

Abstract
The neuronal isoform of nitric oxide synthase (nNOS), the enzyme responsible for the production of nitric oxide in the central nervous system, represents an attractive target for the treatment of various neurodegenerative disorders. X-ray crystal structures of complexes of nNOS with two nNOS-selective inhibitors, (4S)-N-{4-amino-5-[(2-aminoethylamino]pentyl}-N′-nitroguanidine (1) and 4-N-(Nω-nitro-L-argininyl)-trans-4-amino-L-proline amide (2), led to the discovery of a conserved structural water molecule that was hydrogen bonded between the two heme propionates and the inhibitors (Figure 2). Based on this observation, we hypothesized that by attaching a hydrogen bond donor group to the amide nitrogen of 2 or to the secondary amine nitrogen of 1, the inhibitor molecules could displace the structural water molecule and obtain a direct interaction with the heme cofactor. To test this hypothesis, peptidomimetic analogues 3–5, which have either an N-hydroxyl (3 and 5) or N-amino (4) donor group, were designed and synthesized. X-ray crystal structures of nNOS with inhibitors 3 and 5 bound verified that the N-hydroxyl group had, indeed, displaced the structural water molecule and provided a direct interaction with the heme propionate moiety (Figures 4 and 5). Surprisingly, in vitro activity assay results indicated that the addition of a hydroxyl group (3) only increased the potency slightly against the neuronal isoform over the parent compound (1). Rationalizations for the small increase in potency are consistent with other changes in the crystal structures.
Introduction
Nitric oxide (NO),1 a highly reactive free radical, is an essential signaling molecule involved in various physiological processes in humans. Generally, NO acts as a potent activator of soluble guanylate cyclase (sGC), which catalyzes the conversion of guanosine 5′-triphosphate (GTP) to an intramolecular second messenger, guanosine 3′,5′-cyclic monophosphate (cGMP).2 This NO/cGMP signaling pathway is essential in many physiological processes including vasodilation, neurotransmission, and platelet aggregation.3 NO also participates in responses that are not mediated via sGC.4
Nitric oxide is synthesized by a family of enzymes called nitric oxide synthases (NOS, EC 1.14.13.39),5 which catalyze the oxidation of L-arginine to L-citrulline and nitric oxide. Three isozymes of NOS have been identified so far, each associated with a distinct physiological function: neuronal signal transmission (neuronal NOS or nNOS),6 smooth muscle relaxation (endothelial NOS or eNOS),7 and immune response (inducible NOS or iNOS).8 The first two isoforms (nNOS and eNOS) are constitutively expressed and intermittently produce small amounts of NO. In contrast, the third isoform (iNOS) is inducible by cytokines and produces large amounts of NO for both a cytoprotective and cytotoxic effect. All three isozymes are encoded by separate genes and, therefore, differently regulated. Their activity depends on a number of cofactors: NADPH, FAD, FMN, tetrahydrobiopterin (H4B), and heme. The activity of the constitutive enzymes is regulated by phosphorylation and by the Ca2+-binding protein calmodulin,9 while iNOS does not depend on the intracellular calcium levels because iNOS carries a permanently bound molecule of calmodulin.10
The three NOS isoforms share a similar structural architecture.11 The C-terminus is a reductase domain, homologous to cytochrome P450 reductase. NADPH, FAD, and FMN are bound in this domain. The N-terminus is the oxygenase domain, which contains binding sites for the essential cofactors; tetrahydrobiopterin, heme, and the substrate, L-Arg. These two domains are linked by a calmodulin-binding motif and form one monomer. Two of the monomers dimerize to form a homodimer enzyme with the aid of a single intersubunit ZnS4 cluster, which stabilizes dimerization and H4B binding site formation.12 Electrons are transferred from NADPH via FAD and FMN to the heme of the other domain. This flow of electrons during catalysis occurs from the reductase domain of one monomer subunit to the oxygenase domain of the other monomer, so an intact homodimer form of the enzyme is essential for full enzyme activity.13
Overproduction of NO from neuronal isozyme has been associated with harmful effects in the central nervous system. Radical nitric oxide (NO·) reacts rapidly with superoxide (O2·−) in aqueous media to form the highly reactive peroxynitrite anion (ONOO−). Either nitric oxide or peroxynitrite causes damage to neuronal cells and tissues and, therefore, produces neurodegeneration.14 Recently, Uehara et al. found that endogenous NO caused S-nitrosylation on the cysteine residue of protein-disulphide isomerase (PDI) and inhibited its activity as a chaperone, indicating that nNOS was involved in the accumulation of unfolded/misfolded proteins in the brain.15 Because eNOS is important in maintaining normal blood pressure, nNOS inhibitors must not inhibit eNOS. Thus, selective inhibition of the neuronal isoform is essential if nNOS inhibitors are used for therapeutic purposes.
Nitric oxide synthase inhibitors have been reviewed comprehensively.16 Among those inhibitors, Nω-nitro-L-arginine (L-NNA) has been extensively studied for pharmacological effects in vitro and in vivo. One limitation of L-NNA as a therapeutic agent is its poor isoform selectivity.17 This weak selectivity is commonly an issue for substrate arginine analogues such as L-NNA and L-NMA (Nω-methyl-L-arginine) due to the lack of distinctive residues among the different isozymes in the substrate-binding pocket.18 To achieve a highly isoform-selective inhibition, an inhibitor should possess a functional group that can reach into the substrate-access channel remote from the substrate-binding pocket, and the functional group can contribute to increase isoform selectivity as well as binding affinity. We have designed Nω-nitro-L-arginine-containing dipeptide amide inhibitors for the purpose of retaining the advantages of L-NNA and using the second amino acid to identify a region outside of the substrate-binding pocket that would differentiate nNOS from iNOS and eNOS.19 A series of dipeptidomimetic analogues were synthesized and tested for in vitro activity, and 1 and 2 (Figure 1) were found to be superior to L-NNA, particularly, in terms of their remarkable isoform selectivity.20 In a continued effort to investigate the active site of the enzyme, further structure-activity relationship studies along with X-ray crystallographic studies have been performed.21,22 To enhance inhibitor binding further, we present here the first example of a nNOS inhibitor designed with the intention of displacing the heme structural water molecule in the active site.
Figure 1.
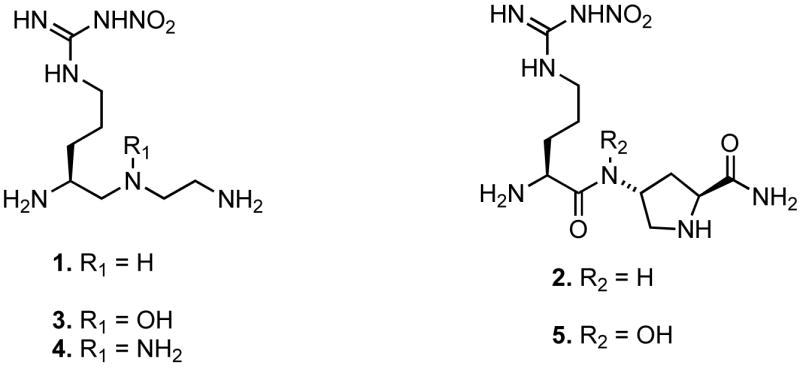
Nω-Nitro-L-Arginine-containing dipeptides and their derivatives.
Chemistry
Synthesis of Hydroxylamine 3
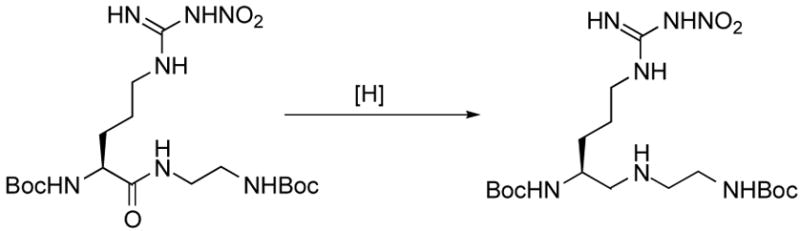
The synthesis of hydroxylamine derivative 3 is depicted in Scheme 1. Secondary amine 7 was prepared via a peptide coupling reaction of Boc-L-Arg(NO2)-OH with N-Boc-ethylenediamine23 followed by amide reduction of the resulting secondary amide (6). A widely exploited protocol for the preparation of secondary amines is reductive amination.24 This method was applied both to the solution- and solid phase syntheses; however, there is a risk of racemization because the proton on the α-carbon becomes acidic during the intermediate iminium ion formation. Alternatively, amides can be converted to thioamides using Lawesson’s reagent, then subsequent reductive desulfurization provides the corresponding reduced amide.25 A third way to synthesize secondary amines is by a direct reduction of an amide to an amine. This approach has been employed less frequently, probably because of the lack of selectivity that the hydride reagents have for various functionalities.26 For the synthesis of 7, we tried the direct reduction method using various conditions (Table 1). LAH, alane, and borane were used as hydride-donor reducing agents. Entries 1–10 show that all three reagents have poor selectivity for the reduction of the amide relative to other functionalities (carbamate and N-nitro) in 6. The milder reagents showed slightly better selectivity (LiAlH4 < BH3 < AlH3). Attempts to obtain a higher yield by raising the reaction temperature resulted in side reactions; usually, the hydride reacted with the carbamates. However, maximum selectivity was achieved when a low reaction temperature (−10 to 0 °C) was used (entry 11). This method was originally reported by Roeske et al.27 for the selective reduction of an amide from an ester using borane. The low temperature conditions suppressed the side reactions, but were adequately effective for secondary amide reduction. At low temperature we isolated 7 in a 56% yield (83% yield based on the consumed starting material).
Scheme 1a.

a Reagents and conditions: (a) N-Boc-ethylenediamine, HBTU, HOBt, DIEA, DMF/CH2Cl2, 78%; (b) BH3-THF, THF, −10 °C, 56%; (c) Benzoyl peroxide, Na2HPO4, Et2O/THF, 70 °C, 68%; (d) 0.3% NaOH, MeOH, ~quant.; (e) 50% TFA, CH2Cl2, ~quant.
Table 1.
Dipeptide Amide Reduction.
 | |||||
|---|---|---|---|---|---|
| Entry | Reagent | Equiva | Time (h) | Temp (°C) | Yield of Product (%) |
| 1 | LiAlH4 | 2 | overnight | r.t. | b |
| 2 | LiAlH4 | 2 | overnight | 70 | c |
| 3 | LiAlH4 | 10 | 2 | r.t. | Traceb |
| 4 | LiAlH4 | 10 | overnight | r.t. | c |
| 5 | AlH3 | 10 | overnight | r.t. | 17%b |
| 6 | AlH3 | 10 | overnight | 70 | c |
| 7 | BH3 | 2 | 72 | r.t. | b |
| 8 | BH3 | 2 | overnight | 70 | b |
| 9 | BH3 | 5 | 2 | 70 | c |
| 10 | BH3 | 10 | 30 | r.t. | 10% |
| 11 | BH3 | 10 | 10 | −10 to 0 | 56%d |
molar equivalents of reagent for relative to starting material
starting material returned
starting material decomposed.
83% based on consumed starting material
Because the oxidation of secondary amines can give N-oxides, which rearrange to the corresponding hydroxylamines, mCPBA was first employed for the direct preparation of hydroxylamine 9 from amine 7.28 However, we found hydroxylamine 9 was very susceptible to further oxidation with mCPBA.29 As evidenced by many side product spots on the TLC plate and M−1, M, and M+1 peaks in the mass spectrum, we concluded that the mCPBA oxidation reaction was complicated by overoxidation. Varying the mCPBA ratio and the reaction temperature did not help circumvent the problem. Therefore, we changed the oxidant to a more controllable reagent, namely, benzoyl peroxide. To trap the byproduct benzoic acid and suppress the N-acylation side reaction, an auxiliary base (Na2HPO4) was added to the reaction mixture,30 and the oxidation reaction proceeded smoothly to provide 8. A mild sodium methoxide solution cleaved the O-benzoyl group successfully, and deprotection of the Boc groups gave 3.
Synthesis of Hydrazine 4
The synthesis of 4 started from intermediate 7 (Scheme 2). An electrophilic N-Boc transfer reaction31 using the commercially available reagent N -Boc-3-(4-cyanophenyl)oxaziridine (10) provided 11 in an excellent yield. Subsequent deprotection of the Boc groups provided 4.
Scheme 2a.
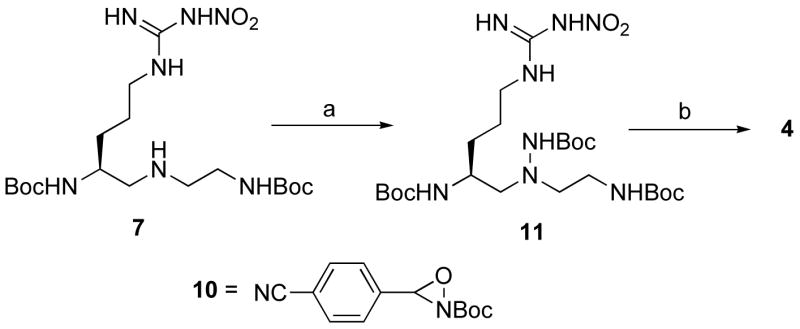
a Reagents and conditions: (a) 10, CHCl3, 97%; (b) 50% TFA, CH2Cl2, ~quant.
Synthesis of Hydroxamate 5
Hydroxamate derivative 5 was synthesized by the synthetic route shown in Scheme 3. The synthesis of an intermediate, hydroxylamine 15, started from commercially available Fmoc protected trans-4-hydroxy-L-proline. An intramolecular Mitsunobu reaction gave 12, and sodium azide-catalyzed methanolysis provided 13 in an 82% yield for two steps.32 The N,O-diBoc-protected hydroxylamine was then incorporated into 13 using Mitsunobu conditions to give 14; subsequent deprotection provided hydroxylamine 15. The hydroxylamine was coupled to Fmoc-Arg(NO2)-Cl to provide hydroxamate 16 in a 74% yield.33 Successful hydroxamate coupling was achieved when the low temperature and anhydrous conditions described above were used. Also, because of its short half-life, it was best to use 15 directly after Boc-deprotection. Prior to basic hydrolysis of the methyl ester, the Fmoc protecting groups were switched to Boc groups (17). The methyl ester was then converted to amide 18 by hydrolysis and amidation reactions. Subsequent deprotection of the Boc groups provided 5.
Scheme 3a.

aReagents and conditions: (a) DIAD, PPh3, THF, 85%; (b)MeOH, NaN3, 40 °C, 96%; (c) Boc-NH-O-Boc, DEAD, PPh3, THF, 45%; (d) 50% TFA, CH2Cl2, 98%; (e) Fmoc-L-Arg(NO2)-Cl, Collidine, THF, 74%; (f) (i) 20% piperidine, DMF; (ii) Boc2O, NaHCO3, dioxane, 83% for two steps; (g) 5% LiOH, THF, 94%; (h) iButylchloroformate, NMM, NH4OH, THF, 65%; (i) 50% TFA, CH2Cl2, ~quant.
Results and Discussion
Inhibitor Design
X-ray crystal structures of the complexes of nNOS with inhibitors 1 and 2 bound reveal the presence of a structural water molecule that is hydrogen bonded to the two heme propionates of the enzyme and to the inhibitor -NH moiety (Figure 2). An analogous water molecule is frequently found in the active site of nNOS complexed with the substrate L-Arg, the catalytic intermediate Nω-hydroxy-L-Arg, or with various inhibitors bound (Table 2). Generally, distances from the water molecule to the heme propionates are between 2.5 to 3.5 Å, and the binding angles are about 110°. This angle is larger than the H-O-H angle (104.5°), but is close to tetrahedral geometry (109°). As is the case of ice, the H-O-H angle of water in the ordered state is close to tetrahedral; the angle increases about 5 degrees from gaseous water to solid water.36 Therefore, the tetrahedral binding angle of the structural water molecule supports the fact that the water molecule is in a highly ordered state, not in bulk aqueous medium. A structural water molecule seems to be highly conserved in heme-containing enzymes when the heme propionates are open to the access of bulk solvent, as is the case with nitric oxide synthases.
Figure 2.
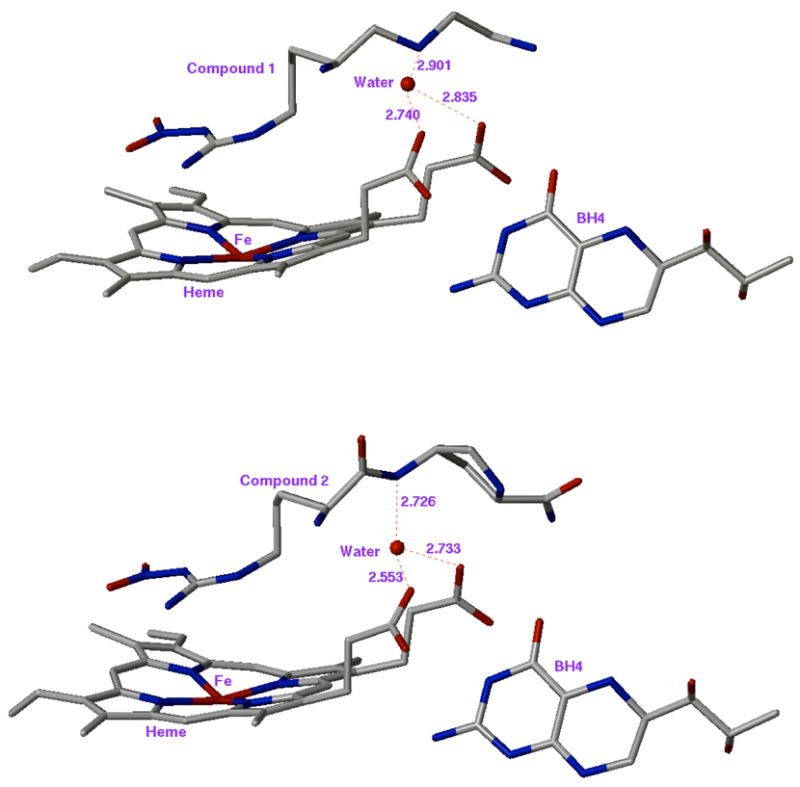
X-ray crystal structure of the active site heme and inhibitor (top: 1, bottom: 2). PDB id of top and bottom structure is 1p6i and 1p6j, respectively. Numbers are in Å.
Table 2.
Heme Structural Water Molecules Found in Rat Brain nNOS
| PDB ida | Molecule | Heme Structural Waterb |
|---|---|---|
| 1zvl | L-Arg | HOH 183 |
| 1lzx | NG-Hydroxy-L-Arg | HOH 66 |
| 1om4 | L-Arg | HOH 81 |
| 1k2r | L-NNA | HOH 82 |
| 1vag | AR-R1747734 | NAc |
| 1k2t | S-ethyl-N-phenylisothiourea | HOH 318 |
| 1qwc | 1400W35 | NAc |
| 1lzz | N-isopropyl-N′-hydroxyguanidine | HOH 83 |
| 1mmw | vinyl-L-NIO | HOH 53 |
| 1mmv | NG-Propyl-L-Arg | HOH 256 |
Protein Data Bank: http://www.rcsb.org/pdb.
Residue number in pdb file.
Structural water molecule was displaced by the inhibitor.
Structural water molecules in enzyme active sites are of interest to medicinal chemists because they modify the active site geometry and contribute to the binding affinity between a ligand and a protein. In addition, the presence of structural water molecules in enzyme-inhibitor binding can provide a possible new direction for the design of more potent inhibitors, which was successfully demonstrated by Lam et al. in their development of a HIV protease inhibitor.37
Our nNOS inhibitor design is based on the following considerations: (1) by extending a hydrogen bond donor group from the inhibitor -NH moiety, displacement of the structural water molecule may occur, which would be entropically favorable;38 (2) direct interaction of the inhibitor and the heme cofactor may occur, which may enhance the binding affinity of the inhibitors. On the basis of these considerations, peptidomimetic modifications of compounds 1 and 2 were made. We chose R = OH and NH2 as the hydrogen bond donor modification groups on 1 and 2 (3–5, Figure 1). These moieties are intended to mimic the hydrogen bonding feature of the structural water molecule and/or provide direct binding of the inhibitor to the heme cofactor. Initially, we carried out a computer modeling study to determine if the displacement of the structural water molecule is feasible. AutoDock 3.0.539 was used to predict the binding mode of designed compound 5 in the nNOS active site. For the docking simulation, the active site binding pocket was defined using the crystal structure of 1 bound to nNOS (pdb id: 1p6i) as described by Ji et al.18 Two different nNOS active sites were employed for the modeling, one with and one without the heme structural water molecule. Binding conformations of 5 obtained by AutoDock calculations are depicted in Figure 3.
Figure 3.
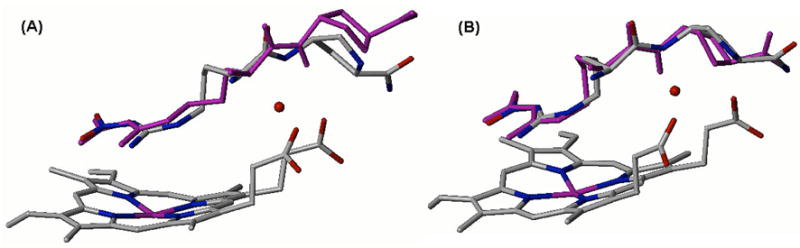
Superimposition of the docked conformations of 5 (purple) and X-ray crystal structure of 2 bound to nNOS (colored by element with carbons in gray, nitrogens in blue, and oxygens in red; pdb id: 1p6j). AutoDock calculations of 5 bound to nNOS active site were performed (A) with or (B) without the structural water molecule.
When 5 was docked into the nNOS active site with the structural water molecule, the N-OH moiety of 5 was placed away from the position of the water molecule; therefore, the proline sidechain was deviated from the conformation of 2 (Figure 3A). In the case of the nNOS active site without the structural water molecule, the N-OH moiety was directed toward the position of the water molecule, and the docked conformation of 5 and the crystal structure of 2 showed similar conformations (Figure 3B). Root mean square deviation (RMSD) values of the docked conformation of 5 from the crystal structure of 2 were (A) 0.37 Å and (B) 0.15 Å. These results led us to conclude that in the minimal binding energy mode between 5 and the nNOS active site, the incorporated N-OH moiety tends to be placed at the position of the structural water molecule. Therefore, displacement of the structural water should be feasible.
X-ray crystal structures of 3 and 5 complexed with nNOS and 3 complexed with eNOS were obtained to 2.00, 2.15, and 1.95 Å resolution, respectively (Figures 4–7). Clearly, the structural water molecule (Water 1 in Figures 5–7) was displaced by the water molecule mimic, the N-OH moiety, and the N-hydroxyl group provided a direct hydrogen bonding interaction with the heme propionate moiety. Therefore, the computer modeling was successful in predicting the desired effect of incorporation of an N-hydroxyl group into the inhibitors. Figure 7 depicts overlapped X-ray crystal structures of the nNOS active sites bound with the inhibitors; (A) 1 and 3 in nNOS, (B) 2 and 5 in nNOS, and (C) 1 and 3 in eNOS. For nNOS, no striking change in the inhibitor/active site residue interactions is observed, and the overall binding conformations of 3 and 5 are quite similar to parent compounds 1 and 2, respectively.
Figure 4.
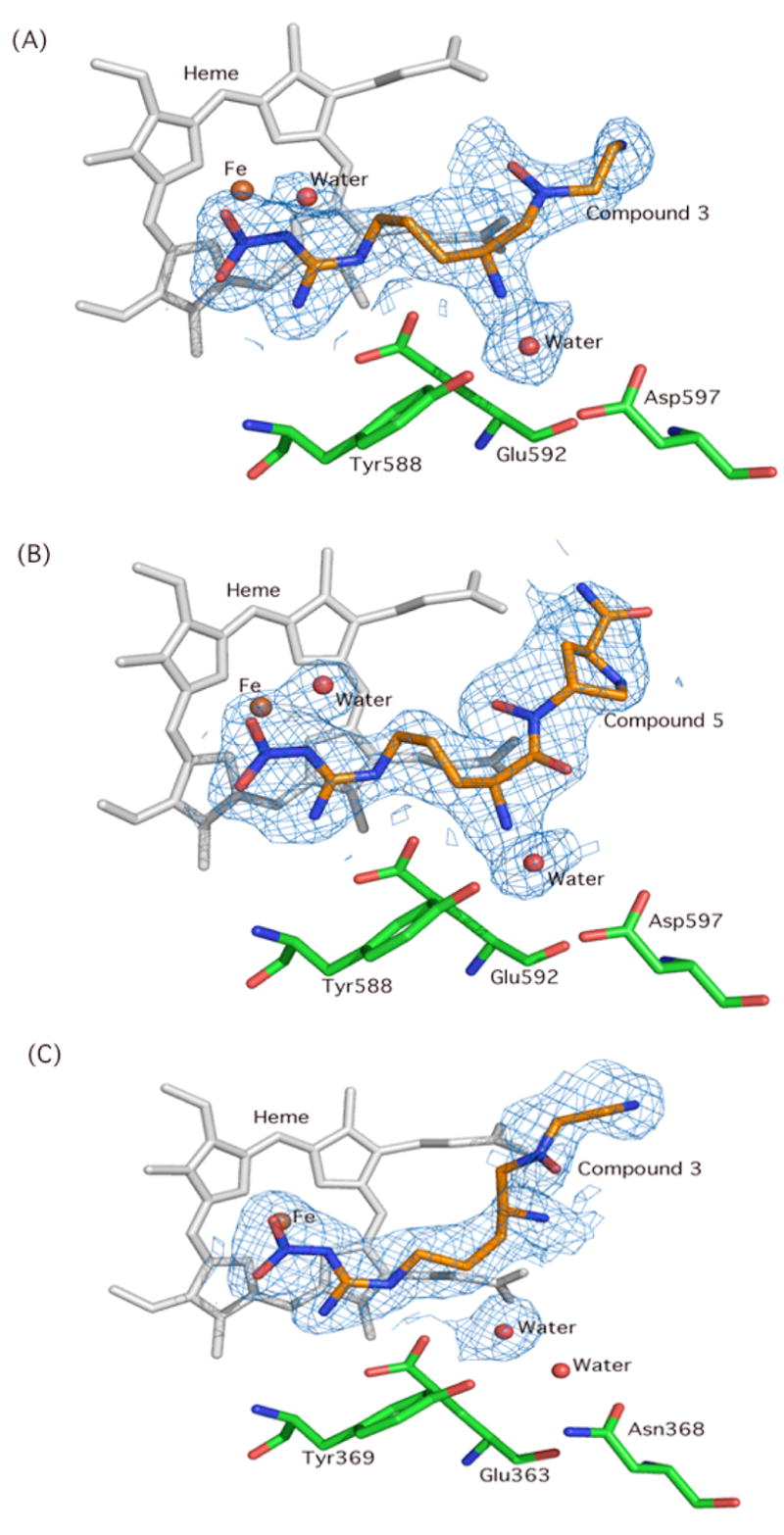
The omit Fo-Fc electron density maps contoured at 3.0s for (A) 3 bound in the nNOS active site; (B) 5 in nNOS; (C) 3 in eNOS. The simulated annealing protocol in CNS with starting temperature at 1000K was used for the calculation.
Figure 7.
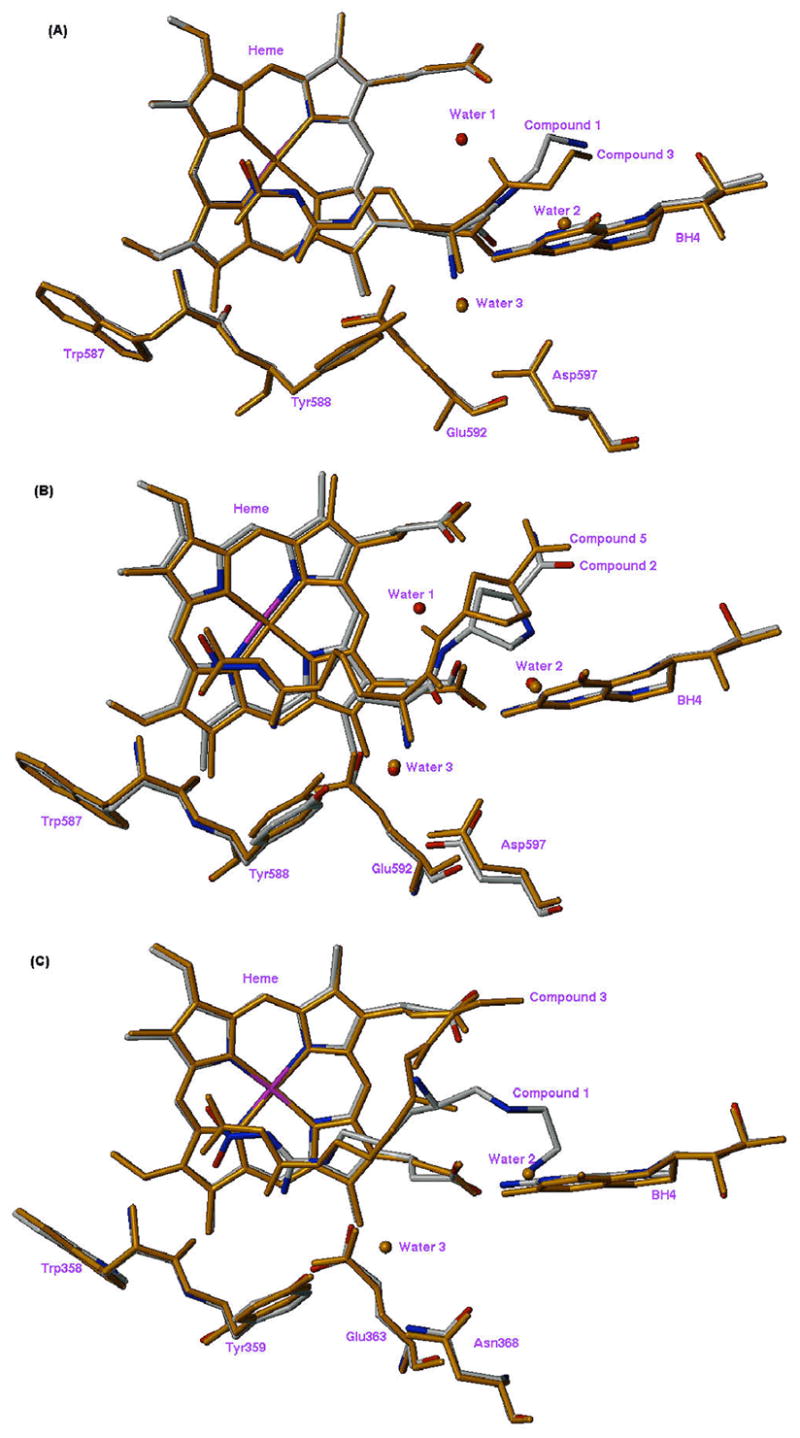
Overlapped X-ray crystal structures of the NOS active site complexed with inhibitors: (A) nNOS (1, gray; 3, orange); (B) nNOS (2, gray; 5, orange); (C) eNOS (1, gray; 3, orange).
Figure 5.
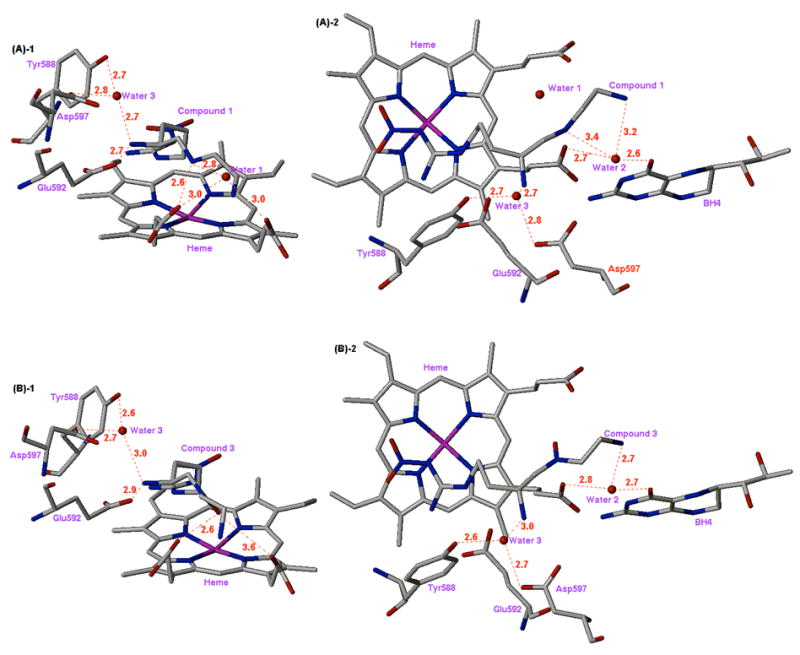
X-ray crystal structures of the nNOS active site with inhibitors 1 (A) and 3 (B) bound (units are Å). (A)-1: Side view of 1; (A)-2: Top view of 1; (B)-1: Side view of 3; (B)-2: Top view of 3.
Compounds 1–5 and L-NNA were tested in vitro against the three isoforms of NOS using the hemoglobin capture assay (see Experimental Section): nitric oxide produced by NOS from L-arginine transforms oxyhemoglobin (Fe2+) to methemoglobin (Fe3+), which is monitored at the maximum absorbance (401 nm); the assay results are shown in Table 3.
Table 3.
NOS inhibition by the N-OH and N-NH2 derivatives and their parent compounds.a
| Compound | Ki (μM) | selectivityd | |||
|---|---|---|---|---|---|
| nNOSb | iNOSc | eNOSc | n/i | n/e | |
| L-NNA | 0.61 ± 0.005 | 4.28 | 0.72 | 7 | 1.2 |
| 1 | 0.17 ± 0.011 | 18 | 191 | 106 | 1124 |
| 2 | 0.33 ± 0.019 | 17 | 72 | 52 | 218 |
| 3 | 0.12 ± 0.017 | 16 | 73 | 133 | 608 |
| 4 | 0.56 ± 0.024 | 37 | 103 | 66 | 184 |
| 5 | 0.31 ± 0.014 | 16 | 35 | 52 | 113 |
The enzyme used for the Ki determinations are bovine brain nNOS, recombinant murine iNOS, and recombinant bovine eNOS. See Experimental Section.
Ki ± SEM. Results are given as a mean of more than or equal to two independent experiments.
The Ki values represent single measurements with 5 data points and correlation coefficients (r2) 0.959–0.996.
The ratio of Ki (iNOS or eNOS) to Ki (nNOS).
Much to our surprise the N-hydroxylated inhibitors 3 and 5 were only marginally more potent than the parent compounds 1 and 2. The N-aminated compound (4) showed more than a three-fold decrease in potency against nNOS. The activity difference between 3 and 4 can be explained by the fact that the N-hydroxyl group is a strong proton donor providing short contacts with carbonyl oxygens whereas the N-amino group is a much weaker proton donor.40 In general, N-hydroxyl modifications (3 and 5) showed little effect on iNOS binding, but tighter binding against eNOS (more than 2-fold), thereby decreasing the nNOS/eNOS selectivity. Similarly, the N-amino compound (4) exhibited increased binding affinity against eNOS, but decreased the affinity against iNOS.
Why should compounds that attain direct interactions with the enzyme not produce much more potent binding interactions? The explanation can be found in other changes in the crystal structures upon binding of 3 and 5. For compound 3 the new interactions between the N-hydroxyl group and the heme propionate causes the inhibitor molecules to move slightly farther from Glu592, weakening somewhat the key hydrogen bonding interaction between the α-amino group of the inhibitors and Glu592.18,22 The distances from the α-amino group to Glu592 are 2.7 Å for 1 and 2.9 Å for 3 ((A)-1 and (B)-1 in Figure 5). The N-OH and heme interaction causes the α-amino groups of 3 to move away from Glu592 by 0.2 Å. As a result, the hydrogen bonding distance from the α-amino group of 3 to another structural water (water 3 in between Tyr588 and Asp597) has also increased (comparison between (A)-1 and (B)-1 of Figure 5). Although there is no direct experimental relationship between hydrogen-bond lengths and hydrogen-bond strengths,41 the bond length difference of approximately 0.25 Å can be significant enough to switch a moderate hydrogen bond (2.65–2.75 Å) to a weak hydrogen bond (2.75–3.00 Å).42 In addition, the decreased basicity of the hydroxylamino nitrogen in 3 compared to the basicity of the secondary amino group in 1 possibly resulted in a weaker interaction of the nitrogen with the heme propionate.
Unlike 3, compound 5 contains a bulkier and conformationally-restricted proline amide side chain. The direct hydrogen bond from N-OH to the heme propionate (pyrrole A) pulls the proline amide tail of 5 closer to the heme compared to 2, which results in a shorter hydrogen bond distance from the inhibitor amide nitrogen to the second heme propionate (pyrrole D). This causes a shift of the proline secondary amine away from another structural water molecule (Water 2) located between the heme propionate (pyrrole A) and the carbonyl oxygen (O-4) of tetrahydrobiopterin ((A)-2 and (B)-2 in Figure 6). When compounds 2 and 5 are compared, their binding interactions involving the nitroguanidino and α-amino groups are similar, and differences are confined to the proline amide tail. However, these do not result in any significant differences in inhibitor-protein interactions that might result in tighter binding. This may explain why the binding affinity of 5 is almost identical to that of 2.
Figure 6.
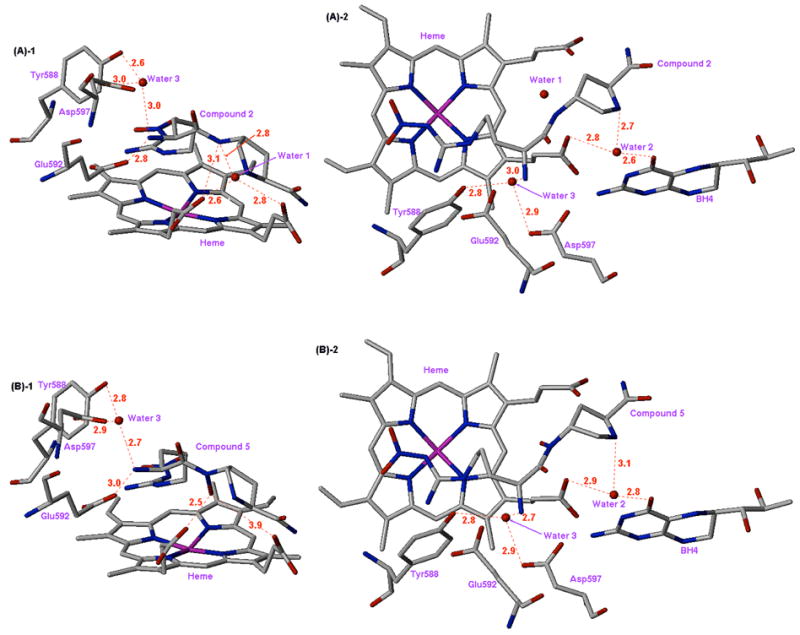
X-ray crystal structures of the nNOS active site with inhibitors 2 (A) and 5 (B) bound (units in Å). (A)-1: Side view of 2; (A)-2: Top view of 2; (B)-1: Side view of 5; (B)-2: Top view of 5.
The differences between 3 and 5 deserve further discussion. The hydroxamate linkage of 5 results in restricted rotation while the ethylamino tail in 3 is small and more flexible. The restricted and bulky proline amide tail in 5 makes more contacts with the heme and the surrounding protein than does 3. These extensive inhibitor-enzyme interactions prevent 5 from undergoing major conformational changes relative to the parent compound 2. In contrast, 3 is more flexible and thus is able to adapt to the addition of the N-OH group, which is even more obvious in the eNOS structures.
In a similar behavior to 1,22 compound 3 binds to wild type eNOS in a different conformation from its binding mode seen in wild type nNOS. Figure 7C indicates that the α-amino group of 3 gains a direct interaction with the heme propionates. This is the likely structural basis for the increased affinity to eNOS in comparison with 1 (Table 3). The common feature for 1 and 3 bound in eNOS is that their α-amino groups stay away from the Glu363 side chain. In contrast, 1 and 3 bound in nNOS have the α-amino group directly hydrogen bonded to Glu592 (Figure 5). As discussed previously22 this isoform-specific binding mode of 1 and 3 originates from one amino acid substitution, Asn368 in eNOS and Asp597 in nNOS, in the carboxylate binding pocket of the substrate binding site. It is interesting to see that the addition of the N-OH group in 3 allows the α-amino group to interact directly with the heme propionate, which was not the case for 1. This makes 3 a better inhibitor for eNOS but a less isoform-selective inhibitor compared to 1 (Table 3).
Conclusions
Compound 3 showed the best potency and selectivity among the N-OH and N-NH2 derivatives. However, a dramatic enhancement in binding affinity as a result of displacement of a structural water molecule and direct interaction with the enzyme was not observed, unlike that observed by Lam et al. for cyclic urea inhibitors of HIV protease.37 In this case the inhibitors had increased potency for multiple reasons: (1) displacement of a structural water molecule; (2) conversion of a flexible and linear inhibitor into a rigid cyclic urea structure with restricted conformation; and (3) a nearly perfect geometry obtained after structural modifications. For compound 3, where the overall hydrogen binding conformation is relatively conserved, a small increase in binding affinity was obtained in nNOS. A larger gain in binding affinity is obtained in eNOS as a result of direct interaction between the α-amino group of 3 and the heme propionate. However, in 5 the energy gain from the structural water displacement did not outweigh the loss of critical hydrogen bonding energy. This indicates that displacement of a structural water molecule itself is not sufficient for a dramatic increase in binding affinity; increased binding requires a highly conserved conformation without disruption of pre-existing hydrogen bonds. Careful structural design of inhibitors is necessary beyond the goal of displacement of a structural water molecule.43 Another reason that there is not much of a change is related to the effects of desolvation. Interchange of an inhibitor-solvent hydrogen bond in the free inhibitor for an inhibitor-protein hydrogen bond may not lead to much gain in terms of binding energy unless the inhibitor-protein interaction is ideal in its properties, such as geometry and distance.
Experimental Section
General Methods
Fisher silica gel 60 (230 – 400 mesh) was used for flash column chromatography with >10% methanol eluent system.; otherwise, Sorbent Technologies silica gel 60 (200 – 400 mesh) was used. Thin-layer chromatography was carried out on E. Merck precoated silica gel 60 F254 plates. Compounds were visualized with a ninhydrin spray reagent or a UV/vis lamp. Combustion analyses were performed by Atlantic Microlab, Inc., Norcross, Georgia. High resolution mass spectroscopy analyses were carried out at the University of Illinois, Urbana-Champaign using a Micromass Quattro spectrometer. Electrospray mass spectra were obtained on a Micromass Quattro II spectrometer. 1H NMR spectra were recorded on a Varian Mercury 400 or Varian Inova 500 NMR spectrometer. Chemical shifts were reported as δ values in parts per million downfield from TMS (δ 0.0) as the internal standard in CDCl3. Melting points were measured on a Buchi melting point B-540 apparatus and are uncorrected. An Orion Research model 701 pH meter with a general combination electrode was used for pH measurements. NOS assays were recorded on a Perkin-Elmer Lambda 10 UV/vis spectrophotometer.
Compounds 1–5 (50 μL injection of a solution of 50 mM sample) were purified by HPLC using a semi-preparative column [Phenomenex, Luna, 250 X 10 mm, C18(2)] with a precolumn [Phenomenex, Luna 50 X 10 mm, C18(2)] at a flow rate of 4 mL/min. Sample elution was detected by absorbance at 254 nm. The mobile phase was as follows: (A, water + 0.1% TFA; B, CH3CN + 0.1% TFA) 10 min using 1% B, then a gradient to 10% B over 10 min, and then a gradient to 1% B over 5 min. The HPLC was performed on a Beckman System Gold chromatograph (Model 125P solvent module and Model 166 detector). Fractions containing the pure product were concentrated in vacuo, and the residue was lyophilized.
Reagents and Materials
All reagents were purchased from Aldrich, Advanced ChemTech, and Nova Biochem. They were used without further purification unless stated otherwise. NADPH, calmodulin, and human ferrous hemoglobin were obtained from Sigma-Aldrich Co.. Tetrahydrobiopterin (H4B) was purchased from Alexis Biochemicals. HEPES, DTT, and conventional organic solvents were purchased from Fisher Scientific. Tetrahydrofuran was distilled under nitrogen from sodium/benzophenone. Diethyl azodicarboxylate (DEAD) was purchased from Lancaster Synthesis, Inc., Pelham, NH. N-Boc-3-(4-Cyanophenyl)oxaziridine was purchased from Acros Organics, NJ (Product number: AC29709-5000).
Nα-(tert-Butoxycarbonyl)-L-nitroarginine 2-(N-tert-butoxycarbonylaminoethyl)amide (6)
To a solution of Boc-L-Arg(NO2)-OH (0.75 g, 2.34 mmol), HBTU (0.89 g, 2.34 mmol), and HOBt (0.32 g, 2.34 mmol) in dry DMF (5 mL) and dry CH2Cl2 (5 mL), was added N-Boc-ethylenediamine (0.25 g, 1.56 mmol) in dry DMF (5 mL) dropwise at 0 °C. DIEA (0.30 mg, 2.34 mmol) in dry CH2Cl2 (5 mL) was added slowly, and stirring was continued for 2 h at room temperature under argon. The solvent was removed under reduced pressure, and the residue was treated with EtOAc (15 mL). The organic mixture was washed successively with 5% NaHCO3, water, 0.5 N HCl, and brine and dried over Na2SO4. The solution was concentrated in vacuo, and the residue was purified by flash column chromatography (EtOAc/MeOH = 19:1, Rf = 0.25) to afford 6 (0.56 g, 78%) as a white solid; mp 98.6–99.6 °C. 1H NMR (500 MHz, acetone-d6) δ 7.67 (s, 1H), 6.26 (s, 1H), 6.14 (s, 1H), 4.17 (s, 1H), 3.41-3.35 (m, 4H), 3.22 (s, 2H), 1.90 (s, 1H), 1.75 (br s, 3H), 1.42 (s, 18H). HRMS (ES) (m/z): M + H+ calcd for C18H36N7O7 462.2676, found 462.2660.
(4S)-4-N-tert-Butoxycarbonylamino-5-[2-(N-tert-butoxycarbonylaminoethyl)aminopentyl]-N′-nitroguanidine (7)
Compound 6 (0.84 g, 1.8 mmol) was treated dropwise with a solution of 1.0 M BH3 (18 mL) in THF at −10 °C under argon. The reaction temperature was allowed to rise to 0 °C. After being stirred at 0 °C for 10 h, the residual BH3 was quenched by cautious addition of MeOH (30 mL) at 0 °C, and the mixture was stirred overnight at room temperature. The solution was evaporated under reduced pressure and treated three times with MeOH (50 mL), evaporating to dryness after each addition, to remove boric acid as trimethyl borate. The residue was purified by flash column chromatography (CH2Cl2/MeOH/Et3N = 12:1:0.1, Rf = 0.3) to afford 7 (0.46 g, 56%) as a transparent oil. 1H NMR (500 MHz, CD3OD) δ 3.69 (s, 1H), 3.37-3.21 (m, 4H), 2.74-2.66 (m, 4H), 1.67 (br s, 2H), 1.61-1.58 (m, 2H), 1.46 (s, 9H), 1.45 (s, 9H); 13C NMR (125 MHz, CD3OD) δ 159.8, 157.5, 157.4, 79.1, 52.8, 49.5, 48.8, 40.9, 39.3, 30.4, 29.0, 27.6; HRMS (ES) (m/z): M + H+ calcd for C18H38N7O6 448.2884, found 448.2888.
(4S)-4-N-tert-Butoxycarbonylamino-5-{[2-(N-tert-butoxycarbonylaminoethyl)benzyoloxyamino]-pentyl}-N′-nitroguanidine (8)
An oven-dried round bottom flask equipped with a magnetic stir bar and reflux condenser was charged with disodium hydrogen phosphate (175 mg, 1.23 mmol), 7 (110 mg, 0.25 mmol), and dry ether/THF (3 mL/3 mL). To this stirred suspension was added dibenzoyl peroxide (121 mg, 0.50 mmol) in dry ether/THF (2 mL/2 mL), and the reaction mixture was stirred at 70 °C for 24 h under argon atmosphere. The solvent was removed under reduced pressure, and the residue was partitioned between EtOAc (5 mL) and water (5 mL). The mixture was washed twice with water and dried over Na2SO4. The solution was concentrated in vacuo, and the residue was purified by flash column chromatography (hexane/EtOAc = 1:4, Rf = 0.4) to afford 8 (96 mg, 68%) as an oil. 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 7.5 Hz, 2H), 7.60 (t, J = 7.5 Hz, 1H), 7.46 (t, J = 8.0 Hz, 2H), 5.56 (s, 1H), 5.19 (br s, 1H), 3.77 (br s, 1H), 3.41 (br s, 1H), 3.21 (br s, 3H), 3.07 (s, 4H), 1.66 (br s, 4H), 1.43 (s, 9H), 1.37 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 166.4, 159.6, 156.4, 134.0, 129.8, 128.9, 128.4, 80.3, 63.4, 60.0, 40.8, 37.8, 31.2, 29.7, 28.7, 28.6, 25.0; HRMS (ES) (m/z): M + H+ calcd for C25H42N7O8 568.3095, found 568.3098.
(4S)-4-N-tert-Butoxycarbonylamino-5-{[2-(N-tert-butoxycarbonylaminoethyl)hydoxyamino]-pentyl}-N′-nitroguanidine (9)
Compound 8 (60 mg, 0.11 mmol) was treated with a solution of 0.3% NaOH in MeOH (3 mL), and the mixture was stirred at room temperature for 4 h. After evaporation of the MeOH in vacuo, the residue was partitioned between water (10 mL) and EtOAc (10 mL), and the aqueous phase was extracted with EtOAc (10 mL). The combined organic layer was washed with brine (20 mL), dried over Na2SO4, and evaporated. The residue was purified by flash column chromatography (hexane/EtOAc = 1:4, Rf = 0.3) to afford 9 (50 mg, 98%) as an oil. 1H NMR (500 MHz, CDCl3) δ 7.11 (s, 1H), 5.38 (s, 1H), 4.99 (s, 1H), 3.86 (s, 1H), 3.43–3.45 (m, 2H), 3.25 (s, 2H), 2.65 – 2.77 (m, 4H), 1.71 (s, 2H), 1.58–1.64 (m, 2H), 1.45 (s, 18H); 13C NMR (125 MHz, CDCl3) δ 159.6, 157.8, 80.2, 63.9, 60.7, 48.3, 41.2, 38.1, 30.9, 29.0, 28.7, 25.3; HRMS (ES) (m/z): M + H+ calcd for C18H38N7O7 464.2833, found 464.2821.
(4S)-N-{4-Amino-5-[(2-aminoethyl)-hydroxyamino]-pentyl}-N′-nitroguanidine (3)
Compound 9 (30 mg, 0.065 mmol) was treated with trifluoroacetic acid/CH2Cl2 (5 mL/5 mL) at 0 °C under nitrogen. The reaction temperature was then allowed to rise to room temperature, and stirring continued for 45 min. Excess TFA and solvent were removed by evaporation in vacuo. The residue was repeatedly dissolved in 10 mL CH2Cl2 and the solvents evaporated to remove traces of TFA. The residue was dissolved in a small amount of water, and the solution was washed with CH2Cl2 and lyophilized to give a yellowish hygroscopic foam (26 mg, 96%). 1H NMR (500 MHz, D2O) δ 3.38 (s, 1H), 3.16 (s, 2H), 3.01 – 3.12 (m, 2H), 2.80 – 2.90 (m, 3H), 2.69 (dd, J = 10.0, 13.5, 1H), 1.58 (s, 4H); [α]24D +8.4 (c = 0.50, MeOH); Anal. Calcd for C8H21N7O3·3.5HCl: C, H, N.
(4S)-4-N-tert-Butoxycarbonylamino-5-{[2-(N-tert-butoxycarbonylaminoethyl)-(N-tert-butoxycarbonylhydrazino)]pentyl}-N′-nitroguanidine (11)
A solution of 7 (16 mg, 0.036 mmol) in dry CHCl3 (2 mL) was treated at 0 °C with a solution of N-Boc-3-(4-cyanophenyl)oxaziridine (10, 13 mg, 0.054 mmol) in dry CHCl3 (2 mL). At the end of the addition the cooling bath was removed, and stirring continued at room temperature overnight. After the starting material was consumed, the mixture was concentrated in vacuo, and the residue was purified by flash column chromatography (hexane/EtOAc = 1:4, Rf = 0.35) to afford 11 (20 mg, 97%) as a syrup. 1H NMR (500 MHz, CDCl3) δ 5.85 (s, 1H), 5.44 (s, 2H), 3.69 (s, 1H), 3.52 (s, 1H), 3.24 (s, 3H), 2.84 (s, 2H), 2.78 (s, 2H), 1.85 (s, 1H), 1.61 – 1.72 (m, 3H), 1.46 (s, 9H), 1.45 (s, 18H); 13C NMR (125 MHz, CDCl3) δ 159.6, 156.5, 80.1, 79.5, 62.9, 58.8, 47.8, 40.8, 38.0, 31.1, 28.7, 28.5, 25.1; HRMS (ES) (m/z): M + H+ calcd for C23H47N8O8 563.3517, found 563.3518.
(4S)-N-{4-Amino-5-[(2-aminoethyl)-hydrazino]-pentyl}-N′-nitroguanidine (4)
Using the procedure described for the preparation of 3, compound 4 was obtained from 11 as a yellowish foam (32 mg, 97%); mp 59.8–61.2 °C. 1H NMR (500 MHz, D2O) δ 3.42 (s, 1H), 3.11 – 3.19 (m, 7H), 2.95 (dd, J = 13.5, 11.0, 1H), 1.57 (s, 4H); Anal. Calcd for C8H22N8O2·4.5HCl: C, H, N.
Compounds 12 to 18 were characterized for structural purity by 1H and 13C NMR spectrometry. The NMR spectra of those compounds showed the characteristic presence of rotamer mixtures originating from the restricted rotation about the tertiary amide bond of the proline ring.44 A high temperature 1H NMR experiment was performed on 13 to see if the rotational energy barrier could be overcome at a higher temperature. At ambient temperature, two distinct peaks from the methyl ester were observed at δ 3.61 and δ 3.55 having a ratio of 1.9 to 1.1, respectively. At 70 °C, the two peaks merged to a singlet at δ 3.64. (500 MHz, DMSO).
N-Fmoc-4-hydroxy-L-proline lactone (12)
To a stirred ice-cold solution of Fmoc-Hyp-OH (3.0 g, 8.5 mmol) and triphenylphosphine (2.7 g, 10.2 mmol) in freshly distilled THF (200 mL) under nitrogen was added DIAD (2.1 g, 10.2 mmol) dropwise. The reaction temperature was then allowed to rise to room temperature and stirring continued for 2 h. The solvent was evaporated under reduced pressure, and the residue was directly purified by flash column chromatography (EtOAc/hexane = 1:1, Rf = 0.35), to give 2.4 g (85%) of 12 as a white foamy solid; mp 106.4–106.8 °C. 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 7 Hz, 2H), 7.64 (m, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.34 (t, J = 7.0 Hz, 2H), 5.14 (s, 1H), 4.67 (s, 1H), 4.39–4.50 (m, 2H), 4.27 (s, 1H), 3.43–3.58 (m, 2H), 2.27 (s, 1H), 2.06 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 171.8, 154.5, 143.9, 141,7, 128.1, 127.4, 124.9, 120.3, 74.8, 67.3, 60.7, 47.4, 39.5, 35.3.
N-Fmoc-cis-4-hydroxy-L-proline methyl ester (13)
A solution of 12 (2.0 g, 6.0 mmol) and sodium azide (1.1 g, 18.0 mmol) in dry MeOH (50 mL) was stirred for 3 h at 40 °C under nitrogen. The solvent was evaporated under reduced pressure, and the residue was partitioned between water and EtOAc. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The compound was purified by flash column chromatography (EtOAc/hexane = 2:1, Rf = 0.3), to give 13 (2.1 g, 96%) as a white solid; mp 48.5–48.8 °C. 1H NMR (400 MHz, DMSO) δ 7.87 (t, J = 8.0 Hz, 2H), 7.58–7.65 (m, 2H), 7.40 (d, J = 6.8 Hz, 2H), 7.32 (d, J = 6.0 Hz, 2H), 4.16–4.28 (m, 5H), 3.49–3.59 (m, 4H), 3.20 (m, 1H), 2.29 (s, 1H), 1.90 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 172.8, 155.4, 143.9, 141.3, 127.6, 126.9, 124.7, 119.8, 69.6, 67.5, 58.0, 54.5, 51.8, 38.8, 37.9; MS (ESI, CH2Cl2) [M+H+] = 368.2.
N-Fmoc-trans-4-(N,O-di-tert-Butoxycarbonyl-hydroxyamino)-L-proline methyl ester (14)
To a stirred ice-cold solution of the 13 (500 mg, 1.36 mmol), triphenylphosphine (393 mg, 1.50 mmol), and Boc-HN-O-Boc (350 g, 1.50 mmol) in anhydrous THF (20 mL) under nitrogen was added diethyl azodicarboxylate (261 mg, 1.50 mmol) dropwise over 15 min. The reaction temperature was allowed to rise to room temperature, and stirring continued for 30 min. The solvent was evaporated in vacuo, and the residue was directly purified by flash column chromatography (EtOAc/hexane = 1:3, Rf = 0.35), to give 14 (332 mg, 42%) as a white foamy solid. 1H NMR (500 MHz, CDCl3) δ 7.71 (m, 2H), 7.54 (m, 2H), 7.35 (t, J = 7.0 Hz, 2H), 7.27 (d, J = 6.0 Hz, 2H), 4.98 (m, 1H), 4.21–4.51 (m, 4H), 3.82–3.94 (m, 1H), 3.57–3.72 (m, 4H), 2.36–2.49 (m, 1H), 2.23 (s, 1H), 1.48 (s, 9H), 1.45 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 172.7, 154.8, 154.3, 152.5, 144.2, 141.5, 127.9, 127.3, 125.3, 120.2, 85.5, 83.5, 68.1, 60.5, 58.1, 56.3, 52.7, 47.3, 32.1, 28.3, 27.7; HRMS (ES) (m/z): M + H+ calcd for C31H39N2O9 583.2656, found 583.2666.
N-Fmoc-trans-4-hydroxyamino-L-proline methyl ester (15)
Compound 14 (300 mg, 0.52 mmol) was treated with trifluoroacetic acid/CH2Cl2 (5 mL/5 mL) at 0 °C under nitrogen. The reaction temperature was then allowed to rise to room temperature, and stirring was continued for 45 min. Excess TFA and solvent were removed by evaporation. The residue was repeatedly dissolved in CH2Cl2 (10 mL) and the solvent was evaporated to remove traces of TFA. Trituration of this brown oil with ether (10 mL) gave 15 (245 mg, 95%) as a yellowish solid, which was used in the next reaction without further purification. TLC (EtOAc/MeOH, 19:1) Rf = 0.50; 1H NMR (400 MHz, CD3OD) δ 7.81 (s, 2H), 7.58–7.64 (m, 2H), 7.40 (s, 2H), 7.32 (s, 2H), 4.48 (s, 2H), 4.37 (d, J = 8.8 Hz, 1H), 4.17–4.28 (m, 1H), 4.08-4.08 (m, 1H), 3.62–3.87 (m, 5H), 2.56 (s, 1H), 2.37 (s, 1H); 13C NMR (125 MHz, acetone- d6) δ 172.6, 154.2, 144.3, 141.4, 128.0, 127.4, 125.5, 120.3, 67.7, 63.0, 58.7, 52.0, 49.0, 47.3, 32.6; HRMS (ES) (m/z): M + H+ calcd for C21H23N2O5 383.1607, found 383.1614.
N-Fmoc-4-N-(Nα-Fmoc-Nω-nitro-L-argininyl)-trans-4-hydroxyamino-L-proline methyl ester (16)
A completely dissolved solution of Fmoc-Arg(NO2)-OH (486 mg, 1.10 mmol) in freshly distilled anhydrous THF (3 mL) was chilled in an ice-acetone bath (−10 °C), and to this was added SOCl2 (262 mg, 2.20 mmol) dropwise. The mixture was stirred under nitrogen for 1 h. Chilled anhydrous ether (20 mL) was introduced to yield a syrupy precipitate. Maintaining low temperature, the solvent was removed under reduced pressure, and the residue was triturated with chilled anhydrous ether (20 mL). A white syrupy solid was obtained after evaporation under reduced pressure and excess thionyl chloride was removed. During the evaporations, an ice-cold solution of 15 (275 mg, 0.55 mmol) and 2,4,6-collidine (67 mg, 0.55 mmol) in THF (5 mL) was prepared and added via cannula to a flask containing the acid chloride. Stirring continued for 20 min at 0 °C then 20 min more at room temperature. The solvent was removed under reduced pressure, and the residue was treated with EtOAc (10 mL). The organic mixture was washed twice with 5% NaHCO3, water, 0.5 N HCl, and brine, and dried over Na2SO4. The solution was concentrated in vacuo, and the residue was purified by flash column chromatography (EtOAc/MeOH = 19:1, Rf = 0.45) to afford 16 (330 mg, 74%) as a white foamy solid. 1H NMR (400 MHz, acetone-d6) δ 7.84 (d, J = 5.6 Hz, 4H), 7.64 (m, 4H), 7.40 (d, J = 7.2 Hz, 4H), 7.32 (d, J = 5.2 Hz, 4H), 6.73 (m, 1H), 5.26 (m, 1H), 4.88 (s, 1H), 4.51 (d, J = 5.6 Hz, 1H), 4.29 (m, 6H), 3.70 (m, 4H), 3.39 (s, 2H), 2.62 (m, 1H), 2.19 (d, J = 6.4 Hz, 1H), 2.05 (q, J = 2.5 Hz, 2H), 1.80 (m, 4H); 13C NMR (125 MHz, acetone-d6) δ 172.73, 170.40, 160.19, 156.60, 154.65, 144.41, 144.24, 141.45, 127.95, 127.43, 127.36, 125.54, 125.32, 120.22, 67.58, 66.71, 59.98, 58.51, 57.94, 53.43, 52.10, 51.87, 49.93, 47.32, 47.22, 40.89, 40.72, 33.88, 32.94, 20.28; HRMS (ES) (m/z): M + H+ calcd for C42H44N7O10 806.3150, found 806.3156.
N-Boc-4-N-(Nα-Boc-Nω-nitro-L-argininyl)-trans-4-hydroxyamino-L-proline methyl ester (17)
Piperidine (1 mL) was added to a solution of 16 (63 mg, 0.078 mmol) in DMF (4 mL), and the mixture was stirred at room temperature for 0.5 h (monitoring by TLC). The volatile components were removed under reduced pressure. To remove excess piperidine completely, the residue was repeatedly dissolved in CH2Cl2, and the solvents were evaporated several times. The residue was then dissolved in 1,4-dioxane (4 mL) and 10% aq. NaHCO3 (3 mL), and the solution of Boc2O (51 mg, 0.23 mmol) in 1,4-dioxane (1 mL) was added dropwise at 0 °C. After being stirred at room temperature overnight, the reaction mixture was partitioned between water (10 mL) and EtOAc (10 mL), and the aqueous layer was extracted with EtOAc (10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, and evaporated. The residue was purified by flash column chromatography (EtOAc/MeOH = 19:1, Rf = 0.3) to afford 17 (36 mg, 83%) as a colorless foam. 1H NMR (500 MHz, CDCl3) δ 5.18 (s, 1H), 4.78 (s, 1H), 4.40 – 4.49 (m, 1H), 3.72 (s, 3H), 3.67 (m, 1H), 3.55 – 3.61 (m, 2H), 3.30 (s, 1H), 2.54 (s, 1H), 2.26 (m, 1H), 1.83 (s, 1H), 1.70 (s, 2H), 1.55 (m, 1H), 1.38 – 1.42 (m, 18H); HRMS (ES) (m/z): M + H+ calcd for C22H40N7O10 562.2837, found 562.2839.
N-Boc-4-N-(Nα-Boc-Nω-nitro-L-argininyl)-trans-4-hydroxyamino-L-proline amide (18)
A mixture of 17 (80 mg, 0.14 mmol) in THF (5 mL) and 5% aq. LiOH (5 mL) was stirred at room temperature for 1 h. After evaporation of THF under reduced pressure, the aqueous solution was washed twice with EtOAc (5 mL), acidified with 1N HCl to pH 3, and extracted twice with EtOAc (10 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, and evaporated. The residue was dried overnight under high vacuum and dissolved in THF (5 mL) at ambient temperature. The solution was cooled to −10 °C. To this solution, N-methylmorpholine (16 mg, 0.15 mmol) and ibutyl chloroformate (21 mg, 0.15 mmol) were added successively, and stirring continued for 30 min at −10 °C, and then 29% aqueous ammonia (0.028 mL) was added. The mixture was stirred at −10 °C for 30 min and at room temperature for 12 h. After addition of water (10 mL), the solution was extracted twice with EtOAc (10 mL), washed with brine, dried over Na2SO4, filtered, and evaporated. The residue was purified by flash column chromatography (EtOAc/MeOH = 10:1, Rf = 0.25) to afford 18 (47 mg, 61% from 17) as a colorless foam. 1H NMR (500 MHz, CD3OD) δ 5.11 (m, 1H), 4.66 (s, 1H), 4.32 (m, 1H), 3.52 – 3.72 (m, 2H), 3.24 (s, 2H), 2.43 (m, 1H), 2.08 (m, 1H), 1.79 (s, 1H), 1.66 (s, 2H), 1.57 (m, 1H), 1.45 (s, 4H), 1.42 (s, 14H); HRMS (ES) (m/z): M + H+ calcd for C21H39N8O9 547.2840, found 547.2837.
4-N-(Nω-nitro-L-argininyl)-trans-4-hydroxyamino-L-proline amide (5)
Using the procedure described for the preparation of 3, compound 5 was obtained from 18 as a white hygroscopic foam (17 mg, 97%). 1H NMR (500 MHz, D2O) δ 5.14 (m, 1H), 4.49 (t, J = 8.5 Hz, 1H), 4.40 (m, 1H), 3.39 – 3.63 (m, 2H), 3.17 (s, 2H), 2.42 – 2.47 (m, 1H), 2.28 – 2.34 (m, 1H), 1.82 (s, 2H), 1.58 (s, 2H); HRMS (ES) (m/z): M + H+ calcd for C11H23N8O5 347.1791, found 347.1801; [α]24D +8.0 (c = 0.20, MeOH); Anal. Calcd for C11H21N8O5·3TFA·H2O: C, H, N.
Docking Analysis
Molecular modeling calculations were performed using the software packages SYBYL 6.8 and AutoDock 3.0.5 running on a Silicon Graphics Octane 2 workstation. The protein structure used in the docking study was prepared as described previously.18 The 3-D structure of the ligand was built in SYBYL 6.8 by modifying the molecular structure of 2, which was extracted from the crystal structure (pdb id: 1p6j). Using the SYBYL program, correct atom types were assigned assuming physiological pH. Energy minimizations were performed following both the addition of polar hydrogen atoms and partial atom charge calculations by the the Gasteiger-Marsilli method.45 The torsion and rotatable bonds in the ligand were defined by AutoTors, an auxiliary program of AutoDock 3.0.5, which also united the nonpolar hydrogens and partial atomic charges to the bonded carbon atoms. Parameters for the docking experiment were used as described in detail previously.18 A flexible docking calculation yielded 100 docked conformations, and appropriate conformations were chosen by visual comparison of the superimposition of the guanidino moiety of the predicted conformations and that of 2.
Enzyme and Assay
All of the NOS isoforms used are recombinant enzymes overexpressed in E. coli from different sources. Murine macrophage iNOS,46 rat nNOS,47 and bovine eNOS48 were expressed and isolated as reported. Nitric oxide formation from NOS was monitored by the hemoglobin capture assay at 30 °C as described previously.49 Briefly, a solution of nNOS or eNOS contained 10 μM L-arginine, 1.6 mM CaCl2, 11.6 μg/mL calmodulin, 100 μM DTT, 100 μM NADPH, 6.5 μM H4B, 3 mM oxyhemoglobin, and specified inhibitor concentration in 100 mM Hepes (pH 7.5) in 600 μL total volume; iNOS contained the same concentrations of cofactors, except CaCl2 and calmodulin were not added. The assay was initiated by addition of enzyme and was monitored at 401 nm on a Perkin-Elmer Lambda 10 UV-vis spectrophotometer.
Determination of Ki Values
The reversible inhibition of NOS by peptidomimetic inhibitors was studied under initial rate conditions using the hemoglobin assay as described above. The apparent Ki values were obtained by measuring percent inhibition in the presence of 10 μM L-arginine with at least four different concentrations of inhibitor. Generally, the inhibitor concentrations of two higher and two lower than 50% inhibition were used. The IC50 values were determined by linear (or logarithmic for a few cases) regression analysis of the percent inhibition data. The apparent Ki values were calculated from the IC50 values using the following inhibition equation:50 % inhibition = 100[I]/{[I] + Ki (1 +[S]/Km)} or Ki = IC50/(1 +[S]/Km). Km values for L-arginine were 1.3 μM (nNOS), 8.3 μM (iNOS), and 1.7 μM (eNOS). The selectivity of an inhibitor was defined as the ratio of the respective Ki values.
Crystal Preparation and Structure Determination
The procedures for preparing nNOS or eNOS crystals complexed with inhibitors 3 and 5 are similar to what were reported previously.22 Cryogenic X-ray diffraction data were collected at either Stanford Synchrotron Radiation Laboratory (Menlo Park, California) or Advanced Light Source (Berkeley, California) using a Q315 CCD detector. Raw data were integrated and scaled with HKL2000.51 The bound inhibitors were revealed by difference Fourier synthesis using CNS.52 Models of inhibitors were built in O53 and structures refined with CNS. Reflections and coordinates of refined structures have been deposited in the RCSB protein data bank. The data collection and structure refinement statistics for the three new structures are summarized in Table S2 of Supporting Information.
Supplementary Material
Details of data collection, structure refinement statistics of the three new structures; elemental analysis data of compounds 3–5; recently reported amide reduction method. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health, GM 49725 to RBS, GM57353 to TLP, and GM52419 to Dr. Bettie Sue Masters, in whose lab PM and LJR work, and Grant No. AQ1192 from The Robert A. Welch Foundation to BSM. We also acknowledge the SSRL and ALS beamline staff for assistance with X-ray data collection. J. S. thanks Dr. Haitao Ji and Dr. Wenxin Gu for helpful discussions.
References
- 1.(a) Kerwin JF, Jr, Lancaster JR., Jr Nitric Oxide; a New Paradigm for Second Messengers. Med Res Rev. 1994;14:23–74. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]; (b) Stuehr DJ. Enzymes of the L-Arginine to Nitric Oxide Pathway. J Nutr. 2004;134(10 Suppl):2748S–2751S. doi: 10.1093/jn/134.10.2748S. [DOI] [PubMed] [Google Scholar]
- 2.(a) Waldman SA, Murad F. Cyclic GMP Synthesis and Function. Pharmacol Rev. 1987;39:163–196. [PubMed] [Google Scholar]; (b) Hobbs AJ. Soluble Guanylate Cyclase: the Forgotten Sibling. Trends Pharmacol Sci. 1997;18:484–491. doi: 10.1016/s0165-6147(97)01137-1. [DOI] [PubMed] [Google Scholar]; (c) Denninger JW, Marletta MA. Guanylate Cyclase and the NO/cGMP Signaling Pathway. Biochem Biophys Acta. 1999;1411:334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 3.(a) Moncada S, Palmer RMJ, Higgs EA. Nitric Oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]; (b) Moncada S, Higgs EA. Molecular Mechanisms and Therapeutic Strategies Related to Nitric Oxide. FASEB J. 1995;9:1319–1330. [PubMed] [Google Scholar]
- 4.(a) Hardy P, Abran D, Hou X, Lahaie I, Peri KG, Asselin P, Varma DR, Chemtob S. A Major Role for Prostacyclin in Nitric Oxide-Induced Ocular Vasorelaxation in the Piglet. Circ Res. 1998;83:721–729. doi: 10.1161/01.res.83.7.721. [DOI] [PubMed] [Google Scholar]; (b) Wink DA, Miranda KM, Espey MG, Mitchell JB, Grisham MB, Fukuto J, Feelish M. The Chemical Biology of Nitric Oxide. Balancing Nitric Oxide with Oxidative and Nitrosative Stress. Handb Exp Pharmacol. 2000;143:7–29. [Google Scholar]
- 5.Moncada S, Higgs A, Furchgott R. XIV. International Union of Pharmacology Nomenclature in Nitric Oxide Research. Pharmacol Rev. 1997;49:137–142. [PubMed] [Google Scholar]
- 6.Schmidt HHHW, Walter U. NO at Work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 7.Forstermann U, Pollock JS, Schmidt HHHW, Heller M, Murad F. Calmodulin Dependent Endothelium-Derived Relaxing Factor/Nitric Oxide Synthase Activity is Present in the Particulate and Cytosolic Fractions of Bovine Aortic Endothelial Cells. Proc Natl Acad Sci US A. 1991;88:1788–1792. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacMicking J, Xie QW, Nathan C. Nitric Oxide and Macrophage Function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 9.Roman LJ, Martásek P, Masters BSS. Intrinsic and Extrinsic Modulation of Nitric Oxide Synthase Activity. Chem Rev. 2002;102:1179–1189. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the Expression of Inducible Nitric Oxide Synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]; (b) Wu C, Thiemermann C. Biological Control and Inhibition of Induction of Nitric Oxide Synthase. Methods Enzymol. 1996;268:408–420. doi: 10.1016/s0076-6879(96)68043-4. [DOI] [PubMed] [Google Scholar]
- 11.(a) Knowles RG, Moncada S. Nitric Oxide Synthases in Mammals. Biochem J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Masters BS, McMillan K, Sheta EA, Nishimura JS, Roman LJ, Martásek P. Neuronal Nitric Oxide Synthase, a Modular Enzyme Formed by Convergent Evolution: Structure Studies of a Cystein Thiolate-Liganded Heme Protein that Hydroxylates L-Arginine to Produce NO as a Cellular Signal. FASEB J. 1996;10:552–558. doi: 10.1096/fasebj.10.5.8621055. [DOI] [PubMed] [Google Scholar]
- 12.(a) Raman CS, Martásek P, Masters BSS. The Porphyrin Handbook. Academic Press; New York: 2000. pp. 293–339. [Google Scholar]; (b) Raman CS, Li H, Martásek P, Král V, Masters BSS, Poulos TL. Crystal Structure of Constitutive Endothelial Nitric Oxide Synthase: a Paradigm for Pterin Function Involving a Novel Metal Center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]; (c) Fishmann TO, Hruza A, Niu XD, Fossetta JD, Junn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Structural Characterization of Nitric Oxide Synthase Isoforms Reveals Striking Active-Site Conservation. Nat Struc Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 13.Siddhanta U, Presta A, Fan B, Wolan D, Rousseau DL, Stuehr DJ. Domain Swapping in Inducible Nitric Oxide Synthases. Electron Transfer Occurs Between Flavin and Heme Groups Located on Adjacent Subunits in the Dimer. J Biol Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 14.(a) Vallance P, Leiper J. Blocking NO Synthesis: How, Where and Why? Nat Rev Drug Discov. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]; (b) Grunewald T, Beal MF. NOS Knockouts and Neuroprotection. Nat Med. 1999;5:1354–1355. doi: 10.1038/70918. [DOI] [PubMed] [Google Scholar]
- 15.Uehara T, Nakamura T, Yao D, Shi Z, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-Nitrosylated Protein-disulphide Isomerase Links Protein Misfolding to Neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 16.(a) Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB. Selective Neuronal Nitric Oxide Synthase Inhibirors. Curr Top Med Chem. 2005;5:603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]; (b) Salerno L, Sorrenti V, Di Giacomo C, Romeo G, Siracusa MA. Progress in the Development of Selective Nitric Oxide Synthase (NOS) Inhibitors. Curr Pharm Des. 2002;8:177–200. doi: 10.2174/1381612023396375. [DOI] [PubMed] [Google Scholar]; (c) Hobbs AJ, Higgs A, Moncada S. Inhibition of Nitric Oxide Synthase as a Potential Therapeutic Target. Annu Rev Pharmacol Toxicol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]; (d) Marletta MA. Approaches Toward Selective Inhibition of Nitric Oxide Synthase. J Med Chem. 1994;37:1899–1907. doi: 10.1021/jm00039a001. [DOI] [PubMed] [Google Scholar]
- 17.Ignarro LJ, Napoli C, Loscalzo J. Nitric Oxide Donors and Cardiovascular Agents Modulating the Bioactivity of Nitric Oxide. Circ Res. 2002;90:21–28. doi: 10.1161/hh0102.102330. [DOI] [PubMed] [Google Scholar]
- 18.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. Computer Modeling of Selective Regions in the Active Site of Nitric Oxide Synthases: Implication for the Design of Isoform-Selective Inhibitors. J Med Chem. 2003;46:5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 19.(a) Silverman RB, Huang H, Marletta MA, Martásek P. Selective Inhibition of Neuronal Nitric Oxide Synthase by Nω-Nitroarginine- and Phenylalanine-Containing Dipeptides and Dipeptide Esters. J Med Chem. 1997;40:2813–2817. doi: 10.1021/jm970200u. [DOI] [PubMed] [Google Scholar]; (b) Huang H, Martásek P, Roman LJ, Masters BSS, Silverman RB. Nω-Nitroarginine-Containing Dipeptide Amides. Potent and Highly Selective Inhibitors of Neuronal Nitric Oxide Synthase. J Med Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 20.(a) Hah J, Roman LJ, Martásek P, Silverman RB. Reduced Amide Bond Peptidomimetics. (4S)-N-(4-Amino-5-[aminoalkyl]aminopentyl)-N′-nitroguanidines, Potent and Highly Selective Inhibitors of Neuronal Nitric Oxide Synthase. J Med Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]; (b) Gomez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Potent and Selective Conformationally Restricted Neuronal Nitric Oxide Synthase Inhibitors. J Med Chem. 2004;47:703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- 21.(a) Hah J, Martásek P, Roman LJ, Silverman RB. Aromatic Reduced Amide Bond Peptidomimetics as Selective Inhibitors of Neuronal Nitric Oxide Synthase. J Med Chem. 2003;46:1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]; (b) Mbadugha BNA, Seo J, Ji H, Martásek P, Roman LJ, Shea TM, Li H, Poulos TL, Silverman RB. Hydroxyl-terminated Peptidomimetic Inhibitors of Neuronal Nitric Oxide Synthase. Bioorg Med Chem. 2006;14:3681–3690. doi: 10.1016/j.bmc.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 22.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah J, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural Basis for Dipeptide Amide Isoform-selective Inhibition of Neuronal Nitric Oxide Synthase. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 23.Jensen KB, Braxmeier TM, Demarcus M, Frey JG, Kilburn JD. Synthesis of Guanidinium-Derived Receptor Libraries and Screening for Selective Peptide Receptors in Water. Chem Eur J. 2002;8:1300–1309. doi: 10.1002/1521-3765(20020315)8:6<1300::aid-chem1300>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 24.Zhao J, Pattaropong V, Jiang Y, Hu L. Facile Synthesis and Cleavage of Imidazolidines in a Novel Protection Strategy for the Preparation of Peptides Containing a Reduced Amide Bioisostere. Tetrahedron Lett. 2003;44:229–232. [Google Scholar]
- 25.Dolbeare K, Pontoriero GF, Gupta SK, Mishra RK, Johnson RL. iso-Lactam and Reduced Amide Analogues of the Peptidomimetic Dopamine Receptor Modulator 3(R)-[(2(S)-Pyrrolidinylcarbonyl)amino]-2-oxo-pyrrolidineacetamide. Bioorg Med Chem. 2003;11:4103–4112. doi: 10.1016/s0968-0896(03)00396-1. [DOI] [PubMed] [Google Scholar]
- 26.Voight EA, Bodenstein MS, Ikemoto N, Kress MH. Efficient Preparation of Chiral Diamines via Red-Al reduction of N-Boc-protected Amino Acid-derived Secondary Amides. Tetrahedron Lett. 2006;47:1717–1720. [Google Scholar]
- 27.Roeske RW, Weitl FL, Prasad KU, Thompson RM. Selective Reduction of the Amide Carbonyl Group in Dipeptides by Borane. J Org Chem. 1976;41:1260–1261. doi: 10.1021/jo00869a041. [DOI] [PubMed] [Google Scholar]
- 28.(a) Hirel C, Vostrikova KE, Pecaut J, Ovcharenko VI, Rey P. Nitronyl and Imino Nitroxides: Improvement of Ullman’s Procedure and Report on a New Efficient Synthetic Route. Chem Eur J. 2001;7:2007–2014. doi: 10.1002/1521-3765(20010504)7:9<2007::aid-chem2007>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]; (b) Tepe JJ, Kosogof C, Williams RM. DNA Interstrand Cross-link Formation by Reductive Activation of Dehydropyrrolizidine Progenitors. Tetrahedron. 2002;58:3553–3559. [Google Scholar]
- 29.Fields JD, Kropp PJ. Surface-Mediated Reactions. 9. Selective Oxidation of Primary and Secondary Amines to Hydroxylamines. J Org Chem. 2000;65:5937–5941. doi: 10.1021/jo0002083. [DOI] [PubMed] [Google Scholar]
- 30.Biloski AJ, Ganem B. Improved Oxidation of Amines with Dibenzoyl Peroxide. Synthesis. 1983;7:537–538. [Google Scholar]
- 31.(a) Vidal J, Damestoy S, Guy L, Hannachi J, Aubry A, Collet A. N -Alkyloxycarbonyl-3-aryloxaziridines: Their Preparation, Structure, and Utilization as Electrophilic Amination Reagents. Chem Eur J. 1997;3:1691–1709. [Google Scholar]; (b) Guy L, Vidal J, Gollet A. Design and Synthesis of Hydrazinopeptides and Their Evaluation as Human Leukocyte Elastase Inhibitors. J Med Chem. 1998;41:4833–4843. doi: 10.1021/jm980419o. [DOI] [PubMed] [Google Scholar]
- 32.(a) Gómez-Vidal JA, Silverman RB. Short, Highly Efficient Syntheses of Protected 3-Azido- and 4-Azidoproline and Their Precursors. Org Lett. 2001;3:2481–2484. doi: 10.1021/ol0161054. [DOI] [PubMed] [Google Scholar]; (b) Gómez-Vidal JA, Forrester MT, Silverman RB. Mild and Selective Sodium Azide Mediated Cleavage of p-Nitrobenzoic Esters. Org Lett. 2001;3:2477–2479. doi: 10.1021/ol016104b. [DOI] [PubMed] [Google Scholar]
- 33.Seo J, Silverman RB. Synthesis of Arginine-containing Hydroxamate Dipeptidomimetics. Tetrahedron Lett. 2006;47:4069–4073. [Google Scholar]
- 34.Fedorov R, Vasan R, Ghosh DK, Schlichting I. Structure of Nitric Oxide Synthase Isoforms Complexed with the Inhibitor AR- R17477 Suggest a Rational Basis for Specificity and Inhibitor Design. Proc Natl Acad Sci US A. 2004;101:5892–5897. doi: 10.1073/pnas.0306588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Lasxlo R, Whittle BJR, Knowles RG. 1400W is a Slow, Right Binding, and Highly Selective Inhibitor of Inducible Nitric Oxide Synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 36.Moriarty NW, Karlstrom G. Geometry Optimization of a Water Molecule in Water. A Combined Quantum Chemical and Statistical Mechanical Treatment. J Chem Phys. 1997;106:6470–6474. [Google Scholar]
- 37.Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang CH, Weber PC, Jackson DA, Sharpe TR, Erickson-Viitanen S. Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors. Science. 1994;263:380–384. doi: 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- 38.Dunitz JD. The Entropic Cost of Bound Water in Crystals and Biomolecules. Science. 1994;264:p670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- 39.Goodsell DS, Morris GM, Olson AJ. Automated Docking of Flexible Ligands: Applications of Autodock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 40.Dupont V, Lecoq A, Mangeot J, Aubry A, Boussard G, Marraud M. Conformational Perturbations Induced by N-Amination and N-Hydroxylation of Peptides. J Am Chem Soc. 1993;115:8898–8906. [Google Scholar]
- 41.Jeffrey GA, Saenger W. Hydrogen Bonding in Biological Structures. Springer-Verlag: Berlin Heidelberg; 1991. p. 51. [Google Scholar]
- 42.Massiah MA, Viragh C, Reddy PM, Kovach IM, Johnson J, Rosenberry TL, Mildvan AS. Short, Strong Hydrogen Bonds at the Active Site of Human Acetylcholinesterase: Proton NMR Studies. Biochemistry. 2001;40:5682–5690. doi: 10.1021/bi010243j. [DOI] [PubMed] [Google Scholar]
- 43.Mikol V, Papageorgiou C, Borer X. The Role of Water Molecules in the Structure-Based Design of (5-Hydroxynorvaline)-2-cyclosporin: Synthesis, Biological Activity, and Crystallographic Analysis with Cyclophilin A. J Med Chem. 1995;38:3361–3367. doi: 10.1021/jm00017a020. [DOI] [PubMed] [Google Scholar]
- 44.Gangamani BP, Kumar VA, Ganesh KN. Synthesis of Nω-(purinyl/pyrimidinyl acetyl)-4-aminoproline Diastereomers with Potential Use in PNA Synthesis. Tetrahedron. 1996;52:15017–15030. [Google Scholar]
- 45.Gasteiger J, Marsili M. Iterative Partial Equalization of Orbital Electronegativity-A Rapid Access to Atomic Charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 46.Roman LJ, Sheta EA, Martasek P, Gross SS, Liu Q, Masters BSS. High-Level Expression of Functional Rat Neuronal Nitric Oxide Synthase in Escherichia coli. Proc Natl Acad Sci US A. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerber NC, Montelloano PR. Neuronal nitric oxide synthase. Expression in Escherichia coli, Irreversible Inhibition by Phenyldiazene, and Active Site Topology. J Biol Chem. 1995;270:17991–17996. doi: 10.1074/jbc.270.30.17791. [DOI] [PubMed] [Google Scholar]
- 48.Martasek P, Liu Q, Roman LJ, Gross SS, Sessa WC, Masters BSS. Characterization of Bovine Endothelial Nitric Oxide Synthase Expressed in Escherichia coli. Biochem Biophys Res Commun. 1996;219:359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
- 49.Hevel JM, Marletta M. Nitric Oxide Synthase Assays. Methods Enzymol. 1994;133:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 50.(a) Cheng YC, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]; (b) Segal IH. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady state enzyme systems. John Wiley & Sons; New York: 1975. p. 106. [Google Scholar]; (c) Burlingham BT, Widlanski TS. An Intuitive Look at the Relationship of Ki and IC50: A More General Use for the Dixon Plot. J Chem Educ. 2003;80:214–218. [Google Scholar]
- 51.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 52.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 53.Jones TA, Zou JY, Cowan SW, Kjeldgaarrd M. Improved Methods for Building Models in Electron Density and the Location of Errors in These Models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of data collection, structure refinement statistics of the three new structures; elemental analysis data of compounds 3–5; recently reported amide reduction method. This material is available free of charge via the Internet at http://pubs.acs.org.