

Abstract
Human cancers have been found to include transformed stem cells that may drive cancer progression to metastasis. Here we report that metastatic colon cancer contains clonally derived tumor cells with all of the critical properties expected of stem cells, including self-renewal and to the ability to differentiate into mature colon cells. Additionally, when injected into mice, these cells initiated tumors that closely resemble human cancer. Karyotype analyses of parental and clonally-derived tumor cells expressed many consistent (clonal), along with unique chromosomal aberrations, suggesting the presence of chromosomal instability in the cancer stem cells. Thus, this new model for cancer origin and metastatic progression includes features of both the hierarchical model for cancerous stem cells and the stochastic model, driven by the observation of chromosomal instability.
Keywords: Chromosomal Instability in Cancer Stem Cells
Introduction
Colon cancer is one of the most common forms of cancer in the industrialized world (1). As with most cancers, colon cancer is a heterogeneous disease that starts locally, but generally spreads through the lymphatic or portal venous system to different organs (2). The liver is the most frequent visceral site of metastatic dissemination (3). It is the initial site of metastasis in the majority of recurring colon cancers, with most patients showing liver involvement at the time of death. It is widely believed that the successful metastatic cancer cell must leave the primary tumor, enter the lymphatic or circulatory systems, survive within the circulation, overcome host defenses, extravasate and then grow as a vascularized metastatic tumor (4). We know that primary tumors shed large numbers of tumor cells, but only a few of these develop into metastases (5). Two models have been proposed to explain the heterogeneous potential of tumor cells and the process of metastasis in general: the stochastic model whereby a distinct population of tumor cells acquires the appropriate set of somatic mutations and develops metastatic capability, and the hierarchical model in which primary tumors and metastatic cancer are initiated by rare cancer stem cells (6, 7). Recently, several studies have identified the presence of cancer initiating cells in breast (8), brain tumors (9–11) and most recently, colon cancer (12–14). With this new insight into the cellular mechanism leading to cancer, it has been suggested that the metastatic potential of tumor cells may be a reflection of the ability of the cancer stem cells to clonally initiate tumorigenicity at distant sites (15, 16). Although strong evidence has been reported for the presence of cancer initiating cells in colon cancer, limited support has surfaced for a possible role of tissue derived stem cells in the initiation of this cancer and its metastatic process. One of the technical difficulties in answering this question is that normal and abnormal colon epithelial cells thus far have proven resistant to growth in culture. To investigate the presence of stem-like tumor cells in metastatic colon cancers, we utilized an in vitro assay, which provides efficient propagation of tumor cells and a functional demonstration of self-renewal and mutipotency at the clonal level.
Results
In vitro selection and expansion of tumor cells derived from metastatic colon cancer
Human colorectal tumor specimens, typically moderately to well-differentiated gland-producing adenocarcinomas, were collected from the metastatic liver site of 13 different Caucasian patients who underwent surgical resection (Table 1). None of the patients in this study had an identified genetic predisposition to colon cancer, such as familial adenomatous polyposis or hereditary nonpolyposis colorectal cancer. Tumor cells were cultured under conditions that favored epithelial cell growth (17). We postulated that in vitro cultures of tumor cells and their expansion would select for a population of tumor cells best adapted for culture and self-renewal. The rationale for this approach is that cancer stem cells have the capacity for self-renewal and this method should allow us to expand these primitive cells in vitro. Within two to four weeks of culture, a minor fraction of tumor cells in each of the 13 tumors studied demonstrated growth into what appeared to be clonally-derived colonies of epithelial cells. The generation of cell colonies from disaggregated tumors provides an index of tumor cell clonogenicity. In our hands, the frequency of tumor cell colony formation was estimated at less than 0.1% of the total cells, with anywhere from a few to tens of colonies per million cells isolated from the tumors. Long-term (more than three passages) patient-specific cultures of tumor cells were established for all 13 cultures (Figure 1A). When observed in vitro, the majority of tumor cells exhibited a similar undifferentiated phenotype, with colonies comprised of hundreds of epithelial cells. In each of the cultures, some of the tumor cell colonies changed gradually over time, from an undifferentiated to a more mature phenotype, ranging from the formation of an organized “ridge” of epithelial cells to large glandular formations resembling aberrant crypts (Figure 1A). This pleiomorphic maturation observed in the cultures of human colon tumor cells provides support for cellular organization in metastatic colon cancer and may have some relevance to the model of cancer stem cells. It appears that a hierarchy of tumor cells is emerging among the cultured cells from undifferentiated primitive cells to more mature cells.
Table 1.
Summary of the patient demographics, treatment, tumor site, histopathology, and culture information. All 13 tissue samples were adenocarcinomas isolated from patients with resectable colorectal hepatic metastases. Long-term cultures were established from most tumor cell samples. Long-term cultures were those ranging from several passages to more than 10 passages. In vitro differentiation was defined as the presence of pleiomorphic maturation and the expression of lineage markers. ND: not determined and chemo: chemotherapy treatment.
| Tumor/Patient | Sex | Age | Origin | Grading/Diagnosis | Treatment | Long-Term Culture | In Vitro Differentiation |
|---|---|---|---|---|---|---|---|
| Tu-7 | male | 55 | colon | Moderately differentiated | none | Yes | Yes |
| Tu-10 | male | 72 | rectal | Moderately well-differentiated | none | Yes | Yes |
| Tu-11 | female | 58 | colon | Moderately differentiated | 6 months systemic chemo | Yes | Yes |
| Tu-12 | female | 65 | colon | Moderately well-differentiated | 3 months systemic chemo | Yes | Yes |
| Tu-14 | female | 55 | rectal | Moderately differentiated | 3 months systemic chemo | Yes | Yes |
| Tu-17 | female | 70 | colon | Moderately differentiated | none | Yes | ND |
| Tu-18 | male | 52 | rectal | Moderately differentiated | 6 months systemic chemo | Yes | Yes |
| Tu-19 | male | 60 | rectal | Moderately differentiated | none | Yes | ND |
| Tu-21 | male | 54 | rectal | Moderately differentiated | 6 months systemic chemo | Yes | Yes |
| Tu-22 | female | 55 | colon | Moderately differentiated | 6 months systemic chemo | Yes | Yes |
| Tu-25 | male | 59 | colon | Moderately differentiated | none | Yes | Yes |
| Tu-27 | female | 84 | colon | Moderately differentiated | none | Yes | Yes |
| Tu-28 | male | 37 | rectal | Moderately differentiated | 3 months systemic chemo | Yes | Yes |
Figure 1. In vitro expansion of metastatic colon cancer in the presence of irradiated stromal cells.
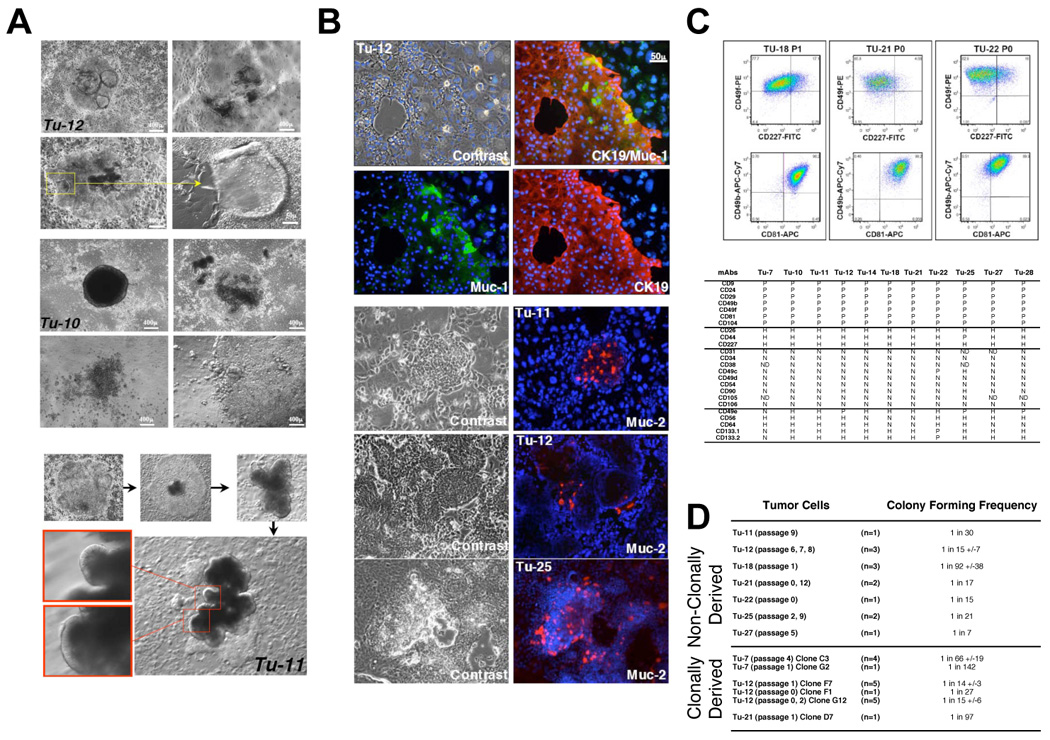
A, tumor cells derived from different patients formed similar organotypic structures, with glandular organization indicating maturation potential. Photomicrographs of metastatic colon tumors Tu-12 and Tu-10 presenting pleiomorphic maturation. For Tu-11, photomicrographs correspond to sequential representation of tumor colony growth and maturation. Tumor cells exhibit an undifferentiated phenotype and differentiation occurs gradually with the formation of organized epithelial structures resembling abnormal crypts. B, Immunohistochemical analyses of Tu-11, Tu-12 and Tu-25 in culture. Immunostaining with anti-CK19 (Alexa-594, red) differentiated human tumor cells from stroma cells. Both Muc-1 (Alexa-488, green) and Muc-2 (Alexa-594, red) positive human cells are found in rare populations of tumor cells. The nuclear counterstain was Hoechst 33342. C, Flow cytometric dot plot profiles of metastatic colon tumor cells isolated from different patients and cultured under identical conditions. Density plots using pseudo-color for Tu-18 (passage 1), Tu-21 (passage 0) and Tu-22 (passage 0). The dot plots depict two-color staining of live tumor cells from three patients on a logarithmic scale. Quadrant markers were positioned to include in the lower left quadrant greater than 98% of control unstained live human tumor cells (not shown). The numbers are the percentage of live cells present in each quadrant after staining. The summary table of Panel C represents flow cytometric analyses of tumor cells. Tumor samples expanded in culture revealed a common flow cytometric profile suggesting the expansion of a similar population of cancer cells. Flow cytometric analyses of multiple passages were performed for each tumor and summarized in this table, Tu-7 (passage 2, 4), Tu-10 (passage 0, 1), Tu-11 (passage 0, 1), Tu-12 (passage 0, 1, 3, 4, 6, 7, 8), Tu-14 (passage 0, 1, 2), Tu-18 (passage 0, 1, 2), Tu-21 (passage 0, 1), Tu-22 (passage 0, 1, 3), Tu-25 (passage 0, 1, 2) Tu-27 (passage 2, 4) and Tu-28 (passage 2). P, positive when all of the tumor cells were positive for the surface marker (seven markers in all of the tumor samples), H, heterogeneous populations of tumor cells with some cells positive and other cells negative among the tumor cell populations (three markers in most of the tumor samples). N, all tumor cells negative for the surface marker (nine markers in most of the tumor samples). Five markers, including CD133 were inconsistent among the tumor samples studied. ND, not determined. Passage 0 corresponds to the first in vitro expansion. D, Summary of colony forming frequency after limiting dilution analyses (LDA) on parental (non-clonally derived) and clonally derived tumor cells. Cultured tumor cells were sorted into 96-well plates at limiting dilutions using positive cell surface markers present on human cells. Cultures were stopped after 2 to 4 weeks and colonies visualized. Regression analyses were performed to determine the frequency of tumor colony forming cells. When n≥3 experiments, standard deviation was calculated using the “nonbiased” method.
Tumor cells derived from metastatic colon cancer display distinct colon cell lineage commitment in vitro
Several markers are associated with more differentiated phenotypes and mature cells of the intestine. These include the epithelial mucins, secretory and non-secretory proteins, within colorectal tissues that have been traced to different lineages (18). Muc-2 is secreted by differentiated intestinal cells and is predominant within colorectal mucus-producing goblet cells, whereas Muc-1 (CD227), a non-secretory and cell membrane-associated glycoprotein, is expressed by certain absorptive enterocytes along the columnar cell apexes in the glandular tissue (18, 19). Villin, a component of the intestinal brush border cytoskeleton, is also a marker of enterocytes, but can be expressed weakly in immature colon cells (20). Lastly, chromogranin A expression is restricted to the rare enteroendocrine cells (21). We first determine whether tumor cells expanded in vitro expressed these distinct lineage-specific markers. Muc-1, Muc-2 (Figure 1B) and Villin positive cells (not shown) were present in all metastatic colon tumor cultures. The mucin-secreting cells were not well–differentiated, with few vacuoles visible. No positive chromogranin A cells could be identified in the cultured tumor cells. Although endocrine differentiation is detected in colorectal carcinomas (21) and in our xenograft model (Figure 3C), only few cell lines have been shown to be capable of such lineage differentiation in vitro (20). Overall, our result indicates the presence of two possible lineage restricted cell types, enterocytes and goblet cells, after in vitro expansion of tumor cells from metastatic colon cancer.
Figure 3. Tumorigenicity and multilineage commitment of clonally derived metastatic colon tumor cells.

A, CEA secretion in vitro in long-term cultures and in vivo in Rag2/γc−/− mouse sera after tumor cell transplantation. Parental and clonal cultures (derived from one single tumor cell) expressed high levels of CEA in vitro. The transplanted animals with engrafted tumor cells were identified by their CEA secretion. Tu-7, Tu-12 and Tu-14 were expanded in vitro before transplantation. Tu-18, Tu-19 and Tu-22 were tumor cells transplanted in mice without previous in vitro expansion. P, passage. B, Xenograft tumors in mice injected subcutaneously with metastatic colon cancer derived from three patients. Frozen sections of Tu-12 (expanded in vitro, 8 weeks after transplantation), Tu-18 (not expanded, 6 weeks after transplantation), and Tu-22 (not expanded, 8 weeks after transplantation) were stained with anti-HLA-ABC (Alexa-488, green) and anti-Muc-2 (Alexa-594, red). Nuclear counterstaining was done with Hoechst 33342 (blue). The observed heterogeneous expression of Muc-2 demonstrates the presence of goblet cells in the moderately to well-differentiated adenocarcinomas. HLA negative cells present around the xenograft represent the murine stromal cells. C, Multilineage commitment in clonally derived tumor cells. Serial sections of Tu-12 clone F7 and non-serial sections of Tu-21 E12 were stained for HLA-ABC (Alexa-488, green) and Muc-1, Muc-2, chromogranin A (CgA) or villin (Alexa-594, red). Nuclear counterstaining was done with Hoechst 33342 (blue). Tumor generated from one cancer cell generated both columnar cells (Muc-1+ and villin+ cells) as well as goblet cells (Muc-2+ cells). Rare CgA+ enteroendocrin cells were detected in Tu-21 but not in Tu-12. D, Goblet cell maturation in xenograft tumors of Tu-12 (clone F7). Left panel, transmission electron micrograph of xenograft section showing the presence in tumor tissues of mucous-containing goblet cells (G) surrounded by columnar cells with apical microvilli and tight junctions. Right upper panel, Muc-2 staining (red) and Hoechst 33342 (blue) showing mature goblet cells. Not every aberrant crypt contains Muc-2 positive goblet cells. Right lower panel, HLA-ABC (green) and Muc-2 (red) staining with Hoechst 33342 (blue) showing both goblet and columnar cells present in xenograft tumors.
Tumor cells derived from metastatic colon cancer share similar flow cytometric profiles after in vitro expansion
We analyzed the flow cytometric profile of 11 of the 13 patient-specific tumor cells (Figure 1C). After in vitro expansion. single-cell suspensions of the various cultured tumor cells were prepared and analyzed by flow cytometry using a panel of 24 different cell-surface markers. Nineteen of the markers showed consistent results among all 11 cultures (7 positive, 9 negative and 3 heterogeneous markers) while 5 markers differed from patient to patient. Analyses of sequential passages per culture revealed a stable phenotype of the cell surface marker repertoire among tumor cell populations in culture (Figure 1C). Based on the recent findings described in the literature, we have included other markers in our study. The cell surface markers, CD133, CD44, CD49f, EpCam and CD166, were recently reported as present on human colon cancer stem cells (12–14). CD44 and CD49f were present in all of our tumor cell cultures, while CD133 antigen (CD133.1 and CD133.2) was not consistently expressed among the cultured samples and was absent in at least one culture. Interestingly, another more recent study (13) also reported that CD133 was not consistently expressed in all colon cancer samples. A limited analysis of several tumor cultures for CD166 and EpCam showed that both markers are positive on our tumor cell cultures (Figure 4A). Based on the above results, cell-surface markers identified on colon cancer stem cells are expressed on the tumor cells propagated in vitro.
Figure 4. Detection of colon cancer stem cells markers after in vitro expansion.
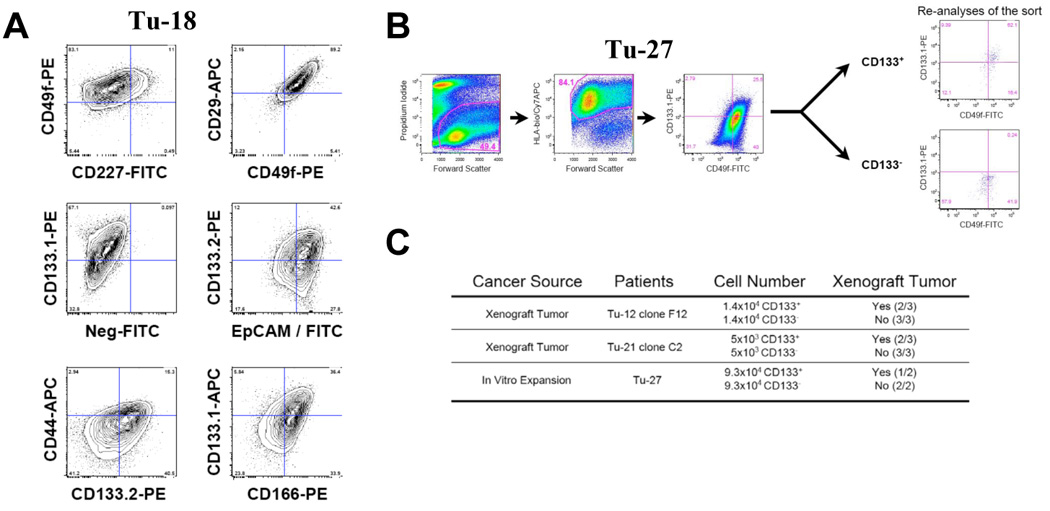
A, Flow cytometric contour plots (5% probability) demonstrating the expression of cancer stem cell markers on one representative culture of tumor tissues from metastatatic colon cancer (Tu-18). The dot plots depict two-color staining of live tumor cells from Tu-18 patient on a logarithmic scale. Quadrant markers were positioned to include in the lower left quadrant greater than 98% of control unstained live human tumor cells (not shown). The numbers are the percentage of live cells present in each quadrant after staining. B, Flow cytometric dot plots indicating the gating strategy and result of the CD133 positive and negative cell isolation. The dot plots depict two-color staining of live tumor cells from Tu-27 on a logarithmic scale. First plot, propidium iodide/forward scatter indicates the gate for live cells (49.4%), second plot indicates the gate for human tumor cells (84.1%, HLA+) and the third plot is the staining for CD133 (and CD49f). Quadrant markers were positioned to include in the lower left quadrant greater than 98% of control unstained live human tumor cells (not shown). The numbers are the percentage of live cells present in each quadrant after staining. The re-analyses of the isolated CD133+ and CD133− subpopulations are presented. C, Table of tumorigenic activity of CD133+ and CD133− human colon cancer cells. For Tu-12 and Tu-21, clones derived from a single cell were expanded in vitro and transplanted in mice to generate xenograft tumors described in the result section. Tumor cells isolated from the xenograft or from the culture (Tu-27) were sorted for CD133+ and CD133− cell populations as described. Cell populations with identical numbers of CD133+ and CD133− cells were transplanted to the left (CD133−) and right (CD133+) flanks of immunodeficient mice. Tumors were collected six to twelve weeks after transplantation.
In vitro self-renewing capacity and clonogenic potential of malignant colon epithelia derived from metastatic colon cancer
The clonogenic capacity of metastatic colon tumor cell cultures was assayed in vitro by limiting dilution analysis (LDA). LDA is a quantitative measurement to study the enrichment of stem cells with stem cell-enriched fractions giving a higher response as measured by the frequency of colony formation (22–25). This classic stem cell assay has been described by others for the presence of hematopoietic stem cells in long-term bone marrow cultures (26) or the presence of neural stem cells in the formation of neurosphere cultures for the central nervous system (27). We applied this stem cell assay to our candidate cancer stem cell population. Tumor cells from seven different cultures were sorted and plated by limiting dilution. Three to four weeks after plating, the colony frequency of tumor cells was calculated by linear regression (25). The frequency of colony forming tumor cells ranged from 1/7 (Tu-27) to 1/92 (Tu-18) (Figure 1D). These data revealed a dramatic enrichment of colony forming cells after in vitro expansion compared to the low number of colonies generated by the initial primary tumor cell cultures. This result suggests that we have enriched for a primitive population of clonogenic tumor cells capable of physiological self-renewal in vitro, as defined by the ability to go through numerous cell division cycles while maintaining the undifferentiated state.
Clonogenic tumor cells propagated in vitro are capable of self-renewal and multilineage differentiation
Next, we determined if, like the parental tumor cell cultures, clonally-derived tumor cell cultures are capable of multilineage differentiation. We expanded 23 clonally derived tumor cell cultures from five different patients (Figure 2A). These cultures, each derived from a single cell, behaved similarly and demonstrated the same pleiomorphic colonies as the parental cultures (Figure 2B). Clonally derived cultures were repeatedly passaged with no sign of replicative senescence, as demonstrated by their high efficiency of forming colonies in vitro (Figure 1D). Distinct tumor cells stained positively for Muc-2 or Muc-1, indicating the presence of both goblets and enterocytes (Figure 2C), similar to the parental cultures (Figure 1B). Villin was weakly positive in numerous undifferentiated cells (not shown), but cells at the apical membrane on the edge of the colonies were often strongly villin-positive, indicating the presence of differentiated enterocytes with organized microvilli and a mature brush border. Staining for Chromogranin A which marks enteroendocrine cells, did not reveal evidence of cells from this lineage, as in the parental cultures, although their presence was detected in xenograft tumors (Figure 3C). This result indicates that heterogeneous tumor cells, with the presence of at least two lineages, enterocyte and goblet cells, can be derived from a single cell in culture. This outcome also suggests that simultaneous activation of differentiation lineage pathways may occur in the pathogenesis of metastatic colorectal cancer as previously reported for primary colon adenocarcinomas (20, 28, 29). Flow cytometry of cultured cells derived from clonogenic tumor cells resulted in profiles identical to those previously seen in the parental cultures (not shown). Clonogenic cultures were repeatedly passaged, demonstrating the physiological capacity for self-renewal as well as lineage maturation potential, properties which are expected from stem cells.
Figure 2. Clonal expansion of metastatic colon cancer cells.

A, Table of the clonally-derived tumor cell cultures. Single tumor cells from sorted populations were isolated and clonally expanded in vitro. No differences in clonal expansion and pleiomorphic differentiation could be detected when compared to parental cultures. B, Phase contrast micrographs of tumor cells obtained after clonal expansion. Glandular organization similar to the parental cultures was observed. a, Tu-7 (passage 3), clone D4 (X10) - 3 month culture. b, Tu-7 (passage 3), clone G2 (X10) - 3 month culture. c, Tu-7 (passage 4), clone C3 (X4) - 2.5 month culture. d, Tu-12 (passage 1) clone F7 (X10) - 1 month culture. e, Tu-12 (passage 1) clone F7 (X4) - 1 month culture. f, Tu-12 (passage 1) clone G12 (X4) - 1 month culture. C, Lineage commitment of clonally derived tumor cells. Tu-12 clones F7, E9 and G12 were stained for HLA-ABC (FITC, green) and Muc-1 or Muc-2 (Alexa-594, red). The nuclear counterstain was Hoechst 33342 (blue). Heterogeneous expression of Muc-2 and Muc-1 indicate the presence of both goblet and enterocyte lineages. D, Illustrative G-banded karyotypes of Tu-12 and Tu-21, parents and clones. Three of twenty metaphase cells analyzed are presented here for each tumor cell culture. The results revealed hypodiploid to near-tetraploid (Tu-12) and near-hexaploid (Tu-21) karyotypes with several clonal and nonclonal numerical and structural chromosomal aberrations in all three tumor cell cultures. Chromosome number ranged from 40 to 94 (Tu-12) and from 38 to 135 (Tu-21). *, abnormal chromosomes; red, chromosomal aberrations common to all three cultures; blue, chromosomal aberrations common only in one or two cultures. The variability in the karyotypes of these tumor cells suggests the presence of chromosomal instability.
Clonogenic, multipotent, self-renewing colon cancer cells are tumorigenic
Because the most critical attribute of any cancer is the capacity to generate and perpetuate tumors, we examined whether the isolated stem cells derived from metastatic colon tumors also have the capacity to initiate tumors. To that extent, we assessed the ability of these cells to give rise to tumors in vivo. Tumor cells isolated from fresh tissues, newly established parental cultures, and clonally-derived cultures from metastatic colon cancer samples were transplanted into immunodeficient Rag-2/γc−/− mice (Figure 3). The dorsal fat pad region under the skin was found to be the most reliable place to grow colorectal tumors, as previously reported by others (30). Our goal was to demonstrate the tumorigenicity of the isolated tumor cell population before and after expansion on feeder cells. Mice transplanted with human tumor cells were tested for xenograft tumors. Carcinoembryonic Antigen (CEA) was screened in the serum of mice by ELISA. CEA is a tumor marker secreted by cells and typically associated with colon cancer (31). We showed that the human tumor cells, expanded in vitro, expressed high levels of CEA in the culture medium (Figure 3A, blue columns). Using the same approach, sera of mice transplanted with different tumor cell cultures were screened for CEA expression. Mice showed positive expression of human CEA, ranging from a few ng/ml to 117ng/ml (Figure 3A, red columns). All of these mice were sacrificed and found to carry human tumors. Tumors derived from the fresh tissues, parental cultures as well as the clonally-derived cultures were morphologically identical to the patient’s isolated tumors. Histopathologic analysis of the xenografts demonstrated a striking adenocarcinoma-like tissue pattern (Figure 3B and Figure 3C). Xenograft tumors were moderately to well-differentiated adenocarcinomas with expansive growth, without evidence of invasion. Both parental and clonally-derived tumors were serially transplanted and shown to be tumorigenic upon secondary transplantation. The secondary tumors expanded faster than the initial xenograft tumors, suggesting that the malignancy might have been exacerbated in both the parental and the clonally-derived tumor cells as a result of selection. When post-transplanted tumor cells were cultured back on stroma, flow cytometric analyses identified the same expression of the 24 cell surface markers as detected on the cells prior to transplantation (not shown).
Xenograft tumors derived from a single expanded tumor cell were further analyzed for lineage markers. Markers for goblet cells, enterocytes, and enteroendocrine cells were present in the xenografts derived from a single expanded tumor cell. Muc-2 expression was present on a limited number of aberrant crypt cells, demonstrating the presence of a few mature goblet cells at the edge of the apical surface (Figure 3B, 3C and 3D). Villin was strongly expressed, indicating the presence of enterocytes. The demonstration of the mature morphology of the clonally-derived tumor cells was determined by transmission electron microscopy of glutaraldehyde fixed tissue. Figure 3D shows the mature morphology of goblet cells with mucus containing vacuoles surrounded by columnar cells with apical microvilli and tight junctions. In conclusion, both well-differentiated goblet cells and columnar cells were observed in clonally-derived xenograft tumors along with undifferentiated tumor cells. These results satisfy all of the criteria necessary to define these tumor cells as multipotent cancer stem cells in both in vitro and in vivo environments.
CD133+ tumor cells mediate tumor growth in xenotransplant
The cancer stem cell theory states that only a subset of prospective tumor cells has the ability to initiate the tumor. To determine if the in vitro culture and expansion system described here could generate similar results as previously reported for colon cancer stem cells (12, 14), we isolated tumor cells from three different patients showing CD133 heterogeneous expression (Tu-12 clone F7, Tu-21 clone C2 and Tu-27). Tumor cells isolated from xenograft tumors in mice (Tu-12 clone F7 and Tu-21 clone C2) and tumor cells expanded in culture (Tu-27) were sorted for CD133.1 positive and CD133.1 negative subpopulations. The CD133.1+ and CD133.1− tumor cell subpopulations were transplanted to the left and right flank of immunodeficient mice. Six to twelve weeks after transplantation, only the site injected with CD133+ cells developed visible tumors, confirming previous reports that CD133-positive colon cancer cells can grow as xenograft transplants (Figure 4 B and C).
Chromosomal instability is present in clonogenic multipotent tumor cells
To assess the presence of chromosomal instability in tumor cells, karyotype analysis was performed on Tu-12 and Tu-21 by examining the parental and two clonally-derived tumor cell cultures (Tu-12 F7, Tu-12 G12, Tu-21 H11, Tu-21 E12). All tumor cells analyzed expressed human karyotypes with clonal and nonclonal numerical and structural aberrations (Figure 2D, and Table 2, see Supplement Material for more information). Many of the same aberrations observed in the parental cells were also seen in the clonally-derived tumor cells, confirming their common origin. Surprisingly, the clonally-derived tumor cells also contained aberrations unique to each culture.
Table 2.
Percentage of cells exhibiting the chromosomal aberrations found in parental and clonal cultures of Tu-12 and Tu-21. Twenty metaphase cells were analyzed per tumor culture. Structural changes that were seen more than once, numerical gains of chromosomes at least seen twice, and numerical chromosome loss seen three or more times were reported in this table. Add, additional unidentifiable chromatin material attached to and replacing the chromatin distal to that breakpoint; del, deletion of a chromosome segment; i, isochromosome; "+1~5 mar", at least two of the cells studied from that sample contained between one and five unidentifiable marker chromosomes. Clonally abnormal chromosomes in all three related cultures confirm the relationship between the parent and her descendants. These would be the chromosomal aberrations found in all three samples. While the nomenclature describing the del(5q) and the marker chromosomes appear the same for the two unrelated tumors, we do not believe that they are actually identical abnormalities in the two tumors at the molecular level (See Supplementary Material for more information).
| Tu-12 | Tu-12 G12 | Tu-12 F7 | Tu-21 | Tu-21 E12 | Tu-21 H11 | |
|---|---|---|---|---|---|---|
| Clonal Aberrations | ||||||
| −1 | 90 | 85 | 85 | |||
| add(1)(q24) | 95 | 80 | ||||
| add(1)(q24)add(1)(p36.1) | 90 | |||||
| del(1)(p13p34) | 100 | 95 | 80 | |||
| del(1)(p36) | 100 | 10 | ||||
| del(1)(p13p34)add(1)(q42) | 20 | |||||
| −2 | 30 | 30 | 20 | 45 | ||
| −3 | 20 | 40 | 25 | |||
| der(3)t(1;3)(q25;q25) | 30 | |||||
| der(3)t(3;5)(q25;q14) | 19 | 95 | ||||
| +4 | 25 | |||||
| −4 | 80 | 65 | 70 | |||
| add(4)(q35) | 20 | 10 | ||||
| del(4)(q31) | 35 | |||||
| −5 | 80 | 95 | 85 | 60 | 95 | |
| del(5)(q11.2q31) | 60 | 90 | 90 | 40 | 15 | 10 |
| −6 | 60 | 15 | 40 | 15 | ||
| +6 | 15 | 10 | ||||
| del(6)(q13q21) | 20 | |||||
| +7 | 40 | 50 | ||||
| −7 | 40 | 40 | ||||
| add(7)(p15) | 90 | 85 | 100 | |||
| del(7)(q22) | 10 | |||||
| +8 | 25 | 10 | 20 | |||
| −8 | 65 | |||||
| der(8)t(1;8)(q25;p12) | 100 | 85 | 100 | |||
| +9 | 25 | |||||
| −9 | 30 | 15 | 40 | 15 | ||
| +10 | 15 | 26 | 25 | |||
| −10 | 20 | 25 | 25 | 20 | ||
| add(10)(p14) | 15 | 20 | ||||
| +11 | 25 | 10 | 10 | |||
| −11 | 30 | |||||
| add(11)(p14) | 30 | |||||
| add(11)(q23) | 15 | 20 | 35 | 15 | ||
| del(11)(q13) | 10 | |||||
| del(11)(q23) | 10 | |||||
| −12 | 25 | 70 | 60 | 80 | ||
| del(12)(p11.2) | 50 | |||||
| +13 | 85 | 75 | 90 | 20 | 20 | 20 |
| −13 | 35 | |||||
| add(13)(p13) | 100 | 80 | 100 | |||
| del(13)(q12q22) | 55 | 40 | 20 | |||
| der(13;13)(q10;q10) | 80 | |||||
| der(13;15)(q10;q10) | 20 | |||||
| −14 | 95 | 95 | 95 | 80 | 65 | 65 |
| der(14;14)(q10;q10) | 10 | |||||
| +15 | 25 | 20 | ||||
| −15 | 100 | 45 | 55 | |||
| der(15;15)(q10;q10) | 40 | 25 | ||||
| +16 | 65 | 45 | 20 | |||
| −16 | 20 | |||||
| add(16)(q24) | 55 | 65 | 25 | |||
| −17 | 100 | 90 | 100 | 50 | 70 | 90 |
| add(17)(p13) | 35 | 45 | 60 | |||
| del(17)(p11.2) | 80 | 65 | 20 | |||
| i(17)(q10) | 100 | 90 | 100 | |||
| −18 | 90 | 75 | 90 | 80 | 70 | 90 |
| +19 | 15 | |||||
| −19 | 45 | 65 | 40 | 65 | ||
| add(19)(q13.4) | 20 | |||||
| add(19)(p13.3) | 20 | 20 | ||||
| +20 | 70 | 40 | 65 | 70 | 75 | 55 |
| add(20)(q13.3) | 70 | 55 | ||||
| del(20)(q11.2q12) | 70 | 15 | ||||
| −21 | 95 | 95 | 30 | 70 | 60 | |
| −22 | 80 | 85 | 85 | |||
| add(22)(q13) | 20 | 15 | 50 | |||
| −X | 25 | 35 | 35 | |||
| add(X)(q28) | 20 | |||||
| −Y | 25 | |||||
| +1~5 mar | 45 | 45 | 40 | 100 | 60 | 55 |
| Non-clonal aberrations | 20 | 10 | 25 | 20 | 25 | 30 |
The variability in the karyotypes of these cells suggests the presence of chromosomal instability with ongoing selection. As a control, normal human fetal liver cells were cultured under the same conditions. After seven passages in culture, no chromosomal abnormalities could be detected in the normal human fetal liver cultures. This result indicates that the in vitro culture conditions are not inducing the chromosomal instability observed in the tumor cell cultures.
Discussion
Colon cancer is a complex and heterogeneous disease. Recently, the hypothesis that cancer stem cells drive the tumorigenic process in colorectal cancer has received support. The cancer stem cell hypothesis suggests that tumors are generated and maintained by a small subset of cancer cells capable of self-renewal and differentiation into bulk tumor cells. Both CD133+ (12, 14) and EpCamhigh/CD44+/CD166+ (13) have been proposed as colon cancer stem cell surface markers. Collectively, these studies show the presence of cancer-initiating cells within colon tumors and raise questions regarding the origin of colon cancer stem cells. It has been anticipated that normal stem cells of the intestinal tract represent the natural target of tumorigenic mutations. Consequently, these cells would be the best candidates for the origin of cancer stem cells. However, not much information has been published on the possibility of cancerous stem cells in colon cancer. Our work reports the identification and isolation of tumor cells from human metastatic colon cancer with the properties of intestinal stem cells as well as tumor initiating cells. The presence of single cells within metastatic tumors, which demonstrate self-renewal, multipotency, and the capacity to differentiate into enterocytes and goblet cells, broadens the current model of colon cancer stem cells by providing additional support for the potential role of natural stem cells in the development of colon cancer. However, the possible role of colon progenitor cells involved in this cancer cannot be ruled out until normal human colon stem cells are identified and characterized. Additionally, our data indicate that all 11 metastatic colon cancer samples extensively analyzed showed a remarkably consistent pattern of antigen staining. This result increases the number of previously proposed cancer stem cell markers and provides a robust and reproducible surface marker profile for colon cancer stem cells. Moreover, the presence of such tumor cells in each of the metastatic tumor samples isolated from patient liver sheds light on a common origin of the metastatic process in colon cancer. Despite extensive heterogeneity in such areas as tissue morphology and somatic mutations (32), evidence for the cellular origin of metastatic colon cancer is consistent with a common origin. The knowledge that tumor cells with similar characteristics are involved in the metastatic process of colon cancer is of vital importance to our understanding as to how these tumors metastasize including, and especially, information on the key cellular and molecular mechanisms responsible for the metastatic process. Finally, the biology of the neoplastic stem cells in patients suffering from metastatic colon cancer is clearly important to future successful therapies and could also serve as a paradigm relevant to other types of malignancies that might originate from transformed stem cells.
Furthermore, chromosome instability has long been recognized as an engine of neoplastic transformation (33, 34) in the development and progression of the majority of colon cancers (35). Chromosomal abnormalities occur at a relatively early stage of colorectal neoplasia (36) and have been proposed as a significant force for clonal dominance in a dividing cell population (37). Our study suggests that chromosomal instability is present in colon cancer stem cells. The presence of genomic instability in cancer stem cells has many biologic and therapeutic implications. From a biologic point of view, genetic instability supports the stochastic model of cancer in the cancer stem cell population. This result points to a heterogeneous cancer stem cell population with an evolutionary process of tumor progression determined by two main events within the cancer stem cells, the generation of genetic heterogeneity in the cancer stem cell pool and the selection of genetic variant cancer stem cells most suited to survival, proliferation, invasion and metastasis (38). This selection should, through the classic stochastic model of cancer origin, generate heterogeneous cancer stem cell populations, with some cancer stem cells better adapted and selected to reach the metastatic stage, while others are not. In this model, not every cancer stem cell would be capable of metastasis. From a therapeutic point of view, genetic instability introduces rapid mutations and adaptation to the selection process and may explain why many experimental therapeutic approaches have poor clinical results (39). Under imatinib therapy, a treatment that causes remission in a majority of patients with chronic myeloid leukemia (CML) due to specific action on BCR-ABL1-positive cells (40), patients relapse due to the inability of the treatment to eradicate CML cancer stem cells (41) and to the instability of BCR-ABL1 gene in the leukemia stem cells (42, 43). Furthermore, compared to normal stem cells, cancer stem cells with genetic instability may have acquired the formidable advantage of tumor cell survival fitness by combining the twin driving forces of mutation and natural selection (38).
This updated model of cancer stem cells once again raises the question of whether or not genetic instability is the initiating event and driving force that may transform colon stem cells or progenitor cells into lethal cancer-initiating cells. Although the ability of cancer stem cells to differentiate despite having chromosomal aberrations supports the development of new therapeutic approaches designed to promote tumor cell differentiation, the apparent capacity of cancer cells to acquire resistance to current treatments begs for a rationalized therapy targeting cells with chromosomal instability (33).
METHODS
Tumor cell acquisition and preparation
Following informed consent, tumor tissues were collected after surgical removal from the liver of metastatic colon cancer patients. Tumor tissues were minced into small fragments with scalpels and then digested using a two-step collagenase method derived from the isolation of liver cells (44). A 20-minute trypsin incubation was added to complete dissociation of the tumor tissue. If enough cells were collected from the tissue, flow cytometric analysis was performed on each tissue fraction. In most cases, tumor cells were plated on a monolayer of previously irradiated (80 Gy) rodent epithelial feeder cells (17).
Flow cytometry
Cells were counted and aliquoted at 106 cells per tube and incubated with monoclonal antibodies at the appropriate dilution. Dead cells were detected with propidium iodide (PI, 10 µg/ml). Post-acquisition analysis of the FACS data and desktop publishing were accomplished using the third-party flow cytometry software, FlowJo (http://www.treestar.com/). CD antibodies, primary and secondary, were obtained from BD Biosciences (San Jose, CA) except CD133 (Miltenyi Biotec, CA) and HLA-ABC (Ancell, MN).
Clonal expansion
Metastatic colon tumor cells from the different patients were stained with cell surface markers and sorted for the desired phenotype. The sorted populations were resuspended in 2 ml of media and deposited at different concentrations in a 6-well plate. To initiate a clonogenic expansion, cells were observed using an inverted microscope at 100x amplification and single cells were individually aspirated using a micropipette. Each single cell was then manually deposited in a unique well of a 96-well plate previously plated with feeder cells. Four weeks later, plates were screened for colonies.
Transplantation in mice
Six to eight week old Rag-2/γc−/− mice were anesthetized and transplanted with tumor cells. In general, tumor cells isolated from primary tissue or culture expansion, were mixed with media/matrigel at a ratio of 1:1 and injected. Tumorigenicity was determined by transplanting tumor cells (106 cells). Six to eight weeks after transplantation, mice were bled and the serum samples were analyzed for the presence of CEA by ELISA (Biomeda, Foster City, CA).
Immunostaining and Transmission electron microscopy
Primary antibodies, Muc-1 (BD Biosciences, CA), Muc-2 (Santa Cruz, CA), villin (Santa Cruz, CA) and chromogranin A (Santa Cruz, CA) were incubated for an hour followed by chicken anti mouse Alexa (488 or 594, Invitrogen, CA). HLA-biotin (Ancell, MN) was followed by Streptavidin-FITC (Pharmingen, CA). Transmission electron microscopy (TEM) was done as described previously (45).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Jeannik M. Littlefield-AACR Grant in Metastatic Colon Cancer Research (C.O., H.F., T.H., L.G., D.A.G. and E.L.), the Commonwealth of Pennsylvania (C.O., H.F., T.H., L.G., and E.L.) and in part by NIH RO1DE14729 (S.M.G. and D.W.L.). Cytogenetic studies were carried out in the University of Pittsburgh Cancer Institute Cytogenetics Facility, supported in part by NIH P30CA047904 to Ronald B. Herberman. We would like to thank Alessandra Calvelli for her technical contribution.
Footnotes
See additional Materials and Methods in Supplementary Material.
References
- 1.Arkenau HT, Chua YJ, Cunningham D. Current treatment strategies in elderly patients with metastatic colorectal cancer. Clin Colorectal Cancer. 2007;6:508–515. doi: 10.3816/ccc.2007.n.016. [DOI] [PubMed] [Google Scholar]
- 2.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 3.Fong Y, Kemeny N, Paty P, Blumgart LH, Cohen AM. Treatment of colorectal cancer: hepatic metastasis. Semin Surg Oncol. 1996;12:219–252. doi: 10.1002/(SICI)1098-2388(199607/08)12:4<219::AID-SSU3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Liotta LA, Kohn EC. Cancer's deadly signature. Nat Genet. 2003;33(1):10–11. doi: 10.1038/ng0103-10. [DOI] [PubMed] [Google Scholar]
- 5.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 6.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 7.Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 8.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 10.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 11.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 12.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 13.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 15.Polyak K, Hahn WC. Roots and stems: stem cells in cancer. Nat Med. 2006;12:296–300. doi: 10.1038/nm1379. [DOI] [PubMed] [Google Scholar]
- 16.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–1115. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Ehmann UK, Peterson WD, Jr., Misfeldt DS. To grow mouse mammary epithelial cells in culture. J Cell Biol. 1984;98:1026–1032. doi: 10.1083/jcb.98.3.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ajioka Y, Allison LJ, Jass JR. Significance of MUC1 and MUC2 mucin expression in colorectal cancer. J Clin Pathol. 1996;49:560–564. doi: 10.1136/jcp.49.7.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tytgat KM, Buller HA, Opdam FJ, Kim YS, Einerhand AW, Dekker J. Biosynthesis of human colonic mucin: Muc2 is the prominent secretory mucin. Gastroenterology. 1994;107:1352–1363. doi: 10.1016/0016-5085(94)90537-1. [DOI] [PubMed] [Google Scholar]
- 20.Henderson K, Kirkland SC. Multilineage differentiation of cloned HRA-19 cells in serum-free medium: a model of human colorectal epithelial differentiation. Differentiation. 1996;60:259–268. doi: 10.1046/j.1432-0436.1996.6040259.x. [DOI] [PubMed] [Google Scholar]
- 21.Kirkland SC, Henderson K. Collagen IV synthesis is restricted to the enteroendocrine pathway during multilineage differentiation of human colorectal epithelial stem cells. J Cell Sci. 2001;114:2055–2064. doi: 10.1242/jcs.114.11.2055. [DOI] [PubMed] [Google Scholar]
- 22.Ploemacher RE, van der Sluijs JP, Voerman JS, Brons NH. An in vitro limiting-dilution assay of long-term repopulating hematopoietic stem cells in the mouse. Blood. 1989;74:2755–2763. [PubMed] [Google Scholar]
- 23.Ploemacher RE, van der Sluijs JP, van Beurden CA, Baert MR, Chan PL. Use of limiting-dilution type long-term marrow cultures in frequency analysis of marrow-repopulating and spleen colony-forming hematopoietic stem cells in the mouse. Blood. 1991;78:2527–2533. [PubMed] [Google Scholar]
- 24.Pettengell R, Luft T, Henschler R, et al. Direct comparison by limiting dilution analysis of long-term culture-initiating cells in human bone marrow, umbilical cord blood, and blood stem cells. Blood. 1994;84:3653–3659. [PubMed] [Google Scholar]
- 25.Waldmann H, Lefkovits I, Quintans J. Limiting dilution analysis of helper T-cell function. Immunology. 1975;28:1135–1148. [PMC free article] [PubMed] [Google Scholar]
- 26.Weilbaecher K, Weissman I, Blume K, Heimfeld S. Culture of phenotypically defined hematopoietic stem cells and other progenitors at limiting dilution on Dexter monolayers. Blood. 1991;78:945–952. [PubMed] [Google Scholar]
- 27.Uchida N, Buck DW, He D, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirkland SC. Clonal origin of columnar, mucous, and endocrine cell lineages in human colorectal epithelium. Cancer. 1988;61:1359–1363. doi: 10.1002/1097-0142(19880401)61:7<1359::aid-cncr2820610714>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 29.Marsh KA, Stamp GW, Kirkland SC. Isolation and characterization of multiple cell types from a single human colonic carcinoma: tumourigenicity of these cell types in a xenograft system. J Pathol. 1993;170:441–450. doi: 10.1002/path.1711700407. [DOI] [PubMed] [Google Scholar]
- 30.De Both NJ, Vermey M, Groen N, Dinjens WN, Bosman FT. Clonal growth of colorectal-carcinoma cell lines transplanted to nude mice. Int J Cancer. 1997;72:1137–1141. doi: 10.1002/(sici)1097-0215(19970917)72:6<1137::aid-ijc32>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 31.Macdonald JS. Carcinoembryonic antigen screening: pros and cons. Semin Oncol. 1999;26:556–560. [PubMed] [Google Scholar]
- 32.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 33.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 34.Gollin SM. Mechanisms leading to chromosomal instability. Semin Cancer Biol. 2005;15:33–42. doi: 10.1016/j.semcancer.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 36.Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61:818–822. [PubMed] [Google Scholar]
- 37.Leedham SJ, Brittan M, McDonald SA, Wright NA. Intestinal stem cells. J Cell Mol Med. 2005;9:11–24. doi: 10.1111/j.1582-4934.2005.tb00333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lagasse E. Cancer stem cells with genetic instability: the best vehicle with the best engine for cancer. Gene Ther. 2008;15:136–142. doi: 10.1038/sj.gt.3303068. [DOI] [PubMed] [Google Scholar]
- 39.Vineis P, Berwick M. The population dynamics of cancer: a Darwinian perspective. Int J Epidemiol. 2006;35:1151–1159. doi: 10.1093/ije/dyl185. [DOI] [PubMed] [Google Scholar]
- 40.Michor F, Hughes TP, Iwasa Y, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 41.Glauche I, Horn M, Roeder I. Leukaemia stem cells: hit or miss? Br J Cancer. 2007;96:677–678. doi: 10.1038/sj.bjc.6603603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang X, Saw KM, Eaves A, Eaves C. Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cells. J Natl Cancer Inst. 2007;99:680–693. doi: 10.1093/jnci/djk150. [DOI] [PubMed] [Google Scholar]
- 43.Jiang X, Smith C, Eaves A, Eaves C. The challenges of targeting chronic myeloid leukemia stem cells. Clin Lymphoma Myeloma. 2007;7:S71–S80. doi: 10.3816/clm.2007.s.005. [DOI] [PubMed] [Google Scholar]
- 44.Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83. doi: 10.1016/s0091-679x(08)61797-5. [DOI] [PubMed] [Google Scholar]
- 45.Wack KE, Ross MA, Zegarra V, Sysko LR, Watkins SC, Stolz DB. Sinusoidal ultrastructure evaluated during the revascularization of regenerating rat liver. Hepatology. 2001;33:363–378. doi: 10.1053/jhep.2001.21998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.