact same carbenes postulated in 1970.[7]
Wanzlick was not the only one to nearly isolate the first stable carbene. Indeed, in the 1970s, Yoshida[8] and Weiss[9] attempted independently the preparation of the bis(diisopropylamino)cyclopropenylidene 4 (Fig. 1). Recently, we isolated the exact same carbene (4) after deprotonation of cyclopropenium salt 2 (X = BPh4) with potassium bis(trimethylsilyl)amide.[10] Among the routes used by Yoshida and Weiss were lithium-halogen exchange from 1 (X = ClO4)[9a] and the deprotonation of 2 (X = ClO4),[8a,b] both with n-BuLi. However, they were not able to isolate the resulting product. Initially, Yoshida claimed the successful synthesis of the free cyclopropenylidene 4,[8a] but several years later he[8b-e] and Weiss[9a] concluded concurrently that the compound in question was more likely the carbene lithium adduct 3 (X = ClO4). More recently, Tamm, Hahn et al.[11] repeated the lithium-halogen exchange reaction of 1 (X = ClO4 and CF3SO3) with n-BuLi, and described the product as stable only at low temperatures. It has also been shown that this compound, generated in situ, effectively transfers the cyclopropenylidene moiety 4 to a number of substrates, including transition metals and main group fragments.[8,9,11,12]
Figure 1.

Schematic representation of bis(diisopropylamino) derivatives of chlorocyclopropenium 1, cyclopropenium 2, cyclopropenylidene-lithium adduct 3, and cyclopropenylidene 4.
Weiss proposed the formation of the carbene lithium adduct 3 (X = ClO4), rather than that of the free carbene 4, based primarily on the observation that no LiClO4 precipitated from the reaction mixture.[9a] The only spectroscopic data that exist for the Weiss-Yoshida (W-Y) reagent is a single report of a lithium NMR chemical shift.[8d] All these experimental observations, including the transfer reactions, do not rule out the possibility that the W-Y reagent could actually be the free carbene 4. Indeed, carbenes that can be isolated as free species do not generally form stable complexes with simple lithium salts (LiX with X = halogen or weaker coordinating anion),[13,14] although there are two exceptions.[15]
This analysis prompted us to re-investigate the exact nature of the W-Y reagent. First, we reproduced the lithium-halogen exchange reaction with n-BuLi, but to avoid any potential explosive hazards due to the perchlorate anion, the chlorocyclopropenium tetrafluoroborate salt 1 (X = BF4) was used as a precursor. After stirring for ten minutes at −78 °C, a very clean reaction occurred and crucially, a 13C NMR signal at 167 ppm was observed. This signal is shifted to high field compared to that of the carbene carbon nucleus of the free cyclopropenylidene 4 (185 ppm),[10] exactly what is expected for a lithium complex. Indeed, it has been shown for the exceptions mentioned above,[15] that such a complexation induces an upfield shift of about 20 ppm. Surprisingly, the 13C NMR spectrum remained unchanged when the solution was warmed to room temperature. After evaporation of the solvent under high vacuum, the residue was washed with hexane to afford a highly air sensitive white powder, which was recrystallized in diethyl ether at −25 °C. The resulting colorless crystals (m.p. 88−90 °C dec., 45% yield) were subjected to a single crystal X-ray diffraction study.[16] In the solid state, 3 is a polymeric chain, with an overall stoichiometry of two LiBF4 per carbene ligand (Figs. 2 and 3). Each cyclopropenylidene moiety is bonded to a lithium cation, which is coordinated by three fluorine atoms from three different tetrafluoroborate anions. The lithium cations that are not complexed by a cyclopropenylidene are tetrahedrally coordinated by fluorine atoms from four different BF4 anions. The carbene-lithium bond length [C1-Li1−2.093 Å] is significantly shorter than those observed in the few other reported carbene-lithium ion adducts (2.135−2.155 Å).[17]
Figure 2.
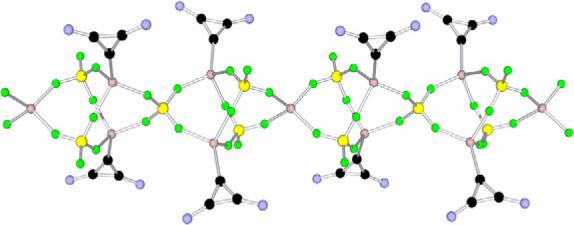
Molecular view of a crystal structure of polymeric cyclopropenylidene-Li adduct 3. The isopropyl groups on nitrogen have been omitted for clarity. Color code: C (black), N (blue), Li (pink), B (yellow), F (green).
Figure 3.
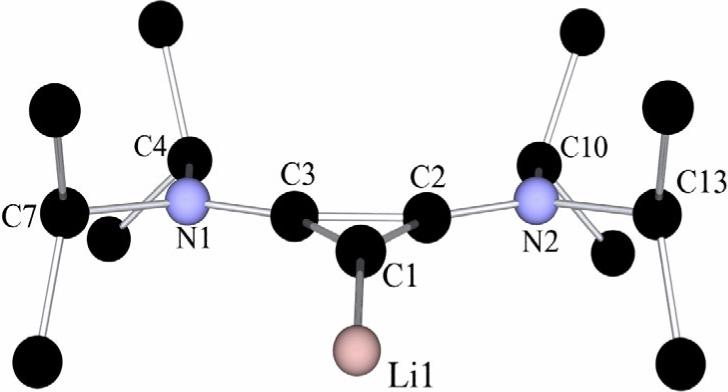
Molecular view of a crystal structure of the cyclopropenylidene-lithium portion of 3. Color code: C (black), N (blue), Li (pink). Selected bond lengths [Å] and angles [°]: Li1-C1 2.139(7), C1-C2 1.399(5), C1-C3 1.418(5), C2-C3 1.389(5), C2-N2 1.334(4), C3-N1 1.320(5); Li1-C1-C2 150.3(3), Li1-C1-C3 148.9(3), C2-C1-C3 59.1(3), C1-C2-N2 146.8(3), C3-C2-N2 152.0(4), C1-C2-C3 61.2(3), C1-C3-N1 147.5(4), C2-C3-N1 152.8(4), C1-C3-C2 59.8(3), C3-N1-C7 117.1(3), C3-N1-C4 121.0(3), C4-N1-C7 121.4(3), C2-N2-C13 116.9(3), C2-N2-C10 122.3(3), C10-N2-C13 120.7(3).
If we view the conjugate acid (2) of the cyclopropenylidene 4 as a proton/carbene complex we can clearly see several structural and spectroscopic trends (Table 1). Upon comparison of the exocyclic C-N bonds, an elongation is observed through the series 2 < 3 < 4, which corresponds to decreased π-donation from the amino groups. This is confirmed by the observed rotational barriers about the NC bond, as deduced from variable temperature NMR experiments. The carbene bond angle contracts along the series 2 > 3 > 4, which suggests an increase in s-character for the exocyclic sp hybrid type orbital. All of the relevant geometric parameters for 3 are closer to those for the free cyclopropenylidene 4 than its proton complex 2, indicating a certain amount of ionic character in the lithium carbene bond.
Table 1.
Comparison of the geometric parameters, rotational barrier about the CringN bonds, and spectroscopic data for compounds 2, 3 and 4.
| Compound | Cring-N Bond Lengtha (Å) |
Carbene Angle (°) |
Rotational Barrierb (kJ/mol) |
13C NMRc (ppm) |
|---|---|---|---|---|
| 2[10] | 1.306 | 62.3 | 75 | 99 |
| 3 | 1.328 | 59.1 | 56 | 167 |
| 4[10] | 1.334 | 57.2 | 53 | 185 |
: average value of the CringN bond lengths.
: Calculated based on variable temperature NMR experiments.
: Chemical shift for the carbene carbon and the corresponding carbon in 2 and 3.
To test the lability of cyclopropenylidene 4 from Li+ we attempted, unsuccessfully, solvent extraction of the free carbene with several non-polar solvents. We then tried to sequester the metal ion into a strong complexing agent. Upon addition of an excess of 12-crown-4-ether to a diethyl ether solution of 3, a yellow solid precipitated immediately. After recrystallization from a THF/diethyl ether solution, the compound was isolated as yellow crystals (mp. 105−107 °C, dec.) in 60 % yield. The 13C NMR spectrum showed signals characteristic of the crown ether. Compared to 3, the chemical shifts for the carbene carbon and the other two ring carbons remained essentially the same (Δδ = 3 and <1 ppm, respectively). These results clearly rule out the presence of the free cyclopropenylidene 4. A single crystal X-ray diffraction study showed the formation of tertiary complex 5 (Fig. 3). Although the structure's disorder precludes detailed discussion of the geometric parameters, it can be seen that it is a monomeric carbene-lithium complex, a type of compound which has eluded isolation thus far.[17,18] These results are in contrast with Alder's observation[15a] that addition of 12-crown-4-ether to the N,N-diisopropyl-tetrahydropyrimid-2-ylidene lithium BF4 complex induces the liberation of the free carbene. This suggests that cyclopropenylidene 4 coordinates lithium cations very strongly compared to N-heterocyclic carbenes.
We then turned our attention to the deprotonation route. When n-BuLi is used as a base, as Yoshida did,[8a,b] but the cyclopropenium 2 with BF4- instead of ClO4- as a counteranion, the reaction appeared to be very clean. The 1H and 13C NMR data of the resulting product are identical to those of the lithium complex 3 obtained by the lithium-halogen exchange reaction. Interestingly, when n-BuLi is replaced by potassium bis(trimethylsilyl)amide, under identical exper imental conditions, multinuclear NMR spectroscopy shows the clean formation of the free cyclopropenylidene 4. After work up, carbene 4 was isolated in 53% yield, which compares advantageously with the 20% yield observed when the cyclopropenium salt 2, with BPh4 as a counteranion, was used for the deprotonation reaction.[10]
From these results as a whole, it appears that free cyclopropenylidene 4 cannot be generated using n-BuLi or any other lithium-containing bases. In contrast to Li+, the presence of potassium cations does not prevent the isolation of the free carbene 4. Moreover, comparing our results with the previous reports, it seems that the counteranion of the cyclopropenium precursors 1 and 2 has a considerable importance with respect to the stability of the cyclopropenylidene lithium complexes. In fact, in our hands, all attempts to isolate the lithium cyclopropenylidene complex featuring BPh4− instead of BF4− failed.
It can be concluded that Weiss and Yoshida were not lucky. They had the right cyclopropenium cation, but they did not choose the right counteranion to isolate the first carbene lithium complex. Moreover, had they chosen the right base and counteranion combination, they would have isolated the first stable carbene. The isolation of both the free carbene and its lithium adduct should allow for the proliferation of cyclopropenylidene chemistry.
Experimental Section
All manipulations were performed under an inert atmosphere of argon using standard Schlenk techniques. Dry, oxygen-free solvents were employed. 1H, 7Li and 13C NMR spectra were recorded on Bruker Avance 300 and 600 spectrometers. 7Li chemical shifts are reported in ppm relative to a solution of LiCl in D2O as external standard. 1 (X = BF4). This compound was obtained from 1 (X = Cl) by the reported procedure,[19] but using NaBF4 for the anion exchange instead of HBF4 (30.00 g, 90%). m.p. 120−121 °C; 1H NMR (CDCl3, 25 °C, 300 MHz): δ = 4.13 (sept, 2H, CHCH3, J = 6.7), 3.87 (sept, 2H, CHCH3, J = 6.8), 1.41 (d, 24H, CHCH3, J = 6.7) ppm; 13C NMR (CDCl3, 25 °C, 75 MHz): δ = 131.9 (Cring), 93.1 (CCl), 57.9 (CH), 48.3 (CH), 22.3 (CH3), 20.4 (CH3) ppm. 2 (X = BF4). This compound was obtained from 2 (X = Cl) by the reported procedure,[10] but using NaBF4 for the anion exchange instead of NaBPh4 (33.50 g, 90%). m.p. 121−122 °C; 1H NMR (CDCl3, 25 °C, 300 MHz): δ = 7.47 (s, 1H, CHring), 4.03 (sept, 2H, CHCH3, J = 6.7), 3.87 (sept, 2H, CHCH3, J = 6.7), 1.39 (d, 12H, CH3, J = 6.7), 1.37 (d, 12H, CH3, J = 6.6) ppm; 13C NMR (CDCl3, 25°C, 75 MHz): δ = 133.6 (Cring), 99.1 (CHring), 57.1 (CHCH3), 48.9 (CHCH3), 20.6 (CH3), 20.6 (CH3) ppm. 3 (X = BF4). To a suspension of 1 (X = BF4) (3.00 g, 8.36 mmol) or 2 (X = BF4) (3.00 g, 9.25 mmol) in diethyl ether (80 mL) was added at −78 °C n-butyllithium (1 eq). The reaction was stirred for 15 minutes and then warmed to room temperature. After concentration under vacuum and washing with hexane (40 mL), a white powder was obtained. The residue was recrystallized from a solution of diethyl ether at −25 °C, and colorless crystals were obtained (1.59 g, 45% from 1, and 1.88 g, 48% from 2). m.p. 88−90 °C, dec.; 1H NMR (C6D6, 25 °C, 300 MHz): δ = 3.62 (broad m, 4H, CH), 1.21 (very broad m, 24H, CH3) ppm; 13C NMR (THF-d8, 25 °C, 75 MHz): δ = 166.8 (CLi), 154.9 (Cring), 50.4 (br, CH), 21.1 (CH3) ppm; 7Li NMR (THF-d8, 25 °C, 233 MHz): δ = 0.27 ppm. 5. At room temperature, to a solution of 3 (2.00 g, 4.72 mmol) in diethyl ether (60 mL) was added 12-crown-4-ether (2 eq) and a precipitate immediately formed. The yellow solid was washed with hexane (40 mL) and dried under vacuum. The residue was recrystallized from a THF/ether solution at −25 °C, and yellow crystals were obtained (1.43 g, 60%). m.p. 105−107 °C, dec.; 1H NMR (THF-d8, 25 °C, 300 MHz): δ = 4.00 (sept, 4H, CH, J = 6.7), 3.57 (s, 16H, CH2), 1.41 (d, 24H, CH3, J = 6.7) ppm; 13C NMR (THF-d8, 25 °C, 75 MHz): δ = 164.6 (CLi), 154.6 (Cring), 52.1 (br, CH), 48.8 (br, CH), 20.9 (CH3) ppm; 7Li NMR (THF-d8, 25 °C, 233 MHz): δ = −0.10 ppm.
Figure 4.
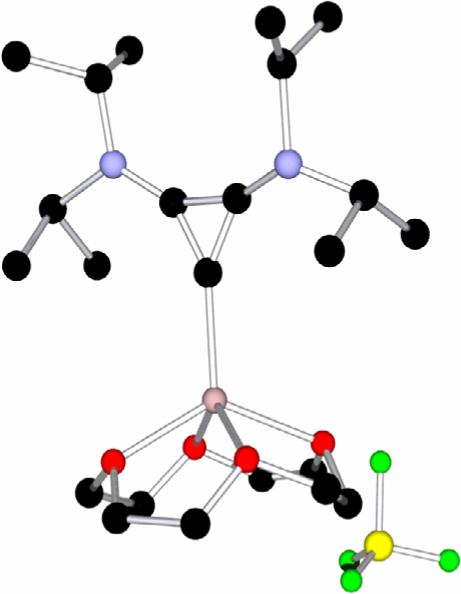
Molecular view of a crystal structure of 5. Color code: C (black), N (blue), Li (pink), B (yellow), F (green), O (red). Due to the presence of thermal disorder, geometric parameters cannot be accurately discussed.
Footnotes
We are grateful to the NIH (R01 GM 68825) and Rhodia for financial support of this work, and to the JSPS for a Fellowship to Y. I.
References
- 1.Breslow R. J. Am. Chem. Soc. 1958;80:3719. [Google Scholar]
- 2.Wanzlick H-W. Angew. Chem. 1962;74:129. [Google Scholar]
- 3.a Hoffmann RW. Angew. Chem. 1968;80:823. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1968;7:754. [Google Scholar]; b Arduengo AJ, III, Krafczyk R. Chem. Unserer. Zeit. 1998;32:6. [Google Scholar]; c Arduengo AJ., III Acc. Chem. Res. 1999;32:913. [Google Scholar]
- 4.a Igau A, Grützmacher H, Baceiredo A, Bertrand G. J. Am. Chem. Soc. 1988;110:6463. [Google Scholar]; b Igau A, Baceiredo A, Trinquier G, Bertrand G. Angew. Chem. 1989;101:617. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1989;28:621. [Google Scholar]
- 5.Arduengo AJ, III, Harlow RL, Kline M. J. Am. Chem. Soc. 1991;113:361. [Google Scholar]
- 6.Arduengo AJ, III, Goerlich JR, Krafczyk R, Marshall WJ. Angew. Chem. 1998;110:2062. [Google Scholar]; Angew. Chem., Int. Ed. 1998;37:1963. [Google Scholar]
- 7.Schönher H-J, Wanzlick H-W. Liebigs Ann. Chem. 1970;731:176. [Google Scholar]
- 8.a Yoshida Z. Abstr. 2nd Int. Symp. Chemistry of Nonbenzenoid Aromatic Compounds, Lindau. 1974:23. [Google Scholar]; b Yoshida Z, Konishi H, Sawada S, Ogoshi H. Chem. Commun. 1977:850. [Google Scholar]; c Konishi H, Matsumoto S, Kamitori Y, Ogoshi H, Yoshida Z. Chem. Lett. 1978:241. [Google Scholar]; d Yoshida Z. Pure Appl. Chem. 1982;54:1059. [Google Scholar]; e Miki S, Ohno T, Iwasaki H, Yoshida Z. J. Phys. Org. Chem. 1988;1:333. [Google Scholar]
- 9.a Weiss R, Priesner C, Wolf H. Angew. Chem. 1978;90:486. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1978;17:446. [Google Scholar]; b Weiss R, Priesner C, Wol H. Angew. Chem. 1979;91:505. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1979;18:472. [Google Scholar]; c Weiss R, Hertel M, Wolf H. Angew. Chem. 1979;91:506. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1979;18:473. [Google Scholar]; d Weiss R, Wolf H. Chem. Ber. 1980;113:1746. [Google Scholar]
- 10.Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Science. 2006;312:722. doi: 10.1126/science.1126675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a Tamm M, Grzegorzewski A, Hahn FE. J. Organomet. Chem. 1995;501:309. [Google Scholar]; a Schumann H, Glanz M, Girgsdies F, Hahn FE, Tamm M, Grzegorzewski A. Angew. Chem. 1997;109:2328. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1997;36:2232. [Google Scholar]
- 12.a Gompper R, Bartmann E. Angew. Chem. 1978;90:490. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1978;17:456. [Google Scholar]; b Gompper R, Wolf U. Liebigs Ann. Chem. 1979:1406. [Google Scholar]
- 13.One of the most classical methods to prepare stable free carbenes is the deprotonation of the conjugate acid; very often lithium salts are present and no complexation occurs.
- 14.a Herrmann WA. Angew. Chem. 2002;114:1342. [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:1290. [Google Scholar]; b Bourissou D, Guerret O, Gabbaï FP, Bertrand G. Chem. Rev. 2000;100:39. doi: 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]; c Kuhn N, Al-Sheikh A. Coord. Chem. Rev. 2005;249:829. [Google Scholar]
- 15.a Alder RW, Blake ME, Bortolotti C, Bufali S, Butts CP, Linehan E, Oliva JM, Orpen AG, Quayle MJ. Chem. Commun. 1999:241. [Google Scholar]; b Otto M, Conejero S, Canac Y, Romanenko VD, Rudzevitch V, Bertrand G. J. Am. Chem. Soc. 2004;126:1016. doi: 10.1021/ja0393325. [DOI] [PubMed] [Google Scholar]
- 16.Structural data for compounds 3 and 5 have been deposited in the Cambridge Crystallographic Data Center under CCDC 613889 (3), and 613890 (5), and can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html.
- 17.a Boche G, Hilfe C, Harms K, Marsch M, Lohrenz JCW. Angew. Chem. 1995;107:509. [Google Scholar]; Angew. Chem., Int. Ed. Engl. 1995;34:487. [Google Scholar]; b Hilf C, Bosold F, Harms K, Lohrenz JCW, Marsch M, Schimeczek M, Boche G. Chem. Ber. 1997;130:1202. [Google Scholar]; c Hilf C, Bosold F, Harms K, Marsch M, Boche G. Chem. Ber. 1997;130:1213. [Google Scholar]; d Arduengo AJ, III, Tamm M, Calabrese JC, Davidson F, Marshall WJ. Chem. Lett. 1999:1021. [Google Scholar]; e Fränkel R, Birg C, Kernbach U, Habereder T, Nöth H, Fehlhammer WP. Angew. Chem. 2001;113:1961. doi: 10.1002/1521-3773(20010518)40:10<1907::aid-anie1907>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]; Angew, Chem., Int. Ed. Engl. 2001;40:1907. [Google Scholar]; f Arnold PL, Mungur SA, Blake AJ, Wilson C. Angew. Chem. 2003;115:6163. doi: 10.1002/anie.200352710. [DOI] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. Engl. 2003;42:5981. [Google Scholar]; g Arnold PL, Rodden M, Davis KM, Scarisbrick AC, Blake AJ, Wilson C. Chem. Commun. 2004:1612. doi: 10.1039/b404614e. [DOI] [PubMed] [Google Scholar]
- 18.All carbene lithium complexes isolated so far[17] are dimers or higher order aggregates.
- 19.Yoshida Z, Tawara Y. J. Am. Chem. Soc. 1971;93:2574. [Google Scholar]