

Abstract
The lymph node vasculature is essential to immune function, but mechanisms regulating lymph node vascular maintenance and growth are not well understood. Vascular endothelial growth factor (VEGF) is an important mediator of lymph node endothelial cell proliferation in stimulated lymph nodes. It is expressed basally in lymph nodes and upregulated upon lymph node stimulation, but the identity of VEGF-expressing cells in lymph nodes is not known. We show that, at homeostasis, fibroblast-type reticular stromal cells (FRC) in the T zone and medullary cords are the principal VEGF-expressing cells in lymph nodes and that VEGF plays a role in maintaining endothelial cell proliferation, although peripheral node addressin (PNAd) pos endothelial cells are less sensitive than PNAdneg endothelial cells to VEGF blockade. Lymphotoxin beta receptor (LTβR) blockade reduces homeostatic VEGF levels and endothelial cell proliferation and LTβR stimulation of murine fibroblast-type cells upregulates VEGF expression, suggesting that LTβR signals on FRC regulates lymph node VEGF levels and, thereby, lymph node endothelial cell proliferation. At the initiation of immune responses, FRC remain the principal VEGF mRNA-expressing cells in lymph nodes, suggesting that FRC may play an important role in regulating vascular growth in stimulated nodes. In stimulated nodes, VEGF regulates the proliferation and expansion of both PNAdpos and PNAdneg endothelial cells. Together, these data suggest a role for FRC as paracrine regulators of lymph node endothelial cells and suggest that modulation of FRC VEGF expression may be a means to regulate lymph node vascularity and, potentially, immune function.
Keywords: Spleen and lymph nodes, Endothelial cells, Stromal cells, Rodent
Introduction
Lymph nodes are dependent on blood and lymphatic vessels for the continuous delivery of blood-borne cells, oxygen and micronutrients and lymph-borne antigens and antigen-presenting cells. Potentially, manipulating lymph node vascularity may be a means to modulate immune function in clinical settings and understanding the mechanisms that regulate homeostatic and immune-stimulated vascular growth can contribute to development of therapeutics that modulate lymph node vascularity. We have recently shown that lymph node endothelial cells have a basal proliferation rate of about one percent and that, upon lymph node stimulation, endothelial cell proliferation is dramatically increased and vascular expansion ensues. Vascular endothelial growth factor (VEGF) is an important mediator of the increased endothelial cell proliferation and is expressed basally and upregulated upon lymph node stimulation (1). These results suggest that modulation of VEGF expression may be a means to modulate lymph node vascularity. The identity of VEGF-expressing cells in lymph nodes, however, remains unresolved.
Blood and lymphatic vessels in lymph nodes reside within the stromal scaffold and are intimately associated with the cellular components of the scaffold. The scaffold consists of a reticular network of collagen-rich fibrils that are ensheathed by fibroblast-type reticular stromal cells (FRC) and that are distributed throughout the lymph node parenchyma. FRC are thought to be of mesenchymal origin and synthesize the basement membrane components of the fibrils (2). FRC also express chemokines that are critical for the migration of B and T cells to and within their respective compartments and express cell adhesion molecules that are likely to be important for the adhesion of resident dendritic cells (2-5). In addition to cellular localization and movement, FRC play an important role in maintaining T cell homeostasis by expressing IL-7 and CCL19 (6). Around high endothelial venules (HEV), FRC form a sheath of concentrically arranged overlapping cells and thus resemble pericytes found around the microvessels of other tissues. This FRC sheath is connected to the rest of the stromal network and directs and regulates the migration of recently transmigrated cells away from HEV into the lymph node parenchyma (4, 7).
FRC express lymphotoxin β receptor (LTβR) and expression of chemokines and cell adhesion molecules is regulated by lymphotoxin β receptor signaling. Blockade of LTβR signaling in-vivo reduces chemokine and adhesion molecule expression in lymphoid tissues (3, 8) and stimulation of LTβR on cultured FRC or other fibroblast-type cells is associated with increased expression of matrix and adhesion molecules (2, 9). LTβR binds LTα1β2 heterotrimer and the related molecule LIGHT expressed primarily by hematopoietic cells (8).
In this study, we identify FRC as the principal VEGF-expressing cells in lymph nodes and show that LTβR regulates VEGF expression and lymph node endothelial cell proliferation. We show that VEGF may be more important for the maintenance of PNAdneg endothelial cell proliferation in homeostatic nodes while proliferation and expansion of both PNADpos and PNAdneg endothelial cells are sensitive to VEGF blockade in stimulated nodes. We also show that reduced vascular growth in stimulated lymph nodes is associated with reduced lymph node cellularity and lymphocyte trafficking. These studies ascribe a previously unrecognized role for lymph node FRC as regulators of the lymph node vasculature and expand our understanding of lymph node vascular regulation and its potential functional consequences.
Materials and Methods
Mice
C57Bl/6 mice between 6-12 weeks of age were obtained from Jackson Labs or Charles River. VEGF-lacZ mice (10) were originally on a CD-1 background and crossed 1-4 generations to B6 mice for our studies. All animal procedures were performed in accordance with the regulations of the Institutional Animal Use and Care Committee of the Hospital for Special Surgery.
Indicated mice were treated with polyclonal goat anti-VEGF (R&D Systems, Minneapolis, MN) or control goat antibody (R&D) as described (1). Indicated mice were injected intraperitoneally with 100ug of LTβR-Ig (11) (kindly provided by Jeff Browning, Biogen, Idec, Cambridge, MA) or monoclonal human IgG1 (Protos Immunoresearch, Burlingame, CA). Indicated mice were injected intraperitoneally with aflibercept or control human-Fc (Regeneron, Tarrytown, NY) (12, 13).
Flow cytometry analysis
Flow cytometric analysis of endothelial cell proliferation is as described in (1). Briefly, mice were pulsed with BrdU for 24 hours before analysis. Lymph nodes were digested with collagenase type II and cells were counted to determine lymph node cellularity. Cells were then stained with antibodies for CD45 (to gate out hematopoietic cells), CD31 (to identify CD45negCD31pos endothelial cells), PNAd (to distinguish PNAdpos and PNAdneg endothelial cells), and BrdU. Cells were analyzed using a FACSCALIBUR (BD Biosciences) and CellQuest (BD Biosciences) software.
For LTβR staining, hamster anti-mouse LTβR clone AFH6 and control clone HA4/8 were used (14). Both antibodies were kindly provided by Jeff Browning, Biogen Idec.
For gp38 staining, hamster monoclonal antibody 8.1.1 (Developmental Studies Hybridoma Bank at U. of Iowa) and anti-hamster-PE (Invitrogen, Carlsbad, CA) were used.
For flow cytometric analysis of β-gal activity in VEGF-lacZ mice, the β-gal substrate fluorescein-di-beta-D-galactopyranoside (FDG)(Invitrogen) was used. Peripheral lymph nodes were pooled from each mouse and collagenase digested in DMEM containing 0.5% BSA (DMEM/BSA). Isolated cells were stained with primary and secondary antibodies for 20 minutes each in DMEM/BSA. Cells were resuspended in 40uL DMEM/BSA and placed in a 37°C water bath for 10 minutes to warm. An equal volume of 2mM FDG in water was added for one minute, after which the cells were placed on ice and 400ul of cold DMEM/5% FBS/7-AAD (E-Biosciences, San Diego, CA) immediately added.
Cryosection staining
LNs from flash-frozen and 7μm sections were cut on a cryostat. Sections were dried for 1 hour, fixed for 5min in 2% paraformaldehyde/0.125% glutaraldehyde, washed three times with 2mM MgCl2 in PBS, and then three times with 2mM MgCl2, 0.02% NP40, 0.01% sodium deoxycholate in PBS. To visualize β-gal activity, sections were incubated at 37°C for 18 hrs in 1M X-gal solution (X-gal (Sigma) in 2mM MgCl2, 0.02% NP40, 0.01% sodium deoxycholate, 0.03% K3Fe(CN)6, 0.03% C6FeK4N6*3H2O in PBS). Sections were then washed in TBS and then stained with additional primary and secondary antibodies as previously described (1).
To highlight the blood vasculature, some mice were injected intravenously with 100ug biotin-conjugated tomato lectin (Vector Laboratories, Burlingame, CA) and euthanized 3 minutes later as in previously described (1). The lectin was visualized by the application of streptavidin- horseradish-peroxidase (Jackson Immunoresearch. West Grove, PA).
Bone marrow chimeras
VEGF-lacZ heterozygote mice or transgene-negative littermates were lethally irradiated using 875 rads from an X-ray source. Mice received about 5×106 donor bone marrow cells intravenously and were allowed to reconstitute for 6 weeks before analysis. Some chimeras were stimulated with footpad injections of 106 LPS-matured bone marrow derived dendritic cells as described in (1).
Cell enrichment and real-time PCR
Pooled peripheral lymph nodes from 3 mice were digested with collagenase as for flow cytometry. Anti-CD45-biotin (BD Biosciences) was added and CD45pos cells were depleted using streptavidin microbeads (Miltenyi Biotec, Auburn, CA) and CS MACS column (Miltenyi Biotec). CD45 neg CD31 pos endothelial cells were separated from CD45 neg CD31 neg stromal cells using anti-CD31-biotin (BD Biosciences) and LS MACS column (Miltenyi Biotec). Approximately 30,000 cells from each fraction was subject to mRNA extraction.
RNA was extracted from cell fractions using RNEasy mini-kit with on-column DNAse digestion (Qiagen). Superscript III First Strand Synthesis System (Invitrogen, Carlsbad, CA) was used to generate cDNA. For real-time PCR, iQ Sybr-Green Supermix Kit (Biorad, Hercules, CA) was used. VEGF primers were as used in (15) and detect all alternatively splice isoforms of VEGF-A. They were 5′GGAGATCCTTCGAGGAGCACTT 3′ and 5′GGCGATTTAGCAGCAGATATAAGAA3′ VE-cadherin primers were 5′GGTATCATCAAACCCACGAAG3′ and 5′GGTCTGTGGCCTCAATGTAGA3′. Cyclophilin primers were 5′CAGACGCCACTGTCGCTTT3′ and 5′TGTCTTTGGAACTTTGTCTGCAA3′. VEGF and ve-cadherin levels were normalized to cyclophilin to generate a measure of relative mRNA levels.
Fibroblast culture and treatment
NIH-3T3 cells were cultured in DMEM (Mediatech, Herndon, VA) containing 10% heat-inactivated calf serum (Hyclone, Logan, UT). Cells were plated at 5×10ˆ3 cells per well in 24-well plates and allowed to settle overnight. Media was changed the next day and LTβR clone AFH6 and control clone HA4/8 (14) were added at 1ug/ml. Fresh media with antibody was added at day 3 and supernatants were taken at day 5. Samples were plated in triplicate.
VEGF measurements
For lymph node VEGF levels, popliteal lymph nodes were solubilized and subject to VEGF measurement using a commercial kit (R&D Systems, DuoSet-mouse VEGF) as previously described (1). VEGF from culture supernatants was measured using the same kit.
Results
FRC are the principal VEGF-expressing cells in homeostatic lymph nodes
We used VEGF-lacZ reporter mice to determine the cell types expressing VEGF in the lymph node. CD31 staining was used to identify blood and lymphatic vessels and biotinylated tomato lectin was injected intravenously to further highlight the blood vasculature in frozen sections. High levels of β-galactosidase (β-gal) activity were found predominantly in cells located near CD31brightlectinpos blood vessels in the T zone and blood and CD31med lymphatic vessels in the medullary cords (Fig. 1A-B). Within the T zone, β-galpos cells were localized both directly adjacent to and in the vicinity of HEV and small blood vessels (Fig. 1C). The medulla is a vessel-rich area and β -galpos cells were highly enriched in this compartment (Fig. 1A-B). As in the T zone, the medullary β-galpos cells were localized near the HEV and the numerous small blood vessels (Fig. 1D). In the medullary cords where HEV were intermingled with the lymphatics, the β -galpos cells were localized to the compartment between the HEV and the lymphatics at the abluminal side of both types of vessels (Fig. 1E). Although the majority of the β-galpos cells were not CD31pos endothelial cells, β-galpos endothelial cells could be found and tended to express less β -gal activity than the non-endothelial β-galpos cells (Fig. 1C, asterik).
Fig. 1. Characterization of VEGF expression using VEGF-lacZ mice.
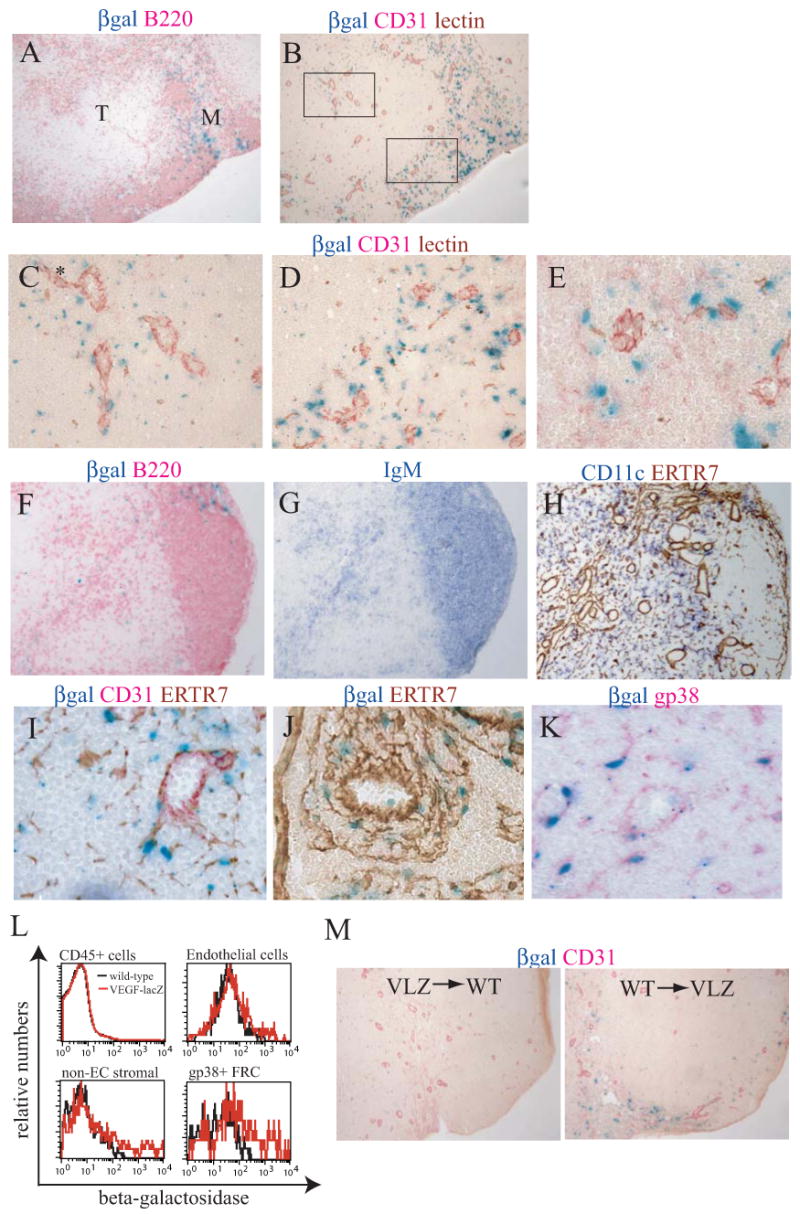
(A-K) Lymph nodes sections from VEGF-lacZ mice were stained for β-gal activity and for indicated markers. Lectin was injected intravenously in some mice to highlight the blood vasculature. (A) β-gal activity relative to B220pos cells in a brachial lymph node. The T zone (marked “T”) and medulla (marked “M”) are shown. 10× magnification. (B) β-gal activity relative to endothelial cells in a nearby section. HEV are distinct large CD31brightlectinpos (red/brown) vessels. The lymphatic vasculature expresses less CD31 and appears light pink. (C) 160× magnification of the T zone highlighted area in (B) showing β-galpos cells around HEV and small microvessels. Asterik indicates an endothelial cell that also demonstrates detectable β-gal expression. (D) 160× magnification of the medullary area highlighted in (B) showing the β-galpos cells around the HEV and numerous small blood vessels. (E) β-gal expression in the medullary cords where HEV are located within lymphatic-rich regions. (F) β-gal expression relative to a B cell follicle and a B220 pos cell-enriched area in the T zone in an inguinal lymph node. 40× magnification. (G) IgM staining in a nearby section to examine presence of B cells in the B220 pos cell-enriched area in the T zone. (H) CD11c and ER-TR7 staining in a nearby section shows enrichment of reticulum and presence of CD11c pos cells in the B cell-enriched area in the T zone. (I) β-gal expression relative to ERTR7pos reticulum in the T zone. 160× magnification. (J) β-gal expression relative to ERTR7pos reticulum in the medullary cords. (K) β-gal expression relative to gp38 pos FRC. (L) Flow cytometric analysis of lymph node β-gal expression by CD45pos hematopoietic cells, CD45neg CD31pos endothelial cells, CD45neg CD31neg non-endothelial stromal cells, and CD45neg CD31neg gp38pos FRC. Data in are representative of 5 experiments. (M) Bone marrow chimeras were generated using VEGF-lacZ donors and wild-type hosts (VLZ→WT) and wild-type donors and VEGF-lacZ hosts (WT→ VLZ). Brachial lymph nodes were harvested and frozen sections were assessed for β-gal activity in conjunction with CD31 staining. Data are representative of 4 pairs of chimeras.
β-gal activity tended to localize in B220pos areas but was only sometimes seen within the B cell follicles (Fig. 1A and 1F). Aside from the B cell-rich medullary cords, β-gal activity was enriched in extrafollicular B220pos areas in the T zone (Fig. 1A and 1F) that were sometimes seen to extend from the interfollicular regions in the cortex (Fig. 1F). These areas were also enriched in IgMpos cells (Fig. 1G), suggesting that these regions were indeed enriched in B cells, although B220pos plasmacytoid dendritic cells may also be enriched in these regions. The ER-TR7 antibody marks the extrafollicular reticular network (2, 16) and ER-TR7 staining showed the enrichment of reticulum and further highlighted the concentration of blood vessels and lymphatic sinuses in the extrafollicular B-rich areas (Fig. 1H). CD11cpos cells were also present in these areas (Fig. 1H). The concentration of blood vessels, lymphatic sinuses and reticulum in these B cell-rich areas suggest that these areas correspond to the borders between deep cortex units described by Belisle and Sainte-Marie (17) or the paracortical lobular units described by Kelly (18) that are areas of blood and lymphatic vessel trafficking (17, 19). In the T zone, then, β-galpos cells are enriched in these border zones.
The localization of β-galpos around and near vessels and in reticulum-enriched areas led us to examine the localization of β-galpos cells relative to the FRC network. β-galpos cells colocalized with the ER-TR7pos fibrils that were both directly apposed to as well as in the vicinity of vessels in the T zone (Fig. 1I) and the medullary cords (Fig. 1J). GP38 is a marker of FRC that are mostly extrafollicular (6, 20, 21). The brightest gp38 staining marked the subcapsular sinus and the lymphatic vessels whereas T zone and medullary FRC demonstrated medium-intensity gp38 staining (data not shown). The majority of β -galpos cells colocalized with the medium-intensity gp38 FRC staining (Fig. 1K). These data suggested that the VEGF-expressing cells within the lymph node could be FRC or perhaps hematopoietic cells closely associated with FRC.
To further establish the identity of β-galpos cells, we used flow cytometry to detect β-gal activity. CD45pos hematopoietic cells showed no detectable β-gal activity and a small subpopulation of CD45negCD31pos endothelial cells showed low levels of β–gal activity. In contrast, CD45negCD31neg non-endothelial stromal cells were more enriched in β-galpos cells. The majority (83% +/-.01%; n=4) of these β-galpos non-endothelial stromal cells were gp38pos, suggesting that these β-galpos cells were FRC. Gating on the gp38pos FRC subpopulation of non-endothelial stromal cells showed that the gp38pos FRC were greatly enriched in β-galpos cells (Fig. 1L). These results, in conjunction with the staining pattern seen in the frozen sections, suggested that FRC were the principal expressers of β-gal.
To further assess the relative contribution of hematopoietic and stromal populations to lymph node VEGF expression, we generated bone marrow chimeras. Transplantation of bone marrow cells from VEGF-lacZ mice into lethally irradiated wild-type hosts yielded no detectable β -galpos cells (Fig. 1M, left panel). In contrast, VEGF-lacZ hosts receiving wild-type donor cells demonstrated a concentration of β -galpos cells around vessels in vessel-rich areas (Fig. 1M, right panel). The β-gal expression, then, segregated with the radioresistant stromal compartment and not with radiosensitive hematopoietic cells and further suggested that FRC were the principal VEGF-expressing cells in homeostatic lymph nodes. Together, our findings in the VEGF-lacZ mice suggested that the pericyte-like FRC that ensheathed the HEV as well as FRC in the T zone and medullary fibrillar network near the vessels were the predominant VEGF-expressing cells in lymph nodes.
To confirm our findings in the VEGF-lacZ reporter mice, we tested for VEGF mRNA levels directly. We enriched for CD45negCD31pos endothelial cells and for CD45negCD31neg non-endothelial stromal cells by magnetic selection (Fig. 2A) and assessed VEGF mRNA levels using real-time PCR. On average, endothelial cells were enriched 260-fold (+/- 90; n=5) and non-endothelial stromal cells were enriched 230-fold (+/- 90; n=5). Of the non-endothelial stromal cells in the non-endothelial stromal cell fraction, 80% were gp38pos (Fig. 2B). In the endothelial cell fraction, about 15% of the cells were also gp38pos, but these cells had higher levels of gp38 and likely corresponded to the gp38bright lymphatic staining seen in the frozen sections (data not shown). Real-time PCR showed that endothelial cells had a 5-fold increase in VEGF mRNA when compared to unseparated lymph node cells while the non-endothelial stromal cells expressed 63-fold more VEGF mRNA (Fig. 2C). As a comparison, we also assayed for mRNA for ve-cadherin, a marker of blood vascular and lymphatic endothelial cells (22, 23). As expected, only the endothelial cell fraction was greatly enriched in ve-cadherin mRNA (Fig. 2D). These results are in agreement with the results from the VEGF-lacZ mice and, together, our results strongly suggested that FRC are principal expressers of VEGF within lymph nodes.
Fig. 2. VEGF mRNA is highly expressed in CD45neg CD31neg stromal cells.

CD45negCD31pos endothelial cells (“EC”) and CD45negCD31neg non-endothelial stromal cells (“non-EC stromal”) were enriched from a suspension of dissociated lymph node cells (“Pre”) using magnetic sorting and real-time PCR was performed on the cell fractions. (A) Representative flow cytometry plots showing relative enrichment of endothelial cells and of non-endothelial stromal cells. Numbers on the plot indicate percentage of live cells that are within the gate. (B) Flow cytometry plot showing gp38 expression on the R1 gated cells that were in the non-endothelial stromal fraction. (C) Relative VEGF mRNA in the pre-selection, endothelial cell, and non-endothelial stromal fractions. (D) VE-cadherin mRNA levels in the cell fractions. Bars represent average of 5 samples; error bars denote SD. *=p<.05 t-test.
VEGF mediates homeostatic lymph node endothelial cell proliferation
VEGF is an important mediator of the increased endothelial cell proliferation seen in immune-stimulated lymph nodes (1), and we asked about the importance of VEGF during homeostasis. Two days after anti-VEGF treatment in unstimulated mice, proliferation of the total endothelial cell population was markedly reduced (Fig. 3A). We asked whether there were differences between HEV endothelial cells and other types of endothelial cells. HEV endothelial cells of peripheral lymph nodes are marked by the expression of peripheral node addressin (PNAd), a group of sulfated glycoproteins that mediate part of the L-selectin-dependent entry of circulating cells. HEV are postcapillary venules, and the endothelial cells of the arterioles and capillaries that are upstream of HEV, and the venules that are downstream of HEV are PNAdneg. Lymphatic endothelial cells are also usually PNAdneg (24-26). We thus used PNAd expression on endothelial cells as a marker of HEV endothelial cells. Both PNAdpos and PNAdneg endothelial cells showed sensitivity to VEGF blockade, although the magnitude of reduction in proliferation tended to be greater in PNAdneg cells (Fig. 3A). As expected, endothelial cell numbers were not reduced with this short-term anti-VEGF treatment (data not shown). However, there was an accompanying modest reduction in lymph node size (Fig. 3B), suggesting that there was altered lymphocyte trafficking or survival, and suggesting that the reduction in endothelial cell proliferation was associated with some other facet of vascular function.
Fig. 3. VEGF blockade reduces homeostatic lymph node endothelial cell proliferation.

(A-B) Mice were injected with 100ug of anti-VEGF, control IgG, or PBS. On day 2, bilateral popliteal lymph nodes were pooled and cells were counted and stained for flow cytometric analysis. (A) Lymph node endothelial cell proliferation as assessed by BrdU uptake. BrdU uptake was determined in the entire CD45neg CD31pos endothelial cell population (“Total”), and in the PNAdpos and PNAdneg fractions of endothelial cells. n=6 per group. *=p<.05 compared to PBS and to Control IgG using t-test. Error bars denote SD. (B) Lymph node cellularity. *=p<.05 compared to Control IgG using t-test. p=.16 for PBS versus anti-VEGF comparison. (C-D) Mice were treated on Day 0 and Day 2 with 100ug of anti-VEGF and popliteal lymph nodes were examined on Day 4. (C) Endothelial cell proliferation. n=5 mice per group. *=p<.05 compared to PBS or Control IgG using t-test. (D) Lymph node cellularity.
We asked if longer treatment with anti-VEGF could amplify the effects on endothelial cell proliferation and lymph node cellularity. While PNAdneg endothelial cells continued to show greatly reduced endothelial cell proliferation after 4 days of anti-VEGF treatment, PNAdpos endothelial cells no longer showed any reduction in endothelial cell proliferation (Fig. 3C). Corresponding to the lack of effect on PNAdpos endothelial cells was the lack of reduction in lymph node cellularity at Day 4 (Fig. 3D), suggesting that the reduction in lymph node cellularity observed at Day 2 may have been linked to alterations in HEV function.
The Day 2 and Day 4 results together pointed to a differential sensitivity between PNAdpos and PNAdneg endothelial cells to VEGF blockade during homeostasis. While VEGF can be sequestered or upregulated in response to VEGF blockade (27-29) and it is possible that complete abolishment of VEGF expression or VEGF signaling can uncover a greater role for VEGF in sustained regulation of PNAdpos endothelial cell proliferation, these blockade experiments suggested that VEGF at homeostasis may play more of a role in maintaining PNAdneg rather than PNAdpos endothelial cell proliferation and numbers.
LTβR blockade reduces homeostatic VEGF and endothelial cell proliferation
FRC express LTβR and LTβR signaling regulates their expression of chemokines, cell adhesion molecules, and extracellular matrix components (2, 3, 8, 9). Flow cytometric analysis showed that gp38pos FRC expressed LTβR (Fig. 4A). We asked whether blockade of LTβR signaling could downregulate lymph node VEGF levels. Treatment of mice with LTβR-Ig fusion protein for 2 days resulted in a 37% reduction in lymph node VEGF levels (Fig. 4B), suggesting that LTβR signaling was required to maintain lymph node VEGF levels.
Fig. 4. LTβR blockade reduces lymph node VEGF and endothelial cell proliferation.

Mice were injected with 100ug of LTβR-Ig (LTβRIg), control human IgG (huIgG), or PBS. Bilateral popliteal lymph nodes were pooled and analyzed on day 2. (A) Flow cytometry histogram showing LTβR expression on lymph node CD45negCD31neg non-endothelial stromal cells (left panel) and on the gp38pos fraction of these cells (right panel). Red=anti- LTβR; black=control antibody. (B) Lymph node VEGF levels as determined by ELISA. Each symbol represents one mouse. *=p<.01 paired t-test. (C) Lymph node endothelial cell proliferation as assessed by BrdU uptake. BrdU uptake was determined in the entire CD45neg CD31pos endothelial cell population (“Total”) and in the PNAdpos and the PNAdneg fractions. n=4 for PBS group and n=6 for human Ig and LTβR-Ig groups. *=p<.05 compared to PBS or to huIgG. (D) Lymph node cellularity. *=p<.05 compared to huIgG using t-test. p=.12 for PBS versus LTβR-Ig comparison.
We tested whether the reduced VEGF levels induced by LTβR blockade was associated with reduced endothelial cell proliferation. Two days of LTβR-Ig treatment led to a significant reduction in proliferation of endothelial cells, although the magnitude of reduction was overall less than with anti-VEGF at 2 days. As with 2 days of anti-VEGF treatment, both PNAdpos and PNAdneg endothelial cells demonstrated reduced proliferation, and PNAdneg cells showed a greater magnitude of reduction (Fig. 4C). Also similar to the effect of 2 days of anti-VEGF treatment was the modest reduction in lymph node size (Fig. 4D). Of note was that, although prolonged LTβR-Ig treatment can reduce PNAd expression and consequent cell trafficking and lymph node cellularity (30), the 2 day treatment used here did not reduce PNAd levels as assessed by flow cytometry (data not shown). Together, these results suggested that LTβR signaling regulates the level of VEGF expression and that this LTβR-VEGF axis can regulate the level of endothelial cell proliferation during homeostasis.
LTβR signals increase VEGF expression by fibroblast-type cells
Our results suggested that the reduction of LTβR signaling on FRC was associated with reduced VEGF expression, but the LTβR signaling may be indirect as other cell types such as endothelial cells and dendritic cells can express LTβR (26, 31, 32). Also, because LTβR-Ig can also block LIGHT-HVEM interactions (8), the LTβR-Ig effect may reflect HVEM signaling. We therefore investigated whether direct modulation of LTβR on cultured fibroblast-type cells could modulate VEGF expression. NIH-3T3 cells are an embryonic fibroblast-derived cell line (33) and we found that they resemble FRC in expressing cell surface LTβR (Fig. 5A) and gp38 (Fig. 5B). Additionally, they were negative for endothelial cell markers such as CD31 and vonWillebrand factor (data not shown). ELISA of culture supernatants showed that VEGF was expressed basally by the NIH-3T3 cells (Fig. 5C). Addition of control antibody to the cells did not alter VEGF levels, but addition of agonist LTβR antibody AFH6 (14) was sufficient to stimulate an increase in VEGF levels (Fig. 5C). These results indicated that VEGF expression by fibroblast-type cells can be directly modulated by LTβR signals and suggested that the downregulation of lymph node VEGF levels with LTβR-Ig could be at least in part attributable to blockade of LTβR signals on FRC.
Fig. 5. LTβR stimulation of fibroblasts increases VEGF expression.

(A) Flow cytometry histogram showing LTβR expression on NIH-3T3 cells. Red=anti-LTβR; black=control antibody. (B) GP38 expression on NIH-3T3 cells. Red=anti-gp38; black=secondary antibody only. (C) Fibroblast VEGF levels upon LTβR stimulation. NIH-3T3 cells were cultured with no antibody (no Ig), control antibody (con Ig), or LTβR agonist antibody (anti-LTβR). VEGF level of culture supernatants were determined by ELISA. Each condition was plated in triplicate wells; bars show average value of the triplicate wells. Experiment is representative of 6 similar experiments. *=p<.05 t-test when compared to “no Ig” and to “control Ig.”
FRC remain the principal VEGF-expressing cells in stimulated lymph nodes
Within one day after immune stimulation, VEGF levels in draining lymph nodes are increased by about 2-fold and this increase in VEGF is abrogated by L-selectin blockade (1). This suggested that blood-borne hematopoietic cells entering stimulated nodes in an L-selectin dependent manner may be induced to upregulate VEGF mRNA expression to levels approaching that of the FRC. Consistent with this model, Angeli et al have demonstrated the induction of VEGF protein staining in B cell zones in stimulated lymph nodes (34). We asked whether we could detect high levels of VEGF expression by B cells or other hematopoietic cells in stimulated nodes. We stimulated VEGF-lacZ bone marrow chimeras with a footpad injection of dendritic cells (1) and examined β-gal expression two days later. With lymph node stimulation, β-gal expression remained undetectable in wild-type recipients reconstituted with VEGF-lacZ bone marrow (Fig. 6A, left panel), suggesting that there was very little or no increase in VEGF mRNA expression in hematopoietic cells. In contrast, VEGF-lacZ recipients reconstituted with wild-type cells continued to demonstrate high β-gal expression near vessels upon stimulation (Fig. 6A, right panel).
Fig. 6. VEGF expression in stimulated lymph nodes.
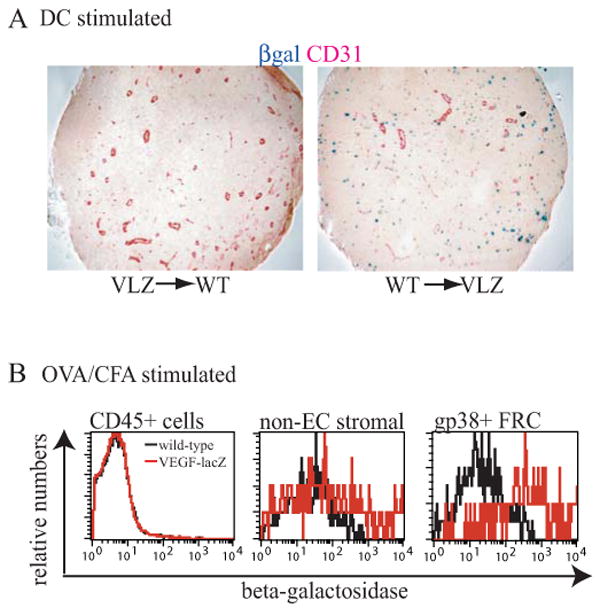
(A) β-gal expression in immune-stimulated VEGF-lacZ chimeras upon stimulation with dendritic cells. VLZ--> WT (left panel) and WT--> VLZ (right panel) chimeras were stimulated by footpad injection of mature dendritic cells and sections from draining popliteal lymph nodes were examined 2 days later for β-gal expression. Data representative of three VLZ--> WT and two WT--> VLZ chimeras. (B) β-gal expression in VEGF-lacZ lymph nodes upon stimulation with OVA/CFA. Mice received footpad injections of 20 microliters OVA/CFA (1mg/ml) and β-gal expression was assayed 2 days later via flow cytometry in CD45pos hematopoietic cells, CD45neg CD31neg non-endothelial stromal cells, and CD45neg CD31neg gp38pos FRC. Data representative of 5 similar experiments.
Flow cytometric analysis of VEGF-lacZ mice corroborated the histochemical findings in the chimeras. CD45pos hematopoietic cells did not show detectable β-gal expression while FRC remained high expressers of β-gal (data not shown). Similarly, in lymph nodes stimulated with ovalbumin in complete Freund adjuvant, gp38pos FRC but not CD45 pos cells showed detectable expression of β-gal (Fig. 6B). Together, these results suggested that FRC remained the principal cell type that expressed VEGF mRNA in stimulated lymph nodes.
VEGF mediates PNAd pos and PNAd neg endothelial cell proliferation and expansion in stimulated lymph nodes
We have previously shown that endothelial cell proliferation is upregulated at the initiation of an immune response, and that 200ug of anti-VEGF reduced the high levels of total endothelial cell proliferation by about 75% at Day 2 (1). In these lymph nodes, there was an 89% reduction in PNAdpos endothelial cell proliferation and a 70% reduction in PNAdneg endothelial cell proliferation in the draining lymph nodes (Fig. 7A), indicating that proliferation of both PNAdpos and PNAdneg endothelial cells was sensitive to VEGF blockade at this time. Lymph node cellularity was not affected in the draining nodes (Fig. 7B), suggesting that cell trafficking in stimulated nodes is regulated differently than in homeostatic nodes.
Fig. 7. VEGF blockade in stimulated lymph nodes.

Mice were stimulated at Day 0 with footpad injections of dendritic cells. (A-B) Mice were injected with 200ug of anti-VEGF, control IgG, or PBS at Day 0 and examined at Day 2. (A) Endothelial cell proliferation. n=4 mice per group. *=p<.05 compared to dendritic cell-stimulated PBS and Control IgG groups using t-test. (B) Lymph node cellularity. (C-D) Mice were treated with 100ug of anti-VEGF on Days 0 and 2 and examined at Day 4. (C) Endothelial cell proliferation. (D) Endothelial cell numbers. (E) Lymph node cellularity. For (C-E), n=4-5 mice per group. *=p<.05 compared to dendritic cell-stimulated PBS and Control IgG groups using t-test. (F-H) Mice were treated with 500ug aflibercept or human Fc control on Day 0 and Day 4 and were examined on Day 1 (D1) or Day 8 (D8). (F) Endothelial cell numbers. (G) Lymph node cellularity. (H) Cell trafficking into lymph nodes. Approximately 5×107 congenically marked CD45.1pos splenocytes were injected intravenously 30 minutes prior to sacrifice and entry of CD45.1pos cells into draining popliteal lymph nodes were enumerated by flow cytometry as in (1). For (F-H), n=5-6 mice per group. *=p<.05 compared to human Fc control at Day 8 using t-test.
Because of the observed differential sensitivity of PNAdpos and PNAdneg cells to VEGF blockade over time during homeostasis, we asked whether the differential sensitivity was observed in stimulated nodes and whether there was an impact on the expansion of endothelial cells. In order to compare the results at homeostasis with results in stimulated mice, we treated dendritic cell-stimulated mice as we did the mice at homeostasis, using 100ug of anti-VEGF at Days 0 and 2. Proliferation of both PNAdpos and PNAdneg cells was reduced in the stimulated nodes at Day 4 (Fig. 7C), although the overall magnitude of reduction was less dramatic than at Day 2 with the higher dose of anti-VEGF (compare with Fig. 7A). In addition to the reduced proliferation, the expansion of endothelial cells from baseline numbers was blunted by 43%, with PNAdpos cell expansion blunted by 40% and PNAdneg endothelial cell expansion blunted by 55% (Fig. 7D). The sensitivity of both PNAdpos and PNAdneg endothelial cells to VEGF blockade suggests that, in contrast to VEGF at homeostasis, VEGF upon immune stimulation is important for the proliferation and expansion of both PNAdpos and PNAdneg endothelial cells. Lymph node cellularity is modestly reduced (Fig, 7E), suggesting that the reduction in vascular expansion could potentially have an impact on lymph node growth and function.
Polyclonal antibodies can have off-target effects and we therefore tested whether lymph node vascular growth and cellularity could be similarly affected in stimulated lymph nodes using another VEGF antagonist, aflibercept, formerly known as “VEGF-Trap.” Aflibercept is a VEGF receptor 1 and VEGF receptor 2 chimeric protein fused to the Fc portion of human IgG1 that acts as a potent VEGF antagonist and also binds other VEGF receptor 1 and 2 ligands such as placental growth factor and VEGF-B (12). We treated mice on Day 0 and Day 4 with aflibercept at a dose similar to the dose used for inhibition of tumor angiogenesis (12, 13) and examined endothelial cell numbers and lymph node cellularity at Day 1 and Day 8. The expansion of endothelial cell numbers at Day 8 was reduced with aflibercept treatment, whereas the baseline endothelial cell number at Day 1 was unaffected (Fig. 7F). Consistent with the effects of polyclonal anti-VEGF on endothelial cell numbers at Day 4, aflibercept treatment reduced the expansion of both PNAdpos and PNAdneg endothelial cells (Fig. 7F). As with anti-VEGF at Day 4, the reduction in endothelial cell numbers with 8 days of aflibercept was associated with reduced expansion of lymph node cellularity (Fig. 7G).
We have previously shown that the increased PNAdpos endothelial cell numbers at Day 8 as compared to Day 1 is associated with increased entry of intravenously injected labeled lymphocytes (1). With the reduction in PNAdpos endothelial cell expansion and lymph node cellularity upon aflibercept treatment, we asked whether there was a corresponding reduction in lymphocyte entry. Indeed, entry of intravenously injected congenically marked CD45.1pos splenocytes was reduced in Day 8 but not Day 1 aflibercept-treated mice, suggesting that the reduced vascular expansion had reduced the number of entry sites for blood-borne lymphocytes (Fig. 7H). This result suggested that reduced expansion of endothelial cell numbers may result functionally in reduced cell entry which may contribute to alterations in lymph node growth and function.
Discussion
The regulation of lymph node vascular homeostasis and growth is not well understood and our results suggest that FRC are a principle source of VEGF and may play a role in regulating lymph node endothelial cell proliferation. Our data in VEGF-lacZ mice show that the β–gal signal colocalizes with gp38pos FRC on sections, that gp38pos FRC are β–gal pos in flow cytometric analysis, and that the β–gal signal segregates with a radioresistant population in bone marrow chimeras. In addition, we show that high levels of VEGF mRNA are expressed by CD45negCD31neg non-endothelial stromal cells, the majority of which are gp38pos FRC. Together, these data strongly suggest that FRC are a principal source of VEGF in lymph nodes.
Our experiments at homeostasis suggest that this local VEGF expression could be important in the regulation of the lymph node vasculature. First, the β-gal signal is located around and near by vessels, suggesting a scenario whereby the locally expressed VEGF could act on nearby vessels. Second, the high concentration of β-gal signal in the medulla correlates with the high vascular density in this region, suggesting that there may be a functional relationship between the amount of VEGF expressed and the quantity of vasculature that is supported. Third, our data in homeostatic lymph nodes show that VEGF-expressing gp38pos FRC express LTβR and LTβR-Ig reduces lymph node VEGF levels. Our in-vitro data indicate that LTβR signals on fibroblast-type cells can modulate VEGF expression, giving support to the concept that LTβR-Ig may have acted on lymph node FRC to reduce locally-expressed VEGF. The reduction in VEGF upon LTβR-Ig treatment correlates with a reduction in endothelial cell proliferation, suggesting that the reduced VEGF had a functional impact on the lymph node vasculature. While we have not excluded the potential role of VEGF generated outside the lymph node, the localization data together with the functional data support a model whereby LTβRpos FRC localized near blood and lymphatic vessels regulate proliferation of local endothelial cells at least in part by expression of VEGF. As LTβR ligands are expressed primarily by hematopoietic cells, the FRC LTβR presumably receives signals delivered by hematopoietic cells, and this hematopoietic cell-FRC interaction regulates the amount of VEGF expressed by the FRC and the consequent level of endothelial cell proliferation. Lymph node FRC are known to play critical roles in regulating lymphocyte compartmentalization and homeostasis; our data suggest that lymph node FRC have an additional role in regulating lymph node vascularity.
The VEGF-producing cells were in proximity to both blood and lymphatic vasculature, suggesting that these cells could regulate both types of vasculature. Our VEGF-blocking studies at homeostasis supported this concept. LYVE-1 lymphatic endothelial cells comprise about 20% of PNAdneg endothelial cells, and the greatly reduced proliferation of PNAdneg endothelial cells upon VEGF blockade suggests that the proliferation of both blood and lymphatic endothelial cells were affected. The idea that one VEGF-expressing cell type can regulate both blood vascular and lymphatic endothelial cells is also consistent with recent studies suggesting a role for VEGF in promoting lymphatic vessel growth (34-37) and with expression of VEGF receptor 2, the major VEGF receptor that mediates endothelial proliferation and growth, on both blood vessels and lymphatic sinuses in lymph nodes (38, 39). During immune responses, expansion of both lymphatic and blood vasculature in the lymph node occurs in the same time frame (1, 26, 34, 40), further suggesting coregulation of blood and lymphatic vessels. VEGF-producing FRC may be pivotal regulatory cells whose activation state dictates the level of both blood and lymphatic vascularity.
Our findings also suggest a new role for LTβR signaling. Prolonged LTβR blockade in homeostatic nodes results in reduced lymph node cellularity. This is at least in part attributable to alterations in HEV gene expression that result in reduced PNAd expression and corresponding reductions in entry of blood-borne cells (30). In addition to this presumably direct effect of LTβR signaling on HEV endothelial cells, our studies would suggest that long-term LTβR blockade may also contribute to reduced lymph node cellularity by reducing VEGF levels, the extent of PNAdneg blood vascular and lymphatic endothelial cell proliferation, and, over time, the number of PNAdneg blood vascular and lymphatic endothelial cells. Reduced blood vascular endothelial cell numbers may lead to reduced cellularity by reducing metabolic support, and reduced lymphatic endothelial cell numbers may contribute to reduced cellularity by reducing accumulation of dendritic cells and other lymph-borne contents which may be important in controlling lymph node cellularity and HEV phenotype (1, 26, 41, 42).
LTβR ligands are expressed primarily by hematopoietic cells (8), and relevant LTβR ligand-bearing candidates include B lymphocytes, which are known to express LTα1β2 at homeostasis (3), and immature dendritic cells, which can express LIGHT and lymphotoxin β (43, 44). We have shown that homeostatic lymph node VEGF levels in lymphocyte-deficient RAG1-/- mice are lower than in wild-type mice (1), suggesting that LTβR ligand-bearing lymphocytes may deliver signals to the VEGF-producing FRC. Also, in inflamed lymph nodes, B cells can regulate lymphangiogenesis (34). Furthermore, B cells are enriched in the areas that are enriched for VEGF-expressing cells, suggesting that they can directly regulate VEGF-expressing FRC. However, it is possible that resident or constitutively arriving dendritic cells (which are not fully mature) are the critical source of LTβR ligand and the effects of B cells are secondary to the effects on dendritic cell accumulation. Indeed, we have observed that homeostatic RAG1-/- lymph nodes have fewer resident/constitutively arriving dendritic cells (B. Webster and T. Lu, unpublished observations). We also observed in this study that resident/constitutively arriving dendritic cells are present in areas of VEGF-expressing cells and they have been shown to be in direct contact with FRC in the lymph node (5), further suggesting that resident/constitutively arriving dendritic cells could directly stimulate FRC. Further work will be required to identify the critical LTβR ligand-bearing cells.
Our results suggested that, at Day 2 in stimulated lymph nodes, FRC remained the principal cells expressing VEGF mRNA. In contrast, Angeli et al detected colocalization of VEGF protein with B cells (20). This apparent difference may reflect increased B cell expression of VEGF protein without a dramatic increase in VEGF mRNA or VEGF protein bound to but not produced by B cells. Our findings also do not exclude potential contributions to lymph node VEGF protein levels by hematopoietic cells such as platelets that may express VEGF protein but not VEGF mRNA or by a remote source such as the skin (45). However, because FRC remain the principle VEGF mRNA-expressing cell type within lymph nodes, it is likely that FRC are an important source of VEGF during immune responses, and that modulating VEGF expression by FRC during immune responses may potentially contribute to the regulation of vascular growth in immune-stimulated nodes. To fully establish the importance of FRC VEGF expression will require greater understanding of lymph node FRC-specific gene expression and the generation of a lymph node FRC-specific-VEGF knockout.
The FRC that expressed high levels of β–gal in the VEGF-lacZ mice were predominantly in the T zone and the medullary cords. FRC in the T zone express CCL19, CCL21, and IL-7 that play critical roles in T cell localization and homeostasis (6, 46-48) and FRC in the medullary cords are thought to express CXCL12 that is critical for plasma cell localization (49). Whether the VEGF-expressing FRC are a subset of these chemokine/cytokine-expressing FRC or whether they represent a distinct subset remains to be determined.
Our results suggested that VEGF blockade produced different effects at homeostasis and during an immune response. At homeostasis, VEGF blockade had a sustained effect on PNAdneg endothelial cell proliferation but only a transient effect on PNAdpos endothelial cell proliferation and lymph node cellularity. In contrast, VEGF blockade in stimulated mice caused more sustained reduction in the proliferation of both PNAdpos and PNAdneg endothelial cells and there was an accompanying reduction in the expansion of PNAdpos and PNAdneg endothelial cells. Although it is possible that off-target effects of anti-VEGF accounted for the reduced proliferation and expansion of both PNAdpos and PNAdneg endothelial cells in stimulated nodes, anti-VEGF and aflibercept had similar effects on PNAdpos and PNAdneg endothelial cell expansion, suggesting that VEGF is indeed an important mediator of PNAdpos and PNAdneg endothelial cell expansion in stimulated nodes. The mechanisms that regulate the differential sensitivity of PNAdpos and PNAdneg endothelial cells to VEGF blockade and the shift in sensitivity of PNAdpos cells in stimulated nodes remain to be determined. Interestingly, Liao and Ruddle recently reported that some vessels appear transiently positive for both the lymphatic marker LYVE-1 and for PNAd at Day 4 after oxazalone stimulation. Double positive vessels are rare at Day 0 and Day 7 (26). Whether these vessels are functionally HEV or lymphatic vessels is unknown, but their finding raises the possibility that some of the PNAdpos endothelial cells in our stimulated lymph nodes could be double positive for LYVE-1 and PNAd and that these cells could potentially contribute to the differential sensitivity to VEGF blockade.
Reduced vascular expansion was associated with reduced lymph node cellularity and reduced lymphocyte entry, suggesting that limiting vascular expansion can potentially have consequences for immune function. Targeting VEGF expression in immune stimulated nodes, then, may potentially be a means to modulate immune responsiveness. Further studies directed at examining the consequences of VEGF-specific blockade on lymphatic and HEV cell trafficking and on T and B cell activation will be required.
Modulating lymph node vascularity may be a means to modulate immune function in autoimmune diseases, transplant rejection, or tumor vaccination. Our results suggest that VEGF-expression by FRC could be a potential target by which to control lymph node vascularity. Better understanding of the nature of the LTβR ligand-expressing cells and the other signals that regulate VEGF-expression by FRC may have implications for the therapeutic control of angiogenesis and immunity.
Acknowledgments
We thank Sanjiv Luther and members of the Lu lab for critical reading of the paper and Jeff Browning for his gift of LTβR-Ig and anti-LTβR.
Funding support: This work was funded by NIAID R01-AI069800, Arthritis Foundation Investigator Award and Lupus Research Institute award (TL), T32-AI007621 (EE), CRI Tumor Immunology Predoctoral Fellowship (TT), and T32-AR007517 (AC).
Abbreviations
- β-gal
β-galactosidase
- FRC
fibroblast-type reticular stromal cells
- HEV
high endothelial venules
- LTβR
lymphotoxin beta receptor
- PNAd
peripheral node addressin
- VEGF
vascular endothelial growth factor
References
- 1.Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT. Regulation of lymph node vascular growth by dendritic cells. J Exp Med. 2006;203:1903–1913. doi: 10.1084/jem.20052272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Lymph Node Fibroblastic Reticular Cells Construct the Stromal Reticulum via Contact with Lymphocytes. J Exp Med. 2004;200:783–795. doi: 10.1084/jem.20040254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 4.Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, Pabst R, Lutz MB, Sorokin L. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity. 2005;22:19–29. doi: 10.1016/j.immuni.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 6.Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007 doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 7.Gretz JE, Anderson AO, Shaw S. Cords, channels, corridors and conduits: critical architectural elements facilitating cell interactions in the lymph node cortex. Immunol Rev. 1997;156:11–24. doi: 10.1111/j.1600-065x.1997.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 8.Gommerman JL, Browning JL. Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat Rev Immunol. 2003;3:642–655. doi: 10.1038/nri1151. [DOI] [PubMed] [Google Scholar]
- 9.Murphy M, Walter BN, Pike-Nobile L, Fanger NA, Guyre PM, Browning JL, Ware CF, Epstein LB. Expression of the lymphotoxin beta receptor on follicular stromal cells in human lymphoid tissues. Cell Death Differ. 1998;5:497–505. doi: 10.1038/sj.cdd.4400374. [DOI] [PubMed] [Google Scholar]
- 10.Miquerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol. 1999;212:307–322. doi: 10.1006/dbio.1999.9355. [DOI] [PubMed] [Google Scholar]
- 11.Browning J, Sizing I, Lawton P, Bourdon P, Rennert P, Majeau G, Ambrose C, Hession C, Miatkowski K, Griffiths D, Ngam-ek A, Meier W, Benjamin C, Hochman P. Characterization of lymphotoxin-alpha beta complexes on the surface of mouse lymphocytes. J Immunol. 1997;159:3288–3298. [PubMed] [Google Scholar]
- 12.Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, Hu-Lowe DD, Shalinsky DR, Thurston G, Yancopoulos GD, McDonald DM. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165:35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph Node Genesis Is Induced by Signaling through the Lymphotoxin [beta] Receptor. Immunity. 1998;9:71–79. doi: 10.1016/s1074-7613(00)80589-0. [DOI] [PubMed] [Google Scholar]
- 15.Shih SC, Robinson GS, Perruzzi CA, Calvo A, Desai K, Green JE, Ali IU, Smith LEH, Senger DR. Molecular Profiling of Angiogenesis Markers. Am J Pathol. 2002;161:35–41. doi: 10.1016/S0002-9440(10)64154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Vliet E, Melis M, Foidart J, Van Ewijk W. Reticular fibroblasts in peripheral lymphoid organs identified by a monoclonal antibody. J Histochem Cytochem. 1986;34:883–890. doi: 10.1177/34.7.3519751. [DOI] [PubMed] [Google Scholar]
- 17.Belisle C, Sainte-Marie G. Tridimensional study of the deep cortex of the rat lymph node. III. Morphology of the deep cortex units. Anat Rec. 1981;199:213–226. doi: 10.1002/ar.1091990206. [DOI] [PubMed] [Google Scholar]
- 18.Kelly RH. Functional anatomy of lymph nodes. I. The paracortical cords. Int Arch Allergy Appl Immunol. 1975;48:836–849. doi: 10.1159/000231371. [DOI] [PubMed] [Google Scholar]
- 19.Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity. 2008;28:122–133. doi: 10.1016/j.immuni.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farr A, Berry M, Kim A, Nelson A, Welch M, Aruffo A. Characterization and cloning of a novel glycoprotein expressed by stromal cells in T-dependent areas of peripheral lymphoid tissues. J Exp Med. 1992;176:1477–1482. doi: 10.1084/jem.176.5.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 22.Lampugnani M, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco L, Dejana E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol. 1992;118:1511–1522. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, Kerjaschki D, Maurer D. Isolation and Characterization of Dermal Lymphatic and Blood Endothelial Cells Reveal Stable and Functionally Specialized Cell Lineages. J Exp Med. 2001;194:797–808. doi: 10.1084/jem.194.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 25.M'Rini C, Cheng G, Schweitzer C, Cavanagh LL, Palframan RT, Mempel TR, Warnock RA, Lowe JB, Quackenbush EJ, von Andrian UH. A novel endothelial L-selectin ligand activity in lymph node medulla that is regulated by alpha(1,3)-fucosyltransferase-IV. J Exp Med. 2003;198:1301–1312. doi: 10.1084/jem.20030182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao S, Ruddle NH. Synchrony of High Endothelial Venules and Lymphatic Vessels Revealed by Immunization. J Immunol. 2006;177:3369–3379. doi: 10.4049/jimmunol.177.5.3369. [DOI] [PubMed] [Google Scholar]
- 27.Carpenter B, Lin Y, Stoll S, Raffai RL, McCuskey R, Wang R. VEGF is crucial for the hepatic vascular development required for lipoprotein uptake. Development:dev.01902. 2005 doi: 10.1242/dev.01902. [DOI] [PubMed] [Google Scholar]
- 28.Kim ES, Serur A, Huang J, Manley CA, McCrudden KW, Frischer JS, Soffer SZ, Ring L, New T, Zabski S, Rudge JS, Holash J, Yancopoulos GD, Kandel JJ, Yamashiro DJ. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proceedings of the National Academy of Sciences. 2002;99:11399–11404. doi: 10.1073/pnas.172398399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kadenhe-Chiweshe A, Papa J, McCrudden KW, Frischer J, Bae JO, Huang J, Fisher J, Lefkowitch JH, Feirt N, Rudge J, Holash J, Yancopoulos GD, Kandel JJ, Yamashiro DJ. Sustained VEGF Blockade Results in Microenvironmental Sequestration of VEGF by Tumors and Persistent VEGF Receptor-2 Activation. Mol Cancer Res. 2008;6:1–9. doi: 10.1158/1541-7786.MCR-07-0101. [DOI] [PubMed] [Google Scholar]
- 30.Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, Fava RA. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity. 2005;23:539–550. doi: 10.1016/j.immuni.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 2005;22:439–450. doi: 10.1016/j.immuni.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, Chen BK, Genden E, Skobe M, Lira SA. Lymphotoxin beta receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc Natl Acad Sci U S A. 2007;104:5026–5031. doi: 10.1073/pnas.0606697104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jainchill JL, Aaronson SA, Todaro GJ. Murine Sarcoma and Leukemia Viruses: Assay Using Clonal Lines of Contact-Inhibited Mouse Cells. J Virol. 1969;4:549–553. doi: 10.1128/jvi.4.5.549-553.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Angeli V, Ginhoux F, Llodra J, Quemeneur L, Frenette PS, Skobe M, Jessberger R, Merad M, Randolph GJ. B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity. 2006;24:203–215. doi: 10.1016/j.immuni.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Wirzenius M, Tammela T, Uutela M, He Y, Odorisio T, Zambruno G, Nagy JA, Dvorak HF, Yla-Herttuala S, Shibuya M, Alitalo K. Distinct vascular endothelial growth factor signals for lymphatic vessel enlargement and sprouting. J Exp Med. 2007;204:1240. doi: 10.1084/jem.20062642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005;201:1089–1099. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Detmar MJ, Lawitts JA, Benjamin L, Tan X, Manseau EJ, Dvorak AM, Dvorak HF. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med. 2002;196:1497–1506. doi: 10.1084/jem.20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruddell A, Mezquita P, Brandvold KA, Farr A, Iritani BM. B lymphocyte-specific c-Myc expression stimulates early and functional expansion of the vasculature and lymphatics during lymphomagenesis. Am J Pathol. 2003;163:2233–2245. doi: 10.1016/S0002-9440(10)63581-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 40.Soderberg KA, Payne GW, Sato A, Medzhitov R, Segal SS, Iwasaki A. Innate control of adaptive immunity via remodeling of lymph node feed arteriole. Proc Natl Acad Sci U S A. 2005;102:16315–16320. doi: 10.1073/pnas.0506190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MartIn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mebius R, Streeter P, Breve J, Duijvestijn A, Kraal G. The influence of afferent lymphatic vessel interruption on vascular addressin expression. J Cell Biol. 1991;115:85–95. doi: 10.1083/jcb.115.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh SL, Nagata S, Ni J, Chen L. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol. 2000;164:4105–4110. doi: 10.4049/jimmunol.164.8.4105. [DOI] [PubMed] [Google Scholar]
- 44.Lu G, Janjic BM, Janjic J, Whiteside TL, Storkus WJ, Vujanovic NL. Innate Direct Anticancer Effector Function of Human Immature Dendritic Cells. II. Role of TNF, Lymphotoxin-{alpha}1{beta}2, Fas Ligand, and TNF-Related Apoptosis-Inducing Ligand. J Immunol. 2002;168:1831–1839. doi: 10.4049/jimmunol.168.4.1831. [DOI] [PubMed] [Google Scholar]
- 45.Halin C, Tobler NE, Vigl B, Brown LF, Detmar M. VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood. 2007;110:3158–3167. doi: 10.1182/blood-2007-01-066811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanabe S, Lu Z, Luo Y, Quackenbush E, Berman M, Collins-Racie L, Mi S, Reilly C, Lo D, Jacobs K, Dorf M. Identification of a new mouse beta-chemokine, thymus-derived chemotactic agent 4, with activity on T lymphocytes and mesangial cells. J Immunol. 1997;159:5671–5679. [PubMed] [Google Scholar]
- 47.Hedrick J, Zlotnik A. Identification and characterization of a novel beta chemokine containing six conserved cysteines. J Immunol. 1997;159:1589–1593. [PubMed] [Google Scholar]
- 48.Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, Zou YR, Littman DR, Cyster JG. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194:45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]