

Abstract
Alzheimer's disease is the leading cause of dementia among the elderly, and with the ever-increasing size of this population, cases of Alzheimer's disease are expected to triple over the next 50 years. Consequently, the development of treatments that slow or halt the disease progression have become imperative to both improve the quality of life for patients as well as reduce the health care costs attributable to Alzheimer's disease. Here, we demonstrate that the active component of marijuana, Δ9-tetrahydrocannabinol (THC), competitively inhibits the enzyme acetylcholinesterase (AChE) as well as prevents AChE-induced amyloid β-peptide (Aβ) aggregation, the key pathological marker of Alzheimer's disease. Computational modeling of the THC-AChE interaction revealed that THC binds in the peripheral anionic site of AChE, the critical region involved in amyloidgenesis. Compared to currently approved drugs prescribed for the treatment of Alzheimer's disease, THC is a considerably superior inhibitor of Aβ aggregation, and this study provides a previously unrecognized molecular mechanism through which cannabinoid molecules may directly impact the progression of this debilitating disease.
Keywords: Cannabinoids, Alzheimer's disease, Acetylcholinesterase
Introduction
Since the characterization of the Cannabis sativa-produced cannabinoid, Δ9-tetrahydrocannabinol (THC) (Figure 1), in the 1960's,1 this natural product has been widely explored as an anti-emetic, anti-convulsive, anti-inflammatory, and analgesic.2 In these contexts, efficacy results from THC binding to the family of cannabinoid receptors found primarily on central and peripheral neurons (CB1) or immune cells (CB2).3 More recently, a link between the endocannabinoid system and Alzheimer's disease has been discovered4 which has provided a new therapeutic target for the treatment of patients suffering from Alzheimer's disease.5 New targets for this debilitating disease are critical as Alzheimer's disease afflicts over 20 million people worldwide, with the number of diagnosed cases continuing to rise at an exponential rate.6,7 These studies have demonstrated the ability of cannabinoids to provide neuroprotection against β-amyloid peptide (Aβ) toxicity.8-10 Yet, it is important to note that in these reports, cannabinoids serve as signaling molecules which regulate downstream events implicated in Alzheimer's disease pathology and are not directly implicated as effecting Aβ at a molecular level.
Figure 1.

Chemical structure of Δ9-tetrahydrocannabinol (THC).
One of the primary neuropathological hallmarks of Alzheimer's disease is deposition of Aβ into amyloid plaques in areas of the brain important for memory and cognition.11 Over the last two decades, the etiology of Alzheimer's disease has been elucidated through extensive biochemical and neurobiological studies, leading to an assortment of possible therapeutic strategies including prevention of downstream neurotoxic events, interference with Aβ metabolism, and reduction of damage from oxidative stress and inflammation.12 The impairment of the cholinergic system is the most dramatic of the neurotransmitter systems affected by Alzheimer's disease and as a result, has been thoroughly investigated. Currently, there are four FDA-approved drugs that treat the symptoms of Alzheimer's disease by inhibiting the active site of acetylcholinesterase (AChE), the enzyme responsible for the degradation of acetylcholine, thereby raising the levels of neurotransmitter in the synaptic cleft.13 In addition, AChE has been shown to play a further role in Alzheimer's disease by acting as a molecular chaperone, accelerating the formation of amyloid fibrils in the brain and forming stable complexes with Aβ at a region known as the peripheral anionic binding site (PAS).14,15 Evidence supporting this theory was provided by studies demonstrating that the PAS ligand, propidium, is able to prevent amyloid acceleration in vitro, whereas active-site inhibitors had no effect.16 Due to the association between the AChE PAS and Alzheimer's disease, a number of studies have focused on blocking this allosteric site.17 Recently, we reported a combined computational and experimental approach to identify compounds containing rigid, aromatic scaffolds hypothesized to disrupt protein-protein interactions.18-20 Similarly, THC is highly lipophilic in nature and possesses a fused tricyclic structure. Thus, we hypothesized that this terpenoid also could bind to the allosteric PAS of AChE with concomitant prevention of AChE-promoted Aβ aggregation.
Experimental Section
Docking procedures
THC was docked to the mouse AChE structure (PDB ID code 1J07) using AutoDock 3.0.5.21 Twenty docking runs (100 million energy evaluations each) were run with a 26.25 Å × 18.75 Å × 26.25 Å grid box with 0.375 Å grid spacing. This grid box was designed to include regions of both the catalytic site and the peripheral anionic site. Otherwise, standard docking settings were used for the AutoDock calculations, as previously detailed.18
Acetylcholinesterase inhibition studies
All assays were performed using a Cary 50 Bio UV-visible spectrophotometer using an 18-cell changer, and conducted at 37 °C, using a Cary PCB 150 Water Peltier System. Solutions of acetylthiocholine iodide (ATCh iodide) and 5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB) were prepared according to the method of Ellman, et al.22 Stock solutions of acetylcholinesterase from E. electricus were prepared by dissolving commercially available enzyme in 1% gelatin. Prior to use, an aliquot of the gelatin solution was diluted 1:200 in water. For the assay, the solution was diluted until enzyme activity between 0.10-0.13 AU/min at 500 μM ACTh iodide was obtained. Compounds were prepared as solutions in methanol.
Assays were performed by mixing AChE, THC, and 340 μM DTNB in 100 mM phosphate buffer, pH 8.0, containing 5% methanol. Solutions were incubated at 37 °C for five minutes before the reaction was initiated by the addition of ATCh iodide (75 – 300 μM). The increase of absorbance at 412 nm was monitored for 2 to 5 min. All assays were run in triplicate. Initial rates were determined by subtracting the average observed initial rate from the non-enzymatic reaction.
Linear regression analysis of reciprocal plots of 1/νo versus 1/[S] for four THC concentrations was performed using Microsoft Excel software. The slope1/v was plotted against [I] to give Ki values. Propagation of error was performed to determine the error, ΔKi.
For studies to determine the mutual exclusivity of THC and propidium iodide, experiments were performed identically to simple THC inhibition studies with a fixed concentration of ACTh iodide (125 μM), and varied concentrations of propidium iodide (0-25 μM) and THC (0-15 μM).
AChE-induced β-amyloid peptide aggregation in the presence of AChE ligands
The aggregation of the β-amyloid peptide was measured using the thioflavin T-based fluorometric assay as described by LeVine23 and Bartolini.16 Assays were measured using a SpectraMAX Gemini fluorescence plate reader with SOFTmax PRO 2.6.1 software. Aβ1-40 stock solutions were prepared in DMSO and HuAChE stocks prepared in distilled water. All stock solutions of Aβ and HuAChE were used immediately after preparation.
In a 96-well plate, triplicate samples of a 20 μL solution of 23 nM of Aβ, 2.30 μM HuAChE and various concentrations of THC in 0.215 M sodium phosphate buffer, pH 8.0 were prepared. These solutions were incubated at room temperature along with triplicate solutions of Aβ alone, Aβ and AChE, and Aβ plus THC at various concentrations. After 48 h, a 2 μL aliquot was removed from each well, placed in a black-walled, clear-bottomed 96-well plate, and diluted with 50 mM glycine-NaOH buffer, pH 8.5, containing 1.5 μM thioflavin T to a total volume of 200 μL. After incubating for 5 min, the fluorescence was measured using λexc = 466 nm and λem = 490 nm with excitation and emission slits of 2 nm. The fluorescence emission spectrum was recorded between 450 and 600 nm, with excitation at 446 nm.
The fluorescence intensities were averaged, and the average background fluorescence of buffer only, or buffer and THC, was subtracted. The corrected fluorescence values were plotted with their standard deviation. The equation, Fi/Fo × 100%, where Fi is the fluorescence of AChE, Aβ, and THC, and Fo is the fluorescence of AChE and Aβ, was used to quantify the extent to which each compound inhibits Aβ aggregation. The student's t-test function of Microsoft Excel was used to determine p values and assess statistical significance between reactions.
Control experiments containing AChE, THC, and thioflavin T or AChE and thioflavin T alone were also performed to ensure that any observed fluorescence decrease was not attributable to the molecular rotor properties of thioflavin T upon binding to AChE. For these reactions, all concentrations were identical to those used in the described Aβ aggregation assays (vide supra).
Results and Discussion
THC binding to AChE initially was modeled in silico using AutoDock 3.0.5.21 Twenty docking runs with 100 million energy evaluations each were performed with a 26.25 Å × 18.75 Å × 26.25 Å grid box with 0.375 Å grid spacing, which included regions of both the catalytic site and the PAS. Examination of the docking results revealed that THC was predicted to bind to AChE with comparable affinity to the best reported PAS binders, with the primary binding interaction observed between the ABC fused ring of the THC scaffold and the Trp86 indole side chain of AChE (Figure 2). Further interactions were also evident between THC and the backbone carbonyls of Phe123 and Ser125. Encouraged by these results, we tested the ability of THC to inhibit AChE catalytic activity. Steady-state kinetic analysis of THC inhibition revealed that THC competitively inhibits AChE (Ki = 10.2 μM) (Figure 3A). This level of inhibition is relatively modest, yet it is important to note that inhibition of acetylcholine cleavage is not a prerequisite for effective reduction of Aβ aggregation; indeed, most PAS binders are moderate AChE inhibitors displaying either non-competitive or mixed-type inhibition.16 While THC shows competitive inhibition relative to the substrate, this does not necessitate a direct interaction between THC and the AChE active site. In fact, given the proximity of the PAS to the protein channel leading to the catalytic triad active site, it is possible to block substrate entry into the active site while bound to the PAS, thus preventing the formation of an ESI complex.18,24 In order to test this hypothesis, additional kinetic experiments were performed to determine the mutual exclusivity of THC and propidium, a well characterized purely noncompetitive AChE inhibitor and PAS binder. Dixon plots of ν-1 versus propidium concentration at varying concentrations of THC returned a series of parallel lines, indicating that THC and propidium cannot bind simultaneously to AChE (Figure 3B). Thus, these studies verify our docking results and demonstrate that THC and propidium are mutually exclusive PAS inhibitors. Additionally, recent reports have suggested that the selectivity of a given inhibitor for AChE over butyrylcholinesterase (BuChE) can be correlated with the ability of a compound to block AChE-accelerated Aβ aggregation.25,26 Kinetic examination of BuChE inhibition revealed a slight reduction in enzymatic activity at high concentrations of THC (IC50 ≥ 100 μM); however, these experiments were limited by the poor solubility of THC in aqueous solution.
Figure 2.
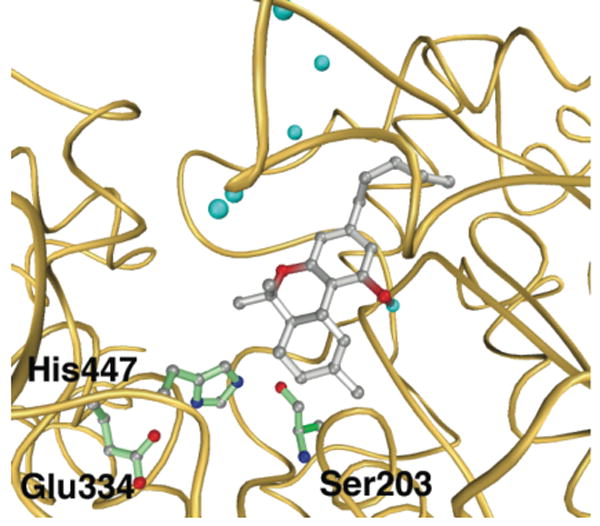
Predicted binding mode of THC (gray) to AChE (orange ribbon). The catalytic triad residues of AChE (green) and water molecules included in the docking calculations (light blue spheres) are shown.
Figure 3.
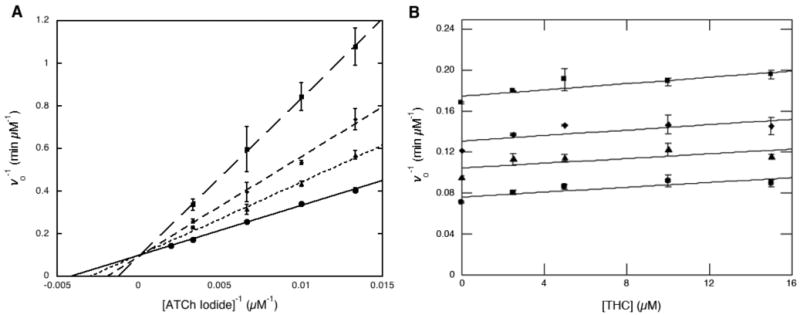
(A) Kinetic analysis of AChE inhibition by 0.0 (●), 6.25 (▲), 12.5 (◆), and 25.0 μM (■) THC. Steady state kinetic analysis was performed using acetylthiocholine (75-300 μM) and Ellman's reagent (340 μM) at 37 °C. (B) Dixon plots of 1/v versus [THC] at different fixed concentrations of propidium iodide: 0 (●), 6.25 (▲), 12.5 (◆), and 25 μM (■).
The activity of THC towards the inhibition of Aβ aggregation was then investigated using a thioflavin T (ThT)-based fluorometric assay to stain putative Aβ fibrils.23 Using this assay, we found that THC is an effective inhibitor of the amyloidogenic effect of AChE (Figure 4). In fact, at a concentration of 50 μM, propidium does not fully prevent AChE-induced aggregation (p = 0.03, student's T-test), while THC completely blocks the AChE effect on Aβ aggregation, with significantly greater inhibition than propidium (p = 0.04, student's T-test), one of the most effective aggregation inhibitors reported to date.16 However, the observed decrease in fluorescence could also be rationalized as a result of a competition between THC and ThT for the same site on AChE. It has been shown that ThT also can bind to the PAS and that this binding leads to an increase in fluorescence. Presumably, this phenomenon results from ThT serving as a molecular rotor in which fluorescence quantum yield is sensitive to the intrinsic rotational relaxation; thus, when molecular rotation is slowed by protein binding, the quantum yield of the molecule can increase dramatically.27,28 In order to ensure that the observed fluorescence decrease was due to fibril inhibition, control experiments were performed using AChE, THC, and ThT. Reactions containing AChE and ThT alone showed the same fluorescence output as those containing AChE, THC, and ThT, providing convincing evidence that any observed reduction in fluorescence can be attributed to fewer Aβ fibrils.
Figure 4.
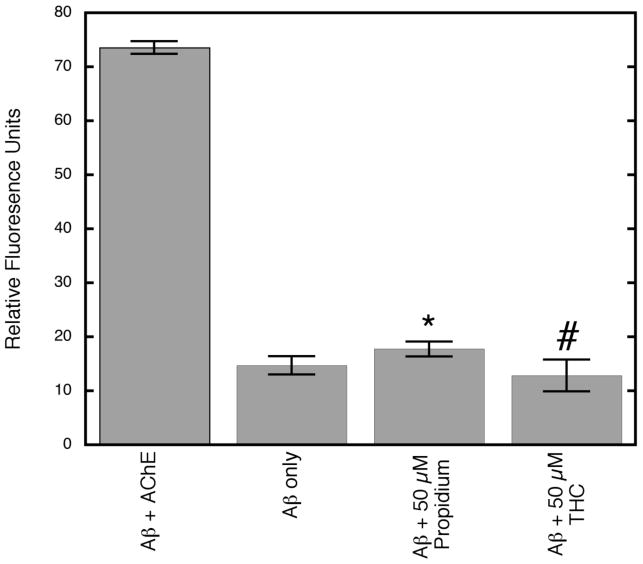
Inhibition of AChE-induced Aβ aggregation by THC and propidium (* p < 0.05 versus Aβ only; # p < 0.05 versus Aβ + propidium).
Conclusion
We have demonstrated that THC competitively inhibits AChE, and furthermore, binds to the AChE PAS and diminishes Aβ aggregation. In contrast to previous studies aimed at utilizing cannabinoids in Alzheimer's disease therapy,8-10 our results provide a mechanism whereby the THC molecule can directly impact Alzheimer's disease pathology. We note that while THC provides an interesting Alzheimer's disease drug lead, it is a psychoactive compound with strong affinity for endogenous cannabinoid receptors. It is noteworthy that THC is a considerably more effective inhibitor of AChE-induced Aβ deposition than the approved drugs for Alzheimer's disease treatment, donepezil and tacrine, which reduced Aβ aggregation by only 22% and 7%, respectively, at twice the concentration used in our studies.7 Therefore, AChE inhibitors such as THC and its analogues may provide an improved therapeutic for Alzheimer's disease, augmenting acetylcholine levels by preventing neurotransmitter degradation and reducing Aβ aggregation, thereby simultaneously treating both the symptoms and progression of Alzheimer's disease.
Acknowledgments
This work was supported by the Skaggs Institute for Chemical Biology and a NIH Kirschstein National Research Service Award to L.M.E.
References
- 1.Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86:1646–1650. [Google Scholar]
- 2.Carlini EA. The good and the bad effects of (−) trans-delta-9-tetrahydrocannabinol (Δ9-THC) on humans. Toxicon. 2004;44:461–467. doi: 10.1016/j.toxicon.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 4.Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, Romero J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer's disease brains. J Neurosci. 2003;23:11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pazos MR, Núñez E, Benito C, Tolón RM, Romero J. Role of the endocannabinoid system in Alzheimer's disease: new perspectives. Life Sci. 2004;75:1907–1915. doi: 10.1016/j.lfs.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 6.Ritchie K, Kildea D. Is senile dementia “age-related” or “ageing-related”?-evidence from meta-analysis of dementia prevalence in the oldest old. Lancet. 1990;346:931–934. doi: 10.1016/s0140-6736(95)91556-7. [DOI] [PubMed] [Google Scholar]
- 7.Evans DA. Estimated prevalence of Alzheimer's disease in the United States. Milbank Q. 1990;68:267–289. [PubMed] [Google Scholar]
- 8.Milton NG. Anandamide and noladin ether prevent neurotoxicity of the human amyloid-β peptide. Neurosci Lett. 2002;332:127–130. doi: 10.1016/s0304-3940(02)00936-9. [DOI] [PubMed] [Google Scholar]
- 9.Iuvone T, Esposito R, Santamaria R, Di Rosa M, Izzo AA. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J Neurochem. 2004;89:134–141. doi: 10.1111/j.1471-4159.2003.02327.x. [DOI] [PubMed] [Google Scholar]
- 10.Ramírez BG, Blázquez C, Gómez del Pulgar T, Guzmán M, de Ceballos ML. Prevention of Alzheimer's disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozemuller JM, Eikelenboom P, Stam FC, Beyreuther K, Masters CL. A4 protein in Alzheimer's disease: primary and secondary cellular events in extracellular amyloid deposition. J Neuropathol Exp Neurol. 1989;48:674–691. doi: 10.1097/00005072-198911000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Bachurin SO. Medicinal chemistry approaches for the treatment and prevention of Alzheimer's disease. Med Res Rev. 2003;23:48–88. doi: 10.1002/med.10026. [DOI] [PubMed] [Google Scholar]
- 13.Racchi M, Mazzucchelli M, Porrello E, Lanni C, Govoni S. Acetylcholinesterase inhibitors: novel activities of old molecules. Pharmacol Res. 2004;50:441–451. doi: 10.1016/j.phrs.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 14.Inestrosa NC, Alvarez A, Pecez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 15.Alvarez A, Alarcon A, Opazo C, Campos EO, Munoz FJ, Calderon FH, Dajas F, Gentry MK, Doctor BP, De Mello FG, Inestrosa NC. Stable complexes involving acetylcholinesterase and amyloid-β peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer's fibrils. J Neurosci. 1998;18:3213–3223. doi: 10.1523/JNEUROSCI.18-09-03213.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartolini M, Bertucci C, Cavrini V, Andrisano V. β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem Pharmacol. 2003;65:407–416. doi: 10.1016/s0006-2952(02)01514-9. [DOI] [PubMed] [Google Scholar]
- 17.Johnson G, Moore SW. The peripheral anionic site of acetylcholinesterase: structure, functions and potential role in rational drug design. Curr Pharm Des. 2006;12:217–225. doi: 10.2174/138161206775193127. [DOI] [PubMed] [Google Scholar]
- 18.Dickerson TJ, Beuscher AE, IV, Rogers CJ, Hixon MS, Yamamoto N, Xu Y, Olson AJ, Janda KD. Discovery of acetylcholinesterase peripheral anionic site ligands through computational refinement of a directed library. Biochemistry. 2005;44:14845–14853. doi: 10.1021/bi051613x. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. A credit-card library approach for disrupting protein-protein interactions. Bioorg Med Chem. 2006;14:2660–2673. doi: 10.1016/j.bmc.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 20.Xu Y, Lu H, Kennedy JP, Yan X, McAllister LA, Yamamoto N, Moss JA, Boldt GE, Jiang S, Janda KD. Evaluation of “credit card” libraries for inhibition of HIV-1 gp41 fusogenic core formation. J Comb Chem. doi: 10.1021/cc0600167. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 22.Ellman GL, Courtney KD, Andres V, Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 23.LeVine H., III Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Science. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szegletes T, Mallender WD, Rosenberry TL. Nonequilibrium analysis alters the mechanistic interpretation of inhibition of acetylcholinesterase by peripheral site ligands. Biochemistry. 1998;37:4206–4216. doi: 10.1021/bi972158a. [DOI] [PubMed] [Google Scholar]
- 25.Piazzi L, Rampa A, Bisi A, Gobbi S, Belluti F, Cavalli A, Bartolini M, Andrisano V, Valenti P, Recanatini M. 3-(4-{[Benzyl(methyl)amino]methyl}-phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: A dual function lead for Alzheimer's disease therapy. J Med Chem. 2003;46:2279–2282. doi: 10.1021/jm0340602. [DOI] [PubMed] [Google Scholar]
- 26.Belluti F, Rampa A, Piazzi L, Bisi A, Gobbi S, Bartolini M, Andrisano V, Cavalli A, Recanatini M, Valenti P. Cholinesterase inhibitors: Xanthostigmine derivatives blocking the acetylcholinesterase-induced β-amyloid aggregation. J Med Chem. 2005;48:4444–4456. doi: 10.1021/jm049515h. [DOI] [PubMed] [Google Scholar]
- 27.De Ferrari GV, Mallender WD, Inestrosa NC, Rosenberry TL. Thioflavin T is a fluorescent probe of the acetylcholinesterase peripheral site that reveals conformational interactions between the peripheral and acylation sites. J Biol Chem. 2001;276:23282–23287. doi: 10.1074/jbc.M009596200. [DOI] [PubMed] [Google Scholar]
- 28.Viriot ML, Carre MC, Geoffroy-Chapotot C, Brembilla A, Muller S, Stoltz JF. Molecular rotors as fluorescent probes for biological studies. Clin Hemorheol Microcirc. 1998;19:151–160. [PubMed] [Google Scholar]