

Abstract
We sought to create a comprehensive catalog of yeast genes whose transcript levels vary periodically within the cell cycle. To this end, we used DNA microarrays and samples from yeast cultures synchronized by three independent methods: α factor arrest, elutriation, and arrest of a cdc15 temperature-sensitive mutant. Using periodicity and correlation algorithms, we identified 800 genes that meet an objective minimum criterion for cell cycle regulation. In separate experiments, designed to examine the effects of inducing either the G1 cyclin Cln3p or the B-type cyclin Clb2p, we found that the mRNA levels of more than half of these 800 genes respond to one or both of these cyclins. Furthermore, we analyzed our set of cell cycle–regulated genes for known and new promoter elements and show that several known elements (or variations thereof) contain information predictive of cell cycle regulation. A full description and complete data sets are available at http://cellcycle-www.stanford.edu
INTRODUCTION
In 1981 Hereford and coworkers discovered that yeast histone mRNAs oscillate in abundance during the cell division cycle (Hereford et al., 1981). To date 104 messages that are cell cycle regulated have been identified using traditional methods, and it was estimated that some 250 cell cycle–regulated genes might exist (Price et al., 1991). There are several reasons why genes might be regulated in a periodic manner coincident with the cell cycle. Such regulation might be required for the proper functioning of mechanisms that maintain order during cell division. Alternatively, regulation of these genes could simply allow conservation of resources. Much of the literature has focused on the posttranscriptional mechanisms that control the basic timing of the cell cycle. However, there is also clear evidence that trans-acting factors play a critical role in the regulation of the abundance of many cell cycle–regulated transcripts.
Most identified cell cycle controls that exert influence over mRNA levels do so at the level of transcription. Three major types of cell cycle transcription factors are known in yeast, the MBF and SBF factors, Mcm1p-containing factors, and Swi5p/Ace2p (Table 1). Many genes expressed at about the G1/S transition contain MCB or SCB elements in their promoters to which MBF and SBF bind respectively (for review, see Koch and Nasmyth, 1994). It is now apparent that SBF is not as specific for SCBs as was originally thought but, rather, can bind, at least in some cases, to motifs more closely matching the MCB consensus (Partridge et al., 1997). MBF and SBF are activated posttranslationally by Cln3p-Cdc28p, and SBF, at least, is inactivated by Clb2p-Cdc28p (Amon et al., 1993). It is this cyclin-dependent activation and inactivation that causes MBF- and SBF-mediated transcription to be cell cycle regulated. Mcm1p can bind with other DNA binding proteins to mediate a specific biological effect. In cooperation with Ste12p, Mcm1p directs the cell cycle expression of some genes in early G1 phase (Oehlen et al., 1996). In cooperation with an uncloned factor called “Swi five factor” (SFF), it induces the expression of CLB1, CLB2, BUD4, and SWI5 in M (Lydall et al., 1991; Sanders and Herskowitz, 1996). Finally, possibly acting without a partner, it induces transcription of CLN3, SWI4, and CDC6 at the M/G1 boundary (McInerny et al., 1997). The Mcm1p + SFF combination is interesting, because it is somehow activated by Clb2p-Cdc28p, and Mcm1p + SFF then induces further transcription of CLB2. Thus, Mcm1p is part of a positive feedback loop for CLB2 transcription. Finally, Swi5p and Ace2p, which are transcriptionally controlled by Mcm1p and SFF, are responsible for the expression of many genes in M and M/G1 (Kovacech et al., 1996). Some of these genes are responsible for inactivating Clb2p and promoting cytokinesis, thus allowing exit from mitosis, and allowing the cycle to begin anew.
Table 1.
Transcription factors that regulate the cell cycle
| Complex | Composition | Site name | Site | Reference |
|---|---|---|---|---|
| SBF | Swi6p + Swi4p | SCB | CACGAAA | Nasmyth, 1985; Andrews and Herskowitz, 1989 |
| MBF | Swi6p + Mbp1p | MCB | ACGCGT | Lowndes et al., 1991; McIntosh et al., 1991; Koch et al., 1993 |
| Mcm1p | Mcm1p | MCM1 | TTACCNAATTNGGTAA | Acton et al., 1997 |
| SFF | SFF | SFF | GTMAACAA | Althoefer et al., 1995 |
| Ace2p | Ace2p | SWI5 | ACCAGC | Dohrmann et al., 1996 |
| Swi5p | Swi5p | SWI5 | ACCAGC | Knapp et al., 1996 |
Many cell cycle–regulated genes are involved in processes that occur only once per cell cycle. Such processes include DNA synthesis, budding, and cytokinesis. Additionally many of these genes are involved in controlling the cell cycle itself, although in most cases it is unclear whether their regulated transcription is absolutely required. The cell division cycle is thus a complex self-regulating program, such that many genes involved in aspects of the cell cycle are also controlled by it.
We present the results of a comprehensive series of experiments aimed at objectively identifying all protein-encoding transcripts in the genome of Saccharomyces cerevisiae that are cell cycle regulated. We used DNA microarrays to analyze mRNA levels in cell cultures that had been synchronized by three independent methods. These data were analyzed by deriving a numerical score based on a Fourier algorithm (testing periodicity) and by a correlation function that identified genes whose RNA levels were similar to the RNA levels of genes already known to be regulated by the cell cycle. This protocol allowed us to include data from a previously published study (Cho et al., 1998). We find that ∼800 genes are cell cycle regulated, which constitutes >10% of all protein-coding genes in the genome. We also find that about one-half of these genes can be controlled by the G1 cyclin CLN3 and/or the mitotic cyclin CLB2. The primary data presented in this article, tools for examining them, and supporting analyses can be found at http://cellcycle-www.stanford.edu.
MATERIALS AND METHODS
Strains
Strains used in this study are shown in Table 2.
Table 2.
Strains used in this study
| Name | Genotype | Source |
|---|---|---|
| DBY7286 | MATa GAL2 ura3 | This study |
| DBY8724 | MATa GAL2 ura3 bar1::URA3 | This study |
| W303a | MATa ade2-1 trp1-1 leu2-3,112 his3-11, 15 ura3 can1-100 [psi+] | Thomas et al., 1989 |
| #31 | MATa leu2 ura3 cln1::HIS3 cln2::TRP1 cdc34-2ts ura3-GAL-CLN3 | Schneider et al., 1996 |
| #245 | W303a clb1::URA3 clb2::LEU2 clb3::TRP1 clb4::HIS3 GAL-CLB2 | Fitch et al., 1992 |
| cdc15-2 | W303α cdc15-2ts | Fitch et al., 1992 |
Media and Growth Conditions
YEP medium (Sherman, 1991) was used in all experiments, supplemented with an appropriate carbon source. Carbon sources are indicated in the descriptions of each experiment and were used at a final concentration of 2% (wt/vol), unless otherwise noted. The pH of the YEP for the α factor experiment was adjusted to 5.5 before use. The medium used for the elutriation was first passed through Whatman #1 filter paper. Cultures of yeast were shaken at 250–300 rpm, in a volume no more than 25% of the vessel maximum at the temperature specified in the description of each experiment.
Microarray Manufacture
Yeast ORFs were amplified using gene PAIRS primers (Research Genetics, Huntsville, AL). One hundred-microliter PCR reactions were performed in 96-well PCR plates using each primer pair with the following reagents: 1 μM each primer, 200 μM each dATP, dCTP, dTTP, and dGTP, 1× PCR buffer (Perkin Elmer-Cetus, Norwalk, CT), 2 mM MgCl2, and 2 U of Taq DNA polymerase (Perkin Elmer-Cetus). Thermalcycling was performed in Perkin Elmer-Cetus 9600 thermalcyclers with a 5-min denaturation step at 94°C, followed by 30 cycles with melting, annealing, and extension temperatures and times of 94°C, 30 s; 54°C, 45 s; and 72°C, 3 min 30 s, respectively. Production of the correct PCR product was verified by gel electrophoresis. Products deemed to have failed were reamplified either by repeating the PCR reaction with the gene PAIRS primers, ordering custom primers, or using the yeast ORF DNA (Research Genetics) as a template. Reamplification of failed PCRs used the same protocol as initial amplification.
DNAs were prepared and printed onto microarrays as described previously (Shalon et al., 1996; DeRisi et al., 1997 [http://cmgm.stanford.edu/pbrown/]; Eisen and Brown, 1999) with 190-μm spacing between the centers of each element. Each microarray was visually inspected, and all microarrays used in this study were estimated to be missing <1% of all elements except for arrays used in the cdc15 experiments, which were missing ∼3% of all elements.
Cell Density and Size Measurements
All cell size measurements were made using a Coulter Counter Multisizer (Coulter Electronics, Hialeah, FL) or a Beckman FACScan workstation (Beckman Instruments, Fullerton, CA). The Coulter Counter was also used to measure the cell density for elutriation. Cell densities in the α factor experiment were measured at OD600 using a Pharmacia Ultrospec III spectrophotometer (Pharmacia, Piscataway, NJ).
Budding Index Calculations
Each sample was sonicated for 30 s with a Virsonic 300 (Virtis, Gardiner, NY) microprobe equipped sonicator at 50% power to separate divided cells. At least 200 cells were counted and scored for the presence of a bud.
DNA Content Determination
Samples were prepared as described previously (Futcher, 1993), and DNA content was measured using a Beckman FACScan workstation.
Nuclear Staining
Cells were washed in water and resuspended in water containing DAPI at 1 μg/ml. Cells were then placed on a glass slide and visualized by fluorescence microscopy, using a Zeiss Axioplan microscope (Carl Zeiss, Thornwood, NY).
α Factor–based Synchronization
Yeast strain DBY8724 was grown to an OD600 of 0.2 in YEP glucose, an asynchronous sample was taken, and α factor (PAN facility, Beckman Center, Stanford University) was added to a concentration of 12 ng/ml. After 120 min the α factor was removed by pelleting the cells for 5 min in a Sorvall (Newtown, CT) S34 rotor at 3000 rpm and decanting the supernatant. The arrested cells were resuspended in fresh YEP glucose to an OD600 of 0.18. Every 7 min, for the next 140 min, 25-ml samples were taken for RNA, and 5-ml samples were taken for FACS analysis. At 91 min after release the OD600 of the culture was reduced to ∼0.2 from ∼0.4 by addition of fresh medium.
Size-based Synchronization
Nine liters of yeast strain DBY7286 were grown in YEP ethanol (2%, vol/vol) at 25°C to a cell density of 1.5 × 107 cells/ml. Cells were pelleted in a Beckman JA-10 rotor for 10 min. The supernatant was saved and is referred to as clarified medium. Cells were resuspended in 300 ml of the clarified medium and sonicated for 2 min with a Virsonic 300 equipped with a microprobe at 50% power. This volume was loaded into a dual-chamber elutriation chamber (Beckman Instruments, Fullerton, CA; catalog numbers 356940 and 356941) in a Beckman J-6 M/E centrifuge equipped with a JE-5.0 elutriation rotor. The elutriator was run with clarified medium at 25°C. Unbudded daughter cells (400 ml at 2.3 × 107 cells/ml) were collected at a modal cell volume of 17.7 fl and grown at 25°C. Samples were take every 30 min for the next 6.5 h with independent samples for DAPI staining (1 ml), FACS analysis (2 ml), budding index (1 ml), and RNA preparation (25 ml). After harvesting, the samples for DAPI, FACS, and budding index were immediately chilled on ice.
Cdc15-based Synchronization
The cdc15-2 (DBY8728) strain was grown to 2.5 × 106 cells/ml in YEP glucose medium at 23°C. The culture was then shifted to an air incubator at 37°C and held at that temperature for 3.5 h. By this time, cell density had reached 6.6 × 106 cells/ml, and 96% of the cells were large dumbbells characteristic of a cdc15 arrest. The cells were then released from the cdc15 arrest by shifting the culture to a 23°C water bath. Samples were taken every 10 min for 300 min, starting at the time of the shift to the 23°C water bath. By 300 min after shift, cell density had reached 4 × 107 cells/ml. Part of the same original culture was grown at 23°C to 1 × 107 cells/ml, and cells were harvested for extraction of the control mRNA. Progress through the cell cycle was monitored by the appearance of new buds.
Because cdc15-2 cells do not quantitatively complete cell separation after a release from a 37°C arrest, FACS analysis is difficult to interpret. We therefore followed the progress of the cdc15-2 cells through the cycle by monitoring the appearance of new buds. The first new buds appeared 50 min after the release to 23°C, when 12% of the dumbbells had small buds (usually, two small buds, one on each half of the dumbbell). The percentage of dumbbells with small buds was 52% at 60 min, 76% at 70 min, 96% at 80 min, and virtually 100% at 90 min, at which time almost all the dumbbells had not just one bud, but two, one on each half of the dumbbell. The second round of small buds appeared at 150 min, when 3% of the cells had small buds. The percentage was 9.7% at 160 min, 32% at 170 min, 68% at 180 min, and 81% at 190 min. The third round of small buds appeared at 270 min, although by this time synchrony was decaying.
Cln3 and Clb2 Experiments
For the Cln3 experiment strain 31 (DBY8725) was grown in YEP raffinose/galactose (1% each) at 23°C to a density of 1 × 107 cells/ml. The cells were then filtered and washed with 2 vol of YEP and resuspended in YEP raffinose at 23°C. These cells arrested because of lack of Cln activity after incubating for 3 h. Cdc34p was then inactivated by shifting the culture to 37°C for 2.5 h. The culture was then split, and galactose was added to one-half at a final concentration of 2% (wt/vol). Cells from this culture were harvested every 10 min for 40 min for RNA. The entire control culture was harvested at time 0. The experiment was performed twice (once for each hybridization in our data set). Data from the 40-min (first experiment) and 30-min (second experiment) postgalactose samples are shown.
For the Clb2 experiment strain 245 (DBY8726) was grown to a density of 5 × 106 at 30°C in YEP raffinose/galactose (1% each) and then centrifuged, and the cells were washed with 2 vol of YEP and then resuspended in YEP raffinose. After 6 h DMSO was added to a final concentration of 1%, and nocodazole was added to a final concentration of 15 μg/ml. The culture was then split, and galactose was added to one-half at a final concentration of 2% (wt/vol). Cells from this culture were harvested every 10 min for 40 min for RNA. The entire control culture was harvested at time 0. The experiment was performed twice (once for each hybridization in our data set). Data from the two 40 min postgalactose samples are shown.
To control the Cln3 and Clb2 experiments for the effects of galactose addition, strain W303a (DBY8727) was grown in 250 ml of YEP raffinose at 30°C to a cell density of 1 × 107 cells/ml. The culture was split in two, and to one-half (the experimental culture) was added galactose to a final concentration of 2% (wt/vol). Forty minutes later both cultures were harvested for preparation of mRNA. The data for this experiment are available on our web site.
RNA Preparation
Samples for RNA isolation were taken by pipetting culture directly into 50-ml Falcon (Lincoln Park, NJ) tubes containing ∼20 g of ice to quickly chill the cells. Cells were collected by spinning for 3 min in a tabletop centrifuge and then frozen by immersion in liquid nitrogen and stored at −80°C until RNA was prepared. RNA was prepared by adding 10 ml of water-saturated phenol, 10 ml of sodium acetate buffer (50 mM sodium acetate, 10 mM EDTA, pH 5.0), and 1 ml of 10% SDS (all prewarmed to 65°C) to each frozen cell pellet. Each mixture was incubated at 65°C for 10 min, vortexing vigorously every 1 min for 10 s. After spinning at 1500 × g for 10 min, the aqueous phase was transferred to another 50-ml conical tube containing 10 ml of water-saturated phenol. Samples were vortexed for 30 s, and the spin was repeated. Aqueous phases were again transferred to a new 50-ml conical tube and 10 ml of phenol:chloroform (1:1) were added, followed by a 15-min spin. RNA was precipitated by adding the aqueous phase to an equal volume of isopropanol and 0.1 vol of 3 M sodium acetate. Samples were spun for 30 min at 1500 × g to pellet the RNA. Pellets were washed with 70% ethanol and dried at room temperature. RNA pellets were dissolved in TE (10 mM Tris, 1 mM EDTA, pH 8.0) to ∼2.5 mg/ml.
Probe Preparation
Total RNA (15 μg) and 6 μg of oligo-dT were combined in a total volume of 15 μl. RNA oligo-dT mixtures were heated to 70°C for 1 min and then cooled on ice. Three microliters of 25 mM Cy3- or Cy5-conjugated dUTP (Amersham, Arlington Heights, IL), 3 μl of 1 M DTT, 6 μl of first-strand buffer (Stratagene, La Jolla, CA), 0.6 μl of dNTPs (25 mM each dATP, dCTP, and dGTP and 15 mM dTTP), and 2 μl of Superscript II (Stratagene) were added. Each sample was then incubated at 42°C for 2 h to generate Cy-labeled cDNA. Starting RNA was degraded by addition of 1.5 μl of stop solution (1 N NaOH, 0.1 M EDTA) and incubation at 70°C for 10 min. Samples were neutralized by addition of 15 μl of 0.1 N HCl and 400 μl of TE (10 mM Tris, 1 mM EDTA, pH 7.4). Labeled cDNA was concentrated and separated from unbound fluor by separation in a Centricon-30 (Amicon, Danvers, MA) until no further fluor was visible in the flow through, and the probe was concentrated to <4 μl.
Microarray Hybridizations
A probe mixture (12 μl) consisting of Cy3- and Cy5-labeled cDNAs, 3× SSC, 0.3% SDS, and 1.8 μg/μl yeast tRNA was applied to each microarray. The microarray was covered by a 22-mm-square coverslip (Fisher Scientific, Pittsburgh, PA) and placed in a custom-manu-factured hybridization chamber (see http://cmgm.stanford.edu/pbrown/). Ten microliters of water were placed inside the hybridization chamber before sealing, and the chamber was placed in a 65°C water bath. The microarrays were allowed to hybridize 4–6 h. Microarrays were removed from the chambers and placed in standard histochemistry slide holders where they were washed by plunging 30 times in each of the following solutions, respectively: 2× SSC, 0.2% SDS; 0.4× SSC; and 0.2× SSC.
Data Acquisition and Processing
Microarrays were scanned using a custom-built scanning laser microscope. Separate 2 × 2-cm images were acquired for each fluor at a resolution of 20 μm/pixel. Data were extracted by manually superimposing a grid of boxes over the combined Cy3–Cy5 image so that each box contained a single array spot. The average fluorescence intensity for each fluor within each box was recorded. The local background was estimated by averaging the intensities of the weakest 12% of the pixels in each box. Fluorescence ratios were computed based on the background corrected values. Spots of poor quality (as assayed by visual inspection) were removed from further consideration. As a measure of the internal consistency of the data for each spot, the pixel-by-pixel correlation coefficient between the Cy3 and Cy5 intensities was computed; spots with low correlation values (i.e., <0.4) were excluded from further analysis.
Identification of mRNAs Regulated in a Cell Cycle–dependent Manner
Data for each gene in the α factor time series were extracted from the database and were normalized so that the average log2(ratio) over the course of the experiments was equal to 0. A Fourier transform (Eq. 1–3) was applied to the data series for each gene, and the resulting vector (C) was stored for each gene, where ω is the period of the cell cycle, t is the time, Φ is the phase offset, and ratio(t) is the ratio measurement at time t. We found that the magnitude of the Fourier transform (D, Eq. 4) was unstable for small variations of ω, so we averaged the vectors of the transform over a range of 40 values, which were evenly spaced around the estimated division t ime for the experiment (66 ± 11). We initially set the value of Φ to 0.
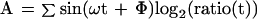 |
1 |
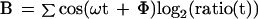 |
2 |
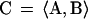 |
3 |
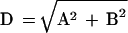 |
4 |
The expression profile of each gene across the experiments was then correlated to five different profiles representing genes known to be expressed in G1, S, G2, M, and M/G1 using a standard Pearson correlation function. The profiles for known gene classes were identified by averaging the log2(ratio) data for each of the genes known to peak in each of the five time periods. The peak correlation score was defined as the highest correlation value between the data series for each gene and each of the profiles. The vector calculated by the Fourier transform was scaled by the peak correlation value.
The above process was repeated for the cdc15 experiment (ω varying between 60 and 80) and for the cdc28 data (ω varying between 80 and 100) from Cho et al. (1998). The cdc28 data set was first converted to ratio style measurements by dividing each measurement by the average value of the measurements for that gene. Before this step it was necessary to exclude some data points that appeared to be aberrant. Any data value where the two values on either side were threefold different in the same direction were excluded. Each gene thus had three vector scores (one for each of the three analyzed data series).
To generate a single vector for each gene, we added the vectors for each experiment together. However, the value of Φ for the three experiments should not be the same, because the experiments start at different points in the cell cycle. Therefore, before combining the vectors from the three experiments, constants, Φcdc15 and Φcdc28 (relative to the α factor experiment), were calculated for the cdc15 and cdc28 experiments, respectively, that maximized, for the known genes, the average magnitude of the summed vectors. The elutriation data were not included, because it was not possible to calculate a Φ that maximized the values of more than a handful of the known genes. The α factor and cdc15 vectors were multiplied by 0.7, so that they would not unduly contribute to the final “aggregate CDC score,” which was calculated by taking the magnitude of this final vector.
Genes were ranked by their aggregate CDC scores, and the list was examined to identify the positions of known cell cycle genes within it. We selected a threshold CDC score that was exceeded by 91% of known cell cycle–regulated genes. Altogether 800 genes met or exceeded this CDC score.
Promoter Analysis
Motifs were identified in the 700 bp upstream of the start codon using a Gibbs sampling strategy. Such a strategy was originally developed by Lawrence et al. (1993) to find patterns in protein sequences and later modified by Neuwald et al. (1995) to take into account the possibility of a repeated motif. We have modified these Gibbs sampling algorithms to allow pattern searches of DNA (Zhang, unpublished data), for which functions such as double-strand search, palindrome symmetry, and submotif inclusion and exclusion were included. Once motifs were established for a group or cluster, we tested their predictive value by searching for the motif consensus (with specified mismatches) in the 700 bp upstream of the ATG for all groups, as well as for a control set of non-cell cycle–regulated genes, and compared the distribution of these sites in different groups.
TAQman Assay
The TAQman assay was performed on the same α factor samples that were used in the microarray hybridization experiments. For each sample, 500 ng of total RNA were incubated for 15 min with 0.1 U/μl DNase I (amplification grade; Life Technologies, Grand Island, NY) in 2 mM MgCl2, 50 mM KCl, 20 mM Tris-HCl (pH 8.4). The reaction was stopped by adding EDTA to 2.5 mM and incubating at 65°C for 10 min. The RNA was reverse transcribed using TAQman reverse transcription reagents (PE Applied Biosystems, Foster City, CA) consisting of 2.5 μM oligo-dT 16 mer, 1.25 U/μl MultiScribe reverse transcriptase, 0.5 mM dGTP, dATP, dTTP, and dCTP, 0.4 U/μl RNase inhibitor, 50 mM KCl, 10 mM Tris-HCl (pH 8.3). The reaction was incubated at 25°C for 10 min, 48°C for 30 min, and then 95°C for 5 min. The resulting cDNA served as a template for real-time quantitative PCR as follows, in which a fluorescent reporter dye (6-carboxy-fluorescein [FAM]) was released and quantitated during each specific replication of the template (Heid et al., 1996). The cDNA was mixed with 2× TAQman universal PCR master mix (PE Applied Biosystems) and then split into separate reaction tubes containing gene-specific forward and reverse primers (900 nM each) and dye-labeled oligonucleotide probes (200 nM). Each resultant PCR (25 μl) contained cDNA generated from 5 ng of RNA. The sequences of the primers and probes were the following: TUB1 primers: forward, 5′-AAAGCCGAAGGGAGGAGAAG-3′; reverse, 5′-CCCTTGGAACGAACTTACCGT-3′; TUB1 probe: 5′(FAM)-CTCCACGTTTTTCCATGAAACCGGC-(6-carboxy-tetramethylrhodamine [TAMRA])p3′; TUB2 primers: forward, 5′-TTGTCCCATTCCCACGTTTAC-3′; reverse, 5′-GATTGAGAGCCAATTGCCGT-3′; TUB2 probe: 5′(FAM)-TTCTTCATGGTCGGCTACGCTCCATT-(TAMRA)p3′; TUB3 primers: forward, 5′-CCTGCGCCTCAATTGTCTACT-3′; reverse, 5′-TTCCAGGGTGGTATGCGTG-3′; TUB3 probe: 5′(FAM)-CGTCGTGGAACCTTACAACACGGTTTTAA-(TAMRA)p3′; PPA1 primers: forward, 5′-TGTCGGTG-CTTCCAATTTGAT-3′; reverse, 5′-CATCGGAAATGGCAGCAGT-3′; and PPA1 probe: 5′(FAM)-CCGGTGATACCGACAGCGATACCA-(TAMRA)p3′. Each gene-specific PCR was done in triplicate or quadruplicate. The tubes were placed in a PE Applied Biosystems Prism 7700 sequence detection system and were incubated with the following parameters: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The computer program Sequence Detector version 1.6.3 (PE Applied Biosystems) provided output, which allowed the average quantities of TUB mRNA relative to PPA1 mRNA to be determined for each RNA sample. The TUB:PPA1 ratio in the asynchronous sample A1 was arbitrarily set at 1, and the results from the other samples were adjusted accordingly.
RESULTS
Experimental Overview
We wished to identify the genes whose RNA levels varied periodically during the cell cycle. We initially obtained microarray data from synchronized cells and suitable controls and analyzed the >400,000 measurements to obtain objective scores based on a Fourier algorithm (which assesses periodicity) and a correlation measurement (which compared our data with those of previously identified cell cycle–regulated genes). We compared scores among the previously known and total gene sets to find a threshold value for deciding the significance of the apparent cell cycle regulation. For completeness, we also reanalyzed the published data of Cho et al. (1998). Using all the data, we arrived at a threshold CDC value above which 91% (95 of 104) of the genes previously shown to be cell cycle regulated are included. This procedure identified a total of 800 yeast genes as being periodically regulated.
Synchronized Cultures
We measured the relative levels of mRNA as a function of time in cell cultures that had been synchronized in three independent ways. First we used α pheromone to arrest MATa cells in G1. Second, we used centrifugal elutriation to obtain small G1 cells. Finally, we used a temperature-sensitive mutation, cdc15-2, which, at the restrictive temperature, arrests cells late in mitosis. We used three methods because each introduces characteristic artifacts. For instance, use of pheromone has regulatory consequences characteristic of mating, whereas use of temperature-sensitive mutants can cause heat shock.
The synchronization experiments differed in major ways. First, they were performed using different carbon sources and at different temperatures, with the consequence that the cells grew at different rates. Second, two different yeast strain backgrounds were used (S288C and W303), and finally, cells were synchronized at different points during the cell cycle. Each method produced significant cell cycle synchrony through one cell cycle (elutriation), two cycles (α pheromone), or three cycles (cdc15), as established by at least one of the following methods for each experiment: bud count, DNA content analysis (FACS) and nuclear staining (DAPI), as described in MATERIALS AND METHODS.
RNA was extracted from each of the samples collected, as well as from a control sample (asynchronous cultures of the same cells growing exponentially at the same temperature in the same medium). Fluorescently labeled cDNA was synthesized using Cy3 (“green”) for all controls and Cy5 (“red”) for all experimental samples. Mixtures of labeled control and experimental cDNAs were competitively hybridized to individual microarrays containing essentially all yeast genes (DeRisi et al., 1997). The ratio of experimental (red) to control (green) cDNA was measured by scanning laser microscopy (Shalon et al., 1996).
Transcription in Response to the Cyclins Cln3p and Clb2p
To gain mechanistic insight into the control of the observed cell cycle regulation, we identified genes whose mRNA levels responded to the induction of two well-characterized cell cycle regulators, Cln3p and Clb2p (see Nasmyth, 1993). Late in G1 phase, the Cln3p-Cdc28p protein kinase complex activates two transcription factors, MBF and SBF, and these in turn promote the transcription of a number of genes important for budding and DNA synthesis (Cross, 1995). Later in the cell cycle, the Clb2p-Cdc28p complex represses the activity of SBF, returning the expression of SBF-regulated genes to low levels (Amon et al., 1993). Furthermore, Clb2p-Cdc28p is known to activate expression of at least four genes, CLB1, CLB2, SWI5, and BUD4 (Althoefer et al., 1995; Sanders and Herskowitz, 1996).
To identify other genes controlled by Cln3p and Clb2p, we arrested cln− or clb− cells in late G1 with cdc34-2 for the CLN3 experiment and in M with nocodazole for the CLB2 experiment. We then induced expression of CLN3 or CLB2 without inducing cell cycle progression. RNA from the G1-phase cells expressing Cln3p (labeled red) was compared with control RNA (labeled green) from the G1-phase cells arrested in the absence of Cln3p. Similarly, for the CLB2 experiment, RNA from M-phase cells expressing Clb2p (labeled red) was compared with control RNA (labeled green) from M-phase cells arrested in the absence of Clb2p. In each case, mRNA levels were quantitatively measured by microarray hybridization. In addition, we performed an experiment to test the effects of galactose to an asynchronous culture with no inducible cyclin (see MATERIALS AND METHODS). Genes identified as strongly affected by galactose addition were not considered further in the Gal cyclin experiments.
Data Analysis and Availability
The total data we collected comprise ∼400,000 individual ratio measurements. The quality and reliability of the data can only be assessed by unrestricted access to all data in forms suitable for further query or computer analysis. Therefore, in addition to the summary printed here, we provide primary data from two locations on the Internet. The numerical data are provided in a table of the actual ratios measured for each gene, on each array. They can be downloaded as a tab-delimited text file from the journal web site (http://www.molbiolcell.org) or from a server at Stanford (http://cellcycle-www.stanford.edu). The Stanford web site also provides images of the arrays, accessory data, and the capability to browse and search the complete data set. Raw data are also available from the authors upon request.
The comprehensive nature of this work has another consequence: in what follows we refer by name to as many as 400 genes. It is impractical to provide detailed literature documentation for each gene every time it appears. Instead, we have provided references selectively, and we encourage readers to use the hyperlinks to the Saccharomyces Genome Database (http://genome-www.stanford.edu/Saccharomyces) and the Yeast Protein Database (http://quest7.proteome.com/YPDhome.html) that will be provided at both the Molecular Biology of the Cell and Stanford web sites.
Identification of Cell Cycle–regulated Transcripts
Combining the data from the synchronization experiments, we were able to identify 800 genes whose expression is cell cycle regulated. We did this by the combination of a Fourier algorithm and a correlation algorithm as described in MATERIALS AND METHODS. This resulted in a score for each gene that we refer to as the aggregate CDC score. To illustrate this, Table 3 provides some summary statistics and examples of the kinds of scores obtained for several genes (including specific examples that are and are not cell cycle regulated).
Table 3.
Example scores and statistics for a collection of genes
| Rank by score | Score gene name | Peak to trough |
|---|---|---|
| 1 | 15.99 PIR1 | 27.3 |
| 9 | 10.90 CLN2 | 12.1 |
| 37 | 8.78 CLB1 | 9.4 |
| 82 | 6.51 BUD9 | 7.0 |
| 177 | 4.25 STE3 | 12.8 |
| 224 | 3.55 TUB4 | 4.8 |
| 255 | 3.29 DUN1 | 4.2 |
| 401 | 2.37 CIN8 | 5.4 |
| 407 | 2.33 TUB2 | 5.5 |
| 585 | 1.71 MET1 | 3.0 |
| 800 | 1.314 STP4 | 5.9 |
| 800 | 1.314 Threshold for inclusion | |
| 844 | 1.28 SEC8 | 4.2 |
| 861 | 1.25 TUB1 | 2.7 |
| 1258 | 0.92 ANP1 | 3.1 |
| 1799 | 0.71 TUP1 | 3.0 |
| 2499 | 0.54 TUB3 | 2.7 |
| 2673 | 0.50 IME2 | 3.5 |
| 6054 | 0.05 RPS8B | 10.9 |
In setting the threshold for the aggregate CDC score by our empirical method, we intended to minimize false-positive assessments while including the vast majority of previously characterized genes that are known to have periodic mRNA levels. Many additional genes showed indications of cell cycle regulation (by visual inspection of the data and by quantitation using our algorithm), although we could not objectively distinguish this behavior from noise.
We estimated the false-positive rate in two ways. First, we randomized the data from each experiment (both by gene and by time point) and performed all of the analyses described above. The randomized data produced 24 “genes” (of nearly 6200) with CDC scores that exceed the threshold we used to classify genes as cell cycle regulated. We assume that this represents a reasonable estimate of the false-positive rate (i.e., ∼3% of all genes identified would be false positives). In a second, more conservative test, we randomized the data set only within genes. The number of genes that had scores above our threshold was about three times higher (75 genes) when we randomly shuffled the data in this way. Thus, the number of false positives (of the 800 genes identified as cell cycle regulated) is likely <10% and perhaps as low as 3%.
Classifying the Cell Cycle–regulated Genes by Pattern of Expression
We used two distinct methods to classify genes by their pattern of expression, which we refer to as “phasing” (by time of peak expression) and “clustering” (by similarity of expression across the experiments, which is described below). There is no simple relationship between these two methods, although there are common features in the results. “Phase groups” were created by determining the time of peak expression for each gene (calculated from the Fourier algorithm) and ordering all genes by this time. We divided this ordered set into five (somewhat arbitrary) groups termed G1, S, G2, M, and M/G1 that approximate those commonly used in the literature. To this end we used the published timing of gene expression for the known genes in determining which genes belonged in which phase group. Figure 1A displays the 800 genes that we identified, sorted according to the phase of expression. Each column represents a time point in an experiment, and each row represents a gene that we identified as cell cycle regulated. The ratio of expression that we measured for each gene in each time point is color coded, reflecting the magnitude of the ratio of expression relative to the average of that gene, with shades of red indicating an increase (on) and shades of green indicating a decrease (off). This display is based on the paradigm of Eisen et al. (1998). Genes expressed during each part of the cell cycle are indicated by the color bar (and phase) on the side, and temporal progress through the cell cycle is indicated on the top.
Figure 1.


Gene expression during the yeast cell cycle. Genes correspond to the rows, and the time points of each experiment are the columns. The ratio of induction/repression is shown for each gene such that the magnitude is indicated by the intensity of the colors displayed. If the color is black, then the ratio of control to experimental cDNA is equal to 1, whereas the brightest colors (red and green) represent a ratio of 2.8:1. Ratios >2.8 are displayed as the brightest color. In all cases red indicates an increase in mRNA abundance, whereas green indicates a decrease in abundance. Gray areas (when visible) indicate absent data or data of low quality. Color bars on the right indicate the phase group to which a gene belongs (M/G1, yellow; G1, green; S, purple; G2, red; M, orange). These same colors indicate cell cycle phase along the top. (A) Gene expression patterns for cell cycle–regulated genes. The 800 genes are ordered by the times at which they reach peak expression. (B) Genes that share similar expression profiles are grouped by a clustering algorithm as described in the text. The dendrogram on the left shows the structure of the cluster.
By phasing there were 300 G1 genes (e.g., CLN2, RNR1, CDC9, RAD27, SMC3, and MNN1), 71 S genes (e.g., the histones), 121 G2 genes (e.g., CLB4, WHI3, and CIS3), 195 M genes (e.g., DBF2, CLB2, CDC5, CDC20, and SWI5), and 113 M/G1 genes (e.g., ASH1, SIC1, CDC6, and EGT2). This is a crude classification with many disadvantages (e.g., the last gene in the G2 group and the first gene in the M group are expressed at virtually the same time yet are in different groups), but nevertheless it is useful for discussing the results.
Identification of DNA Binding Sites
We searched through the 700 bp immediately upstream of the start codon of each of the 800 genes in our list to identify potential binding sites for known or novel factors that might control expression during the cell cycle. We found that the majority of the genes have good matches to known cell cycle transcription factor binding sites relevant to the time of peak expression. Furthermore, we examined the distribution of these elements within the upstream sequences, and found that both the site and its position relative to the ATG contain information that is predictive of the phase group of the gene. Figure 2 shows the frequency of six sites in promoters of the G1, S, G2, M, and M/G1 phase groups and a control set of non-cell cycle–regulated genes. These sites are the previously published SCB and MCB as well as four extensions and modifications of published sites (MCM1 + SFF, extended SWI5, SCB variant, and degenerate MCB). Full results of all promoter searches are available on our web site.
Figure 2.

Binding site frequencies. The distribution of various promoter elements in the upstream regions of each the five cell cycle–regulated groups and a control group of 279 non-cell cycle–regulated genes that do not respond to either Cln3p or Clb2p induction are graphically displayed. In each case the numbers on the x axes represent distance from the start codon, and the bars represent the frequency of a particular site per residue per gene at that position in the upstream promoter regions. N is any base; Y is C or T; W is A or T; R is A or G; and M is A or C. (A) SCB. (B) A variant on the SCB, which is also similar to an MCB. (C) MCB. (D) MCB_d, a degenerate MCB sequence. (E) MCM1/SFF site. (F) Swi5e, an extended Swi5 site.
Clusters and Their Regulation
Clusters were established using the clustering algorithm of Eisen et al. (1999). This algorithm sorts through all the data to find the pairs of genes that behave most similarly in each experiment and then progressively adds other genes to the initial pairs to form clusters of apparently coregulated genes. As will be discussed below, the clustering algorithm successfully identifies coregulated genes, because analysis of the 5′ regions of the genes in a cluster shows that such genes share common promoter elements, many of which are identifiable based on the published literature. Thus, these clusters provide a foundation for understanding the transcriptional mechanisms of cell cycle regulation. Figure 1B shows the entire clustergram of our cell cycle–regulated genes; a larger version with gene names attached is available at our web site. The same color-coded presentation is used, with the addition, on the extreme left, of the similarity tree (dendrogram) calculated by the clustering algorithm. Many portions of the clustergram (subclusters) are described below, and those that we discuss are summarized in Table 4. The locations of these subclusters in the main cluster are indicated on Figure 1B.
Table 4.
Cluster summary
| Cluster | No. of genes | Binding site | Regulator | Peak expression |
|---|---|---|---|---|
| CLN2 | 119 | ACGCGT | MBF, SBF | G1 |
| Y′ | 26 | Unknown | Unknown | G1 |
| FKS1 | 92 | ACRMSAAA | SBF, (MBF?) | G1 |
| Histone | 10 | ATGCGAAR | Unknown | S |
| MET | 20 | AAACTGTGG | Met31p, Met32p | S |
| CLB2 | 35 | MCM1 + SFF | Mcm1p + SFF | M |
| MCM | 34 | MCM1 | Mcm1p | M/G1 |
| SIC1 | 27 | RRCCAGCR | Swi5p/Ace2p | M/G1 |
| Total | 363 |
The FKS1 set of genes is not a cluster, and the CLN2 cluster includes genes outside the core cluster that is shown. Consensus binding sites defined in the literature for various factors are as follows: MCM1, TTACCNAATTNGGTAA; SFF, GTMAACAA.
The G1 Clusters
The “CLN2” cluster is the largest subcluster and contains 76 genes. Genes in this cluster include CLN1, CLN2, CLB6, RNR1, CDC9, CDC21, CDC45, POL12, POL30, SWE1, and many other genes involved in DNA replication. A portion of this cluster is shown in Figure 3A. The key features of these genes are that expression is strongly cell cycle regulated (i.e., large peak-to-trough ratios); peak expression occurs in mid-G1 phase (∼10 min before budding in the cdc15 experiment); and they are strongly induced by GAL-CLN3 but are strongly repressed by GAL-CLB2. Fifty-eight percent of the 5′ regions of these genes had at least one copy of the motif ACGCGT (vs. 6% of control genes), which is a perfect MCB element. Fifty-two percent had at least one copy of CRCGAAA (vs. 13% of control genes), a degenerate SCB element. In addition, 16 had the motif AGAAGAAA, which is similar to a functionally important sequence found upstream of CLN3 (AAGAAAAA) (Parviz et al., 1998). Finally, 17 had the motif CCACAK, which we do not recognize. Outside the core of this cluster are at least 43 additional genes that are less tightly clustered but nevertheless appear to be coregulated with the CLN2 cluster (119 total genes).
Figure 3.

The G1 clusters. The transcription profiles are displayed as described in the legend to Figure 1. (A) CLN2 cluster. A fraction of the genes regulated similarly to the G1 cyclin CLN2, which reaches peak expression in the G1 phase of the cell cycle. To view the full cluster of all the cell cycle–regulated genes, visit http://cellcycle-www.stanford.edu. (B) Y′ cluster. Thirty-one genes that are located within the Y′ elements show cell cycle regulation of mRNA levels that peak in G1.
The “Y′” cluster (Figure 3B) contains 31 ORFs that all share DNA sequence similarity. There are 38 ORFs that share this similarity in the genome and we identify 36 of them as cell cycle regulated. All of these 38 ORFs are found in Y′ elements, located at chromosomes ends. It should be noted that these results may not represent 36 independent observations, because the cDNAs corresponding to these ORFs are almost certain to cross-hybridize on the microarrays. We do not know how these ORFs are regulated or the functional significance.
There is a set of 92 genes, containing ALG7, FKS1, GAS1, GOG5, PMT1, and PMI40, as well as other genes involved in cell wall synthesis (Klis, 1994), that are not a cluster on the clustergram but that are substantially coregulated. These genes can be seen on our web site as Figure 3C. Expression is strongly cell cycle regulated, and peak expression is nearly coincident with budding (∼10 min later than the CLN2 cluster in the cdc15 experiment). These genes are induced by GAL-CLN3 and repressed by GAL-CLB2. The majority of these genes had the motif ACRMSAAA (where R is A or G, M is A or C, and S is C or G), which may be an extension and variation of the SCB motif (CACGAAA). Comparison of the CLN2 cluster with this set suggests that expression from MCB motifs may be activated somewhat before expression from SCB motifs, but both kinds of expression are induced by CLN3 (consistent with previous studies) and repressed by CLB2. Earlier studies demonstrated that repression of SCB-driven expression requires CLB2, whereas repression of MCB-driven expression did not (Amon et al., 1993). Our results extend this by showing that CLB2 can repress MCB-driven expression, even though there may be additional repressive mechanisms. Many of the genes in this set also had the motif AARAARAAG, which is similar to a motif found in the CLN2 cluster (see above). However, because promoters generally are rich in such sequences, the significance of this motif is unclear.
The S and M Clusters
The histone cluster in Figure 4A forms the tightest cluster of any of the cell cycle genes. These nine genes have very high peak-to-trough ratios and give aggregate scores of ∼10. The histones have three known modes of regulation: first, there are negative elements repressing transcription; second, there is an element in the 3′ region of the mRNAs that destabilizes the message except during S phase; and third, there is a repeated positive element, which activates transcription (Freeman et al., 1992). Part of the core motif of the positive element is ATGCGAAR, which is similar to our degenerate SCB motif (ACRMSAAA). Consistent with this, histone expression is induced by GAL-CLN3. However it has been shown that the level and periodicity of HTA2/HTB2 mRNA accumulation are not noticeably affected by single mutation of SWI4, SWI6, or MBP1 (Lowndes et al., 1992; Cross et al., 1994). Additionally, histone levels are unaffected by GAL-CLB2. The sharpness of the peak in histone regulation is worth noting, both because it gives a good impression of the degree of synchronization and because the histones were the first genes for which periodic regulation was discovered (Hereford et al., 1981).
Figure 4.

The S and M clusters. The transcription profiles are displayed as described in the legend to Figure 1. (A) Histone cluster. The eight genes encoding histones and the yeast histone H1 homologue cluster very tightly and are expressed during S phase of the yeast cell cycle. (B) MET cluster. The expression of many of the members of the methionine pathway peaks just after the histones. (C) CLB2 cluster. A subcluster of genes that are expressed similarly to CLB2 highlights genes that peak during M phase.
The “MET” cluster (20 genes, Figure 4B) was completely unexpected. It contains 10 genes involved in the biosynthesis of methionine. Furthermore, two of the unnamed genes in this cluster show sequence similarity to human methionine synthetase, two are likely to be amino acid transporters (with unidentified specificities), one is similar to MET17, and one is on the opposite strand of MET2. Finally, ECM17, the only previously characterized gene in the cluster that is not known to be part of the methionine biosynthetic pathway, is similar to a sulfite redoxin from human. Thus, nearly all of the genes in this cluster are likely to be involved in methionine metabolism. Expression of the genes in this cluster peaks just after the histones, and at least some are inducible by CLN3. We searched the upstream region of the genes in the MET cluster and found that 15 of the genes had the consensus AAACTGTGG, which is identical to the consensus found for Met31/Met32 binding (Blaiseau et al., 1997).
The “CLB2” cluster (Figure 4C) contains 35 genes and includes many genes involved in mitosis such as CLB2, CDC5, CDC20, and SWI5. There are also many other less tightly clustered genes that appear to be regulated in a similar manner, including WSC4, PMP1, and the major plasma membrane proton pumps PMA1 and PMA2. The CLB2 cluster is highly regulated with a peak in M, and the genes are very strongly induced by GAL-CLB2, whereas GAL-CLN3 appears somewhat repressive. It was previously known that four of the genes found in this cluster, CLB1, CLB2, SWI5, and BUD4, are regulated by a combination of two transcription factors, Mcm1p and SFF (Althoefer et al., 1995; Sanders and Herskowitz, 1996). Mcm1p binds to the consensus TTACCNAATTNGGTAA (Acton et al., 1997), whereas, on the basis of three of these genes, SFF was thought to bind to the consensus sequence GTMAACAA. Furthermore, transcription of CLB1, CLB2, and SWI5 was known to be induced by Clb2p activity, possibly because of posttranslational activation of SFF (Amon et al., 1993). We compared the upstream regions of genes in the CLB2 cluster and certain other coregulated genes (e.g., ASE1, also thought to be a possible target of SFF [Pellman et al., 1995]) and found that most of them contain an easily recognizable MCM1 + SFF motif. Of the 35 genes in the cluster, only 9 genes (KIP2, MOB1, NUM1, YCL012W, BUD3, CHA1, YCL063W, YLR057W, and YML033W) did not have an easily recognizable near match to the MCM1 + SFF consensus. An alignment of the genes that contained this site can be viewed on our web site, and on the basis of this alignment, we deduce a new consensus for MCM1 + SFF binding, shown in Figure 5.
Figure 5.

The MCM1 + SFF consensus. By aligning promoter elements of several coregulated genes found in the CLB2 cluster (see our web site for the alignment), we developed a matrix for a new MCM1 + SFF consensus. The number of times each base was found at each position in the site was tallied and is displayed. The consensus was determined by examining the nucleotide frequencies at each position.
The M/G1 Clusters
The “MCM” cluster (Figure 6A) contains 34 genes, including all six MCM genes that are directly involved in DNA replication (MCM2, MCM3, CDC54, CDC46, MCM6, and CDC47; reviewed by Chevalier and Blow, 1996) as well as FAR1, DBF2, SPO12, and KIN3. These genes peak late in the cycle, at about the M/G1 boundary, and are induced by CLB2 and somewhat repressed by CLN3. This cluster has similarities to the CLB2 cluster, except that peak expression is slightly later. Searches of the upstream regions reveal that the majority of these genes contain binding sites for Mcm1p, as was previously shown for some members of the cluster (McInerny et al., 1997). Some, but not all, of these MCM1 sites have nearby sites for SFF (e.g., in FAR1, SPO12, KIN3, and CDC47), although these presumptive SFF sites are of varying quality. It has been suggested that some of the genes in this cluster are regulated through the “ECB,” a variant of the Mcm1p binding site (McInerny et al., 1997).
Figure 6.

The M/G1 clusters. The transcription profiles are displayed as described in the legend to Figure 1. (A) MCM cluster. The MCM genes are involved in initiation of DNA replication and are coregulated during the M/G1 transition of the cell cycle. (B) SIC1 cluster. Twenty-seven genes that peak at the M/G1 boundary form a subcluster. (C) MAT cluster. This is a cluster of 13 coregulated genes expressed at the M/G1 boundary, many of which are involved in mating.
The “SIC1” cluster comprises 27 genes, including EGT2, PCL9, TEC1, ASH1, SIC1, and CTS1. These genes are strongly cell cycle regulated (Figure 6B) and peak in late M or at the M/G1 boundary. GAL-CLN3 may repress some of these genes, whereas GAL-CLB2 has no consistent effect on the expression of these genes. Several of these genes are known to be regulated by the transcription factor Swi5p, which itself is a member of the CLB2 cluster (Dohrmann et al., 1992; Bobola et al., 1996; Knapp et al., 1996). Swi5p is thought to bind to a site with the consensus ACCAGC (Knapp et al., 1996), and indeed, when we searched for common motifs in the 5′ regions of the SIC1 cluster, we found the consensus RRCCAGCR in many of the 27 genes. When all cell cycle–regulated genes were examined for the presence of either the original Swi5p consensus, or this new extended consensus, the extended consensus was found to be much more specific for late M-phase genes. This comparison is shown on our web site. The motif GCSCRGC was also found in ∼40% of the genes in this cluster.
The “MAT” cluster contains 13 genes and is shown in Figure 6C. Some of these genes (MFα1, MFα2, and STE3) are specific for MATα cells (Jarvis et al., 1988) and so are significantly expressed only in the cdc15 experiment, which was done with a MATα strain. Other genes in the cluster (KAR4, AGA1, SST2, and FUS1) are induced by α factor and so are very strongly expressed at the beginning of the α factor experiment. However, these four genes oscillate in the other experiments when no α factor is present. We found MCM1 binding sites in the upstream regions of several of these genes, including MFα1 and MFα2. Furthermore, as discussed below, we found MATα1, the transcription factor that cooperates with Mcm1p to induce α-specific genes, is itself cell cycle regulated, and this may largely explain the oscillation of the α specific genes in this cluster.
Other Genes and Regulators
The nine clusters or near clusters summarized in Table 4 account for about half of the cell cycle–regulated genes. The remaining genes tend to be less strongly cell cycle regulated and cluster less tightly. We have attempted to find novel elements in the promoters of the remaining genes without great success. The best of these elements was the consensus GCAGNRNCCW, which we found in the upstream regions of CLB4, BUD3, CPR8, PRO2, YCL012W, YCL063W, YGL217C, YNL043C, YDR130C, and YOL030W; these genes appear to be moderately well coregulated (peak expression occurs in G2). There may be additional, novel, upstream elements that we are unable to find.
It is likely that many of the remaining genes are actually coregulated with members of the clusters we have described, and their transcription may be controlled by the same types of elements. Indeed, we know that some of the remaining genes have recognizable elements (e.g., MCBs and SCBs), whereas in other cases, the elements may be highly degenerate versions of the known elements. This may explain why the cell cycle regulation we observe is relatively weak and why the genes do not cluster tightly. Finally, mRNA levels could oscillate, not because of transcriptional control, but because of cell cycle control of mRNA stability; the histone mRNAs are controlled partly in this way (Wang et al., 1996).
For the clusters we have identified, some of the genes in the cluster do not contain an obvious element; for instance, nine of the genes in the CLB2 cluster do not contain an obvious MCM1 + SFF site. We do not know whether these genes contain cryptic, degenerate sites that our algorithms fail to recognize, or whether these genes are regulated by an unknown factor.
The Functions of the Cell Cycle–regulated Genes
The major functions of the cell cycle regulated genes we identified are cell cycle control, DNA replication, DNA repair, budding, glycosylation, nuclear division and mitosis, structure of the cytoskeleton, and mating. In Figure 7 we arrange 294 named genes in our set, according to both a functional class and the phase group to which they belong.
Figure 7.

Cell cycle–regulated genes with characterized functions. Two hundred ninety-seven of the cell cycle–regulated genes are grouped by both function and phase of peak expression. Several functional groups are split into subgroups, which reflect the nature of the function. Those highlighted in red were previously known to be cell cycle regulated. Many functional categories display strong biases toward gene expression during particular intervals of the cell cycle. Obvious examples include genes involved in DNA synthesis and DNA repair (G1), mating (M, M/G1, and G1), chromatin structure (G1 and S), and methionine biosynthesis (S and G2).
DNA Replication, Repair, and Chromosome Assembly
It is instructive to look at the pattern of expression of genes involved in a particular process. For instance, we can trace the expression of many genes somehow involved in DNA replication (as shown in Figure 7). Of the genes that peak in G1 there are 23 genes with known functions in DNA replication. These genes include subunits of the DNA polymerases and their accessory factors (e.g. CDC2, POL1, and POL2), genes involved in nucleotide synthesis (e.g. CDC21), and genes involved in initiation of DNA synthesis (e.g. CDC45). Many genes involved in DNA repair such as PMS1 and MSH2 reach peak expression in G1 phase, suggesting that repair of DNA lesions may be a normal part of S phase.
Later, when S phase is actually occurring, the histone genes reach peak expression. In late M phase or M/G1 all six MCM genes important for prereplicative complex formation (MCM2, MCM3, CDC54, CDC47, MCM6, CDC47, and CDC54) and CDC6 reach their peaks, presumably to help set up origins for the next cell cycle. Thus, many genes needed for replication and repair reach peak expression just before they are needed, the histones peak exactly at the time they are needed, and a few genes important for regulation of DNA synthesis peak well in advance of the next round of S phase. Only two known initiator genes, CDC45 and DBF4 (which we did not identify in our analysis; see below) peak just before S phase, suggesting these may be particularly important to trigger replication.
Bud Initiation and Bud Growth
Budding is a major metabolic activity for the cell and involves several subprocesses. The cell must choose a site for the new bud (initiation) and make components for an ever-increasing surface area consisting of a new cell membrane (which requires lipids and integral membrane proteins) and a new cell wall (composed largely of glucan, chitin, and mannoproteins). All of these processes require delivery of components, via the secretory apparatus, to the sites of new membrane and cell wall synthesis, which, in normal conditions, occurs exclusively in the bud (Kaiser et al., 1997; for reviews, see Lew et al., 1997; Orlean, 1997).
We found 17 genes that involved in bud site selection and cell polarization (e.g., BUD3, BUD4, BUD8, BUD9, BEM1, GIC1, MSB1, and MSB2). As indicated in Figure 7, none of these genes had been reported to be cell cycle regulated. Some of these (BUD9, CDC10, and RSR1) show peak expression in G1, consistent with roles in bud initiation. Others, (BUD4, BUD8, and BEM1) peak in M phase, suggesting roles in the following cell cycle, i.e., earlier in the budding pathway than the G1 group. We also identified many genes needed for secretion, glycosylation (needed for making mannoproteins), synthesis of lipids, and cell wall synthesis.
Cell Division and Mitosis
Another fundamental process of cell division, in which a large number of the genes involved have their messages regulated by the cell cycle, is the process of mitosis (for review of microtubule-related topics, see Botstein et al., 1997). During the cell cycle many events occur that allow mitosis to progress in a timely manner. This process begins in G1 when the spindle pole body (SPB) replicates. To facilitate this process six known components of the SPB reach peak expression in G1 (CNM67, NUF1, SPC42, SPC97, SPC98, and TUB4), one (SPC34) peaks during S, and one (NUF2) peaks during M phase. Some of these genes were already known to be cell cycle regulated (NUF1, and SPC42) (Kilmartin et al., 1993; Donaldson and Kilmartin 1996).
Once the mitotic program is entered the cell must create a spindle, which is responsible for moving the nucleus to the bud neck so that nuclear division can occur. This process requires microtubules and many accessory proteins (to form the spindle) as well as kinesins (for movements of the nucleus and the SPB). These genes reach peak expression largely during the first half of the cell cycle. In G1 BIM1, BUB1, IPL1, KAR3, and SLK19 reach peak expression, and during S, five genes (CIN8, KAR9, KIP1, STU2, and VIK1) peak. Five genes peak during G2 (BUB2, CIK1, KIP2, KIP3, and NUM1), as well as the major β tubulin TUB2. Finally, one gene (ASE1) reaches peak expression during M.
It was somewhat unexpected that tubulin messages would be regulated by the cell cycle; unfortunately the microarrays that we used for the α factor and elutriation experiments did not contain DNA complementary to either the major (TUB1) or minor (TUB3) α tubulins. Our data set suggested that that TUB1 might be cell cycle regulated because it had a score just below our cutoff. We wished to verify that the major tubulins were regulated in the cell cycle by an independent method (quantitative real-time PCR [Heid et al., 1996]). This method allows determinations of relative mRNA levels with excellent reproducibility. We performed the analysis as detailed in MATERIALS AND METHODS with the result that, as we suspected, TUB1 and TUB2 are moderately cell cycle regulated, but TUB3 appears less so (Figure 8). This suggests that the low score for TUB1 may have been caused by their absence from some of the arrays. It should be noted that TUB2 with a score of 2.33 is clearly above the threshold we set for cell cycle regulation, but that TUB1 with a score of 1.25 is just below, and TUB3 (score 0.53) is considerably below the threshold. Comparison with Figure 8 illustrates the point that a score near the threshold can be the result either of inadequate data or weak regulation.
Figure 8.

Tubulin message levels. The mRNA levels for TUB1, TUB2, and TUB3, relative to those of PPA1, were determined during synchronous division after release from an α factor arrest, using the TAQman assay as described in MATERIALS AND METHODS.
One relatively small class of genes that displays tight temporal regulation is a group of genes involved in chromatid cohesion. Five of these genes (SMC1, SMC3, MCD1, PDS1, and PDS5 [Strunnikov et al., 1993; Yamamoto et al., 1996; Guacci et al., 1997; Michaelis et al., 1997]) have peak expression during G1 just before the next round of DNA synthesis.
At the end of the cell cycle the cell must exit mitosis so that the next round of division can occur. To do this, a system of proteins acts to inhibit the activity of Clb-Cdc28p. One of these proteins is Sic1p, whose expression is known to peak at this time (Donovan et al., 1994). Many of the proteins that inhibit Clb-Cdc28p or prepare the cell to exit from mitosis are known to be cell cycle regulated and peak in M phase. We also find that DBF20 (which is functionally related to DBF2) is cell cycle regulated and peaks in G2.
Mating
At least 19 genes directly involved in mating are cell cycle regulated. These include both mating pheromones (a-factor and α-factor) and, perhaps most interestingly, include the central mating-type transcription factor MATα1 itself. MATα1 binds to DNA in cooperation with Mcm1 (Sengupta and Cochran, 1991) and induces expression of α-specific genes. It was previously shown that some genes involved in mating were cell cycle regulated, and this regulation was shown to be due to cooperative binding between Mcm1 and Ste12. The fact that the MATα1 transcription factor itself oscillates provides yet another mechanism by which genes involved in mating might be cell cycle regulated. We found Mcm1 sites in the upstream regions of several of these genes, including MATα1. The regulation of genes involved in mating is clearly complex, and several transcription factors are involved. However, it seems that most of these transcription factors cooperate in one way or another with Mcm1. The fact that so many mating functions are cell cycle regulated, including an α-specific transcription factor, helps explain the deep connection between mating, start, and the cell cycle. For instance, if genes involved in mating are turned off at start by multiple mechanisms, it helps explain how passage through start precludes mating.
Cell Cycle Control Genes
Of the 19 genes involved in cell cycle control we identified, 17 were already known to be cell cycle regulated. This set mainly includes cyclins and transcription factors, whose activities and time of action are well documented (see Koch and Nasmyth, 1994; Andrews and Measday, 1998). The only two cell cycle control genes that we identified newly as regulated were WHI3 and HSL7.
Methionine Biosynthesis
It was an unexpected and somewhat surprising result that many genes involved in methionine biosynthesis are cell cycle regulated. A number of possibilities suggest themselves. First, the pool of available cellular methionine is smaller than virtually any other amino acid; thus, methionine is likely to be limiting (Jones and Fink, 1982). Indeed, Unger and Hartwell (1976) noted that starvation for sulfur or for methionine effectively causes G1 arrest, suggesting that cell cycle progression is particularly sensitive to the availability of methionine. They also found that a temperature-sensitive allele of methionine tRNA synthetase causes G1 arrest, even in the presence of methionine. These observations suggest that the cell cycle regulation of methionine genes ensures sufficient capacity for protein synthesis in that biosynthetic pathway for the next cell cycle; if there are insufficient resources, G1 arrest ensues.
It is known that the more than 20 genes that constitute the sulfur amino acid biosythesis pathway are coordinately regulated at the level of transcription. This transcription is repressed in response to an increase in the intracellular concentration of S-adenosylmethionine, an end product of the pathway (methionyl tRNA is another end product) (Thomas et al., 1989). A second possibility therefore is that the concentration of S-adenosylmethionine is depleted as cells enter S phase, causing derepression of these genes, which results in cell cycle regulation.
A third possibility is that the protein that actually represses these genes, Met30p, is available in limiting amounts and for some reason is titrated during or just before S phase, causing coordinate derepression of this set of genes. Data supporting this idea are that Met30p is involved in cell cycle regulation as an F-box protein that targets Swe1 for degradation (Kaiser et al., 1998; Patton et al., 1998). SWE1 transcription is cell cycle regulated (Ma et al., 1996) (our analyses recapitulate this observation), peaking at the G1/S phase boundary. Therefore, the concentration of a known Met30p substrate increases just before the derepression of genes involved in methionine biosynthesis that Met30p is known to repress. Thus, Met30p may become limiting, allowing expression of the MET genes. We do not know whether the cell cycle regulation of these genes is important for their function.
Interestingly, another F-box protein, Grr1, which is also involved in cell cycle regulation, regulates the expression of the HXT genes (hexose transporters). The HXT genes are members of a cluster of very weakly cell cycle–regulated genes peaking in M/G1 that also includes PHD1 and RGA1 (visible in Figure 1B on our web site). Thus, two different F-box proteins involved in cell cycle control also regulate genes involved in providing nutrients, and these nutrient-related genes are weakly cell cycle regulated. It is possible that these F-box proteins somehow coordinate nutrient availability with the cell cycle.
Other Nutritional Genes
A very large fraction of the genes involved in nutrition that are cell cycle regulated are involved in transport of essential minerals and organic compounds across the cell membrane. Some of the compounds that are moved by these transporters are amino acids (GAP1), ammonia (AUA1 and MEP3), sugars (e.g., HXT1 and RGT2), and iron (FET3 and FTR1). We also identified the acid phosphatases (e.g., PHO3 and PHO8). Nearly all of these genes reach peak expression late in the cell cycle during M and M/G1.
Developmental Pathway Genes: Sporulation and Pseudohyphal Growth
A number of genes associated with functions in specialized developmental pathways show cell cycle regulation. These include the apparently sporulation-specific genes SPS4 and SSP2, which have peak expression in M and M/G1, respectively. These might represent cases such as SPO12, which has known function in both mitotic and meiotic pathways (Klapholz and Esposito, 1980; Toyn and Johnston, 1993).
The Y′ Genes
Although not strictly a functional category, the Y′ genes form an interesting group of coregulated genes. The Y′ sequences are repeated sequences found just centromere proximal to the telomere itself on many chromosomes. Within the Y′ elements are two open reading frames, and there are appropriate splicing signals that suggest that they form one large product, although it has not been shown experimentally that these sites are functional (Louis and Haber, 1992; Louis, 1995). The larger (telomere proximal) of these two ORFs shows similarity to RNA helicases, containing all the motifs known to be necessary for helicase activity (Louis and Haber, 1992). However, sequence similarities among these ORFs are very high, and we cannot distinguish whether one, a few, or all of these elements are cell cycle regulated.
The GAL-CLN3 and GAL-CLB2 Experiments
Our experiments to investigate the transcriptional effects of Cln3p and Clb2p provide an excellent corroborative data set that supports cell cycle regulation for more half of the genes in our list. Of the genes that are cell cycle regulated, there are 116 genes that are induced more than twofold by Cln3p, and are repressed by Clb2p. Eighty-seven percent of these peak in either G1 or S phase. In contrast there are only eight cell cycle–regulated genes that are induced by Cln3p and not repressed by Clb2p. There are 33 genes induced greater than twofold by Clb2p that are repressed by Cln3p, whereas only five genes induced by Clb2 are not repressed by Cln3p. All cell cycle–regulated genes responsive to Clb2p peak in either M or M/G1 phases.
There were also genes that responded to Clb2p or Cln3p (or both) that we did not identify as cell cycle regulated. For instance there are 53 genes induced by Cln3p and repressed by Clb2p that are not on our cell cycle–regulated list. Many of these are involved in functions for which we know many genes are cell cycle regulated, e.g., secretion (PMT2, PMT4, SEC53, and SEC21), chitin synthesis (CHS3), and nucleotide biosynthesis (ADE3, RNR2). However, we have no other evidence to suggest that these may be false negatives. Indeed, by visual inspection, none of these genes displays convincing signs of periodicity. This observation reinforces the notion that using many types of experiments is crucial to drawing legitimate conclusions.
Our experiments on the transcriptional effects of Cln3p and Clb2p help us dissect the transcriptional regulators of each gene (see above). In addition, they support the notion that mechanistically two opposing oscillators drive the cell cycle. This is particularly well illustrated in some of the subclusters, for instance, the CLN2 cluster, where the effects of CLN3 induction are almost exactly mirrored by the opposite effects of CLB2 induction. For other subclusters we see that the genes respond to only one of the cyclins, (e.g., the Y′ cluster is induced by CLN3 yet relatively unchanged by CLB2).
Finally, we found that CLN3 can repress the transcription of certain genes, particularly a group of genes involved in mating. This was not entirely unexpected, because it had previously been demonstrated that FAR1 transcription (McKinney et al., 1993) is negatively regulated by start, although a direct link to Cln3p activity has not been previously demonstrated.
DISCUSSION
Reliability of the Methods for Assessing Cell Cycle Regulation
Our identification of a gene as cell cycle regulated was objective in the sense that it was entirely quantitative. Nevertheless, it should be clear that the setting of the threshold was in the end arbitrary. We can ask whether genes below the threshold might still be cell cycle regulated in a biologically significant way, and we can consider whether there are any circumstances in which our experiments might have failed to reveal the cell cycle pattern of regulation. In this context it is relevant that we do not identify 9 of the 104 genes (CDC8, DBF4, CHS3, PRI1, TIR1, CDC14, CLB3, DPB3, and RAD17) previously reported to be cell cycle regulated (White et al., 1987; Chapman and Johnston, 1989; Siede et al., 1989; Johnston et al., 1990b; Araki et al., 1991; Fitch et al., 1992; Wan et al., 1992; Igual et al., 1996; Caro et al., 1998). Of these, only RAD17 appears visually to vary with the cell cycle in our data and then only in the cdc15 experiment. We believe that most of the remaining false negatives are due to noise in our data set that dampens their signal. It is worth noting that some of these genes showed only very weak cell cycle regulation in the original publications or in other studies done by traditional methods. For instance, DBF4 oscillated by 2.5-fold in the original publication (Chapman and Johnston, 1989), and in some Northern blotting experiments, oscillation is not seen (Sclafani, personal communication).
This analysis convinces us that relatively few genes with significant cell cycle regulation have not been identified. However, there are many plausible causes of false-positive identifications. Random fluctuations in the data could appear as a cell cycle oscillation, but as described in MATERIALS AND METHODS, we expect this to be a relatively rare event.
Cross-hybridization between genes whose DNA sequences are similar can produce false positives when only one of the genes is actually cell cycle regulated. It has been estimated that cross hybridization in our system can become significant at or above 75% DNA sequence identity (DeRisi, Iyer, and Brown, personal communication). For instance, our data set includes both DBF2 and DBF20, which are 75% identical over the last 800 bp of each gene; it has previously been published that only DBF2 is cell cycle regulated (Johnston et al., 1990a; Toyn et al., 1991). DBF20 may appear in our set because DBF2 cDNA is cross-hybridizing with the DNA sequence of DBF20 on the microarray. These two genes show somewhat different regulation, however, so this could be an instance of strain differences or errors in the published literature.
A third manner in which false positives can occur is when an unregulated gene overlaps the mRNA for a cell cycle–regulated gene. The cDNA corresponding to the regulated gene would hybridize with the unregulated gene’s DNA, generating a false positive. In our list there are 42 pairs of genes that overlap, and 39 additional pairs in which the distance between the two genes is <300 bp. A full list of all genes that are near each other by chromosomal position can be found at the web site.
Comparisons with Previous Analyses
One part of our method for identifying genes regulated by the cell cycle relied on the examining the large body of data already that has been published. Cho et al. (1998) performed a study similar to ours using different technology and methods. Our methods can aggregate data from many experiments and thereby improve the signal-to-noise ratio in the total data set. It illustrates as well the value and necessity for making available primary data from genome scale experiments of this kind. The data from Cho et al. (1998) aided in our ability to identify some genes as being cell cycle regulated. Figure 9 shows several points located very near the x axis that represent genes whose identification as cell cycle regulated clearly depends on the data of Cho et al. The assessment of cell cycle regulation for the remainder of the genes, as shown in Figure 9, is essentially the same whether the data of Cho et al. are included.
Figure 9.

Aggregate CDC score is largely independent of the cdc28 data set. Scatter plot for the scores that were derived using either the cdc15 and α factor experiments only (plotted on the y axis) or using the cdc15, the α factor, and the cdc28 experiments (x axis). A single dot is plotted for each gene. It should be noted that adding data increases the absolute magnitude of the scores; it is the relative magnitude, however, that indicates cell cycle regulation.
The most obvious difference between our results and those of Cho et al. (1998) is in the number of genes identified as cell cycle regulated. With a manual decision process, they found 421 genes to be cell cycle regulated. Our set of 800 genes includes 304 of these, but the other 117 do not appear significantly cell cycle regulated in our data. Our set of 800 therefore contains 496 genes not identified by Cho et al.
There were many technical differences in the way the two studies were carried out, and it is difficult to say how these may have contributed to the differences between the results. One significant advantage of our analysis was the diversity of experiments from which we could identify the characteristic pattern of cell cycle regulation. This allowed us to distinguish cell cycle regulation from confounding patterns such as those caused by the heat shock response when a culture is shifted from one temperature to another.
One of the largest discrepancies between the two analyses regards genes that may peak twice per cell cycle. We identified 10 genes as cell cycle regulated that according to Cho et al. (1998) showed more than one peak but had no single prominent peak in expression. Our algorithm is designed to find genes with single expression peaks; it significantly penalizes more than one peak. Furthermore, visual inspection of the aggregate data does not show multiple peaks in any of these cases. Therefore, we believe the aggregate data set does not support the existence of multiple peaks of expression for these genes. This leaves open the possibility that there may be other genes with more than one peak per cell cycle.
Many of the genes differing between the two data sets had low peak-to-trough ratios and relatively poor aggregate CDC scores. It is perhaps natural that we identified a larger number of modestly regulated genes, because we had a larger number of experiments and a statistical rather than manual approach to identification. It is important to note, however, that some of the differences were in genes with very strong cell cycle regulation. Cho et al. (1998) failed to find FKS1, GOG5, EGT2, two histone genes, and several other genes with very strong regulation. Before including the data set of Cho et al., we had failed to find HO. Quite a few of the strongly regulated genes not identified by Cho et al. were genes known to be regulated fairly directly by Cdc28p (in combination with either Cln3p or Clb2p). The expression of such CDC28-dependent genes may have been altered in the cdc28-13 block–release experiment of Cho et al.
Mechanisms of Transcriptional Regulation
The 800 genes in our list were examined for the binding sites of known cell cycle transcription factors, and for a little more than half of these we found good matches to known sites relevant to the phase of peak expression. Moreover, nearly 70% of these same genes showed a significant response to Cln3p or Clb2p induction (or both). In addition, we identified as cell cycle regulated other sets of genes that form functional pathways and are known to be coregulated (e.g., the methionine biosynthesis genes) or about whose regulation something is known (e.g., the hexose transporters). Thus, there are ∼500 genes for which we understand, at some level, the molecular mechanism of cell cycle control, a gratifying number.
This, however, leaves ∼300 genes for which we do not have good binding sites for appropriate cell cycle transcription factors and have not identified any compelling novel sites. Because these 300 genes tend to be the less strongly regulated ones, it may be that some of them contain degenerate sites for known factors and were therefore missed in our searches (which generally did not allow mismatches). Alternatively, some of these genes may be controlled by novel sites that we failed to identify; others may be controlled by known factors in a combinatorial manner such that peak expression is at an unexpected time. Another possibility is that some of these transcripts may be controlled in some other way, such as by mRNA stability. Our data set provides fertile ground for further investigation of these questions.
These data give us a partial picture of the logical circuitry of transcriptional controls in the cell cycle. A large number of genes (≥200) are induced in G1 and S by the action of Cln3p-Cdc28p on MBF and SBF. Furthermore, by M they become repressed by the action of Clb2p-Cdc28p. At the same time, Clb2p-Cdc28p, acting through MCM1 + SFF, induces its own constellation of genes, which may number >50. These genes include the important transcription factor Swi5p; once its transcription has been induced, and it is allowed to enter the nucleus (perhaps because of a dip in Clb2p-Cdc28p activity) (Moll et al., 1991), another set of genes is turned on. Among these is the CDK inhibitor SIC1, which is important for deactivating Clb2p-Cdc28p. The loss of Clb2-Cdc28 activity causes a collapse in the transcription of all Clb2p-dependent transcripts and, moreover, allows Cln3p-Cdc28p to reactivate MBF and SBF to begin a new cell cycle. A gap in this picture is that we do not understand the oscillation in the genes expressed in M/G1 phase from an MCM1 site (the ECB). That is, what is it about an ECB or about Mcm1p that makes expression of these genes cell cycle regulated? A second omission is that we do not understand very clearly how CLB2 is induced in the first place, although part of the answer certainly lies in regulation of Clb2p protein stability and Clb2p-Cdc28p activity.
Functional Significance of Cell Cycle Regulation
Studies of cell cycle regulation have focused on genes with cell cycle–specific functions. That is, they are genes whose functions are only needed for a part of the cycle. These genes are directly involved in, for instance, DNA replication, budding, and mitosis. For some such genes, transcriptional regulation may be a matter of conserving resources. For instance, it would probably do no great harm to express CDC9 (DNA ligase) constitutively. However, by expressing it, and hundreds of other similar genes, only when needed, the cell can achieve a small advantage. Moreover, yeast often encounter what one might call a “Sleeping Beauty” situation: they lie dormant for weeks or months, and then are required to resume life where they left off. Genes needed for cycling are evidently not needed during the dormant period, but very much needed immediately afterward. Thus, cell cycle–regulated expression may ensure that necessary gene products are always available to cycling cells. In this regard, it is interesting to note that the purine-rich motif AAGAAAAA (Parviz et al., 1998) is thought to be important for response to glucose; this motif may be important in the switch from stationary phase to rapid growth, and we find similar motifs enriched in the promoters of several types of cell cycle–regulated genes. That is, these genes may be growth regulated as well as cell cycle regulated.
Other genes with cell cycle–specific functions act as regulators or switches. For these genes, it is not only important when exactly they are on but also when they are off. An excellent example is Clb2p, which is important for mitotic events but antagonistic toward G1 events. Clb2p is highly regulated: its transcription is regulated; the stability of the protein is regulated; and the activity of the Clb2p-Cdc28p complex is regulated by phosphorylation, dephosphorylation, and the binding of inhibitors. Any one of these regulatory mechanisms is dispensable for viability, but if several are lost, the deregulated Clb2p is lethal. Thus, transcriptional regulation of a gene controlling a switch can be central to its function.
Cell cycle–regulated transcription can be used to build a structure in a highly controlled way. This can be illustrated with some parallels between the strategy of the cell for regulating DNA replication and its strategy for regulating budding. Both processes occur once and only once per cycle; that is, the cell must both initiate the process and also prevent reinitiation. In both cases the cell builds an initiation structure, which for prereplication complexes contains origin recognition complex components, Mcm proteins, and Cdc6p, and for the prebud site contains Bud proteins (and others). In both cases transcriptional controls provide key components of the initiation complexes at certain times so that the complexes can be built in an orderly manner; however, the complexes cannot easily later be rebuilt at an inappropriate time, partly because the components are no longer available. The building of the replication and budding initiation complexes occurs long before replication or budding actually occur, and accordingly, key components of the complexes (e.g., Mcms, Cdc6p, Bud4p, and Bud8p) are provided in M phase, long before they are used.
These types of cell cycle–regulated genes are well known. However, because our identifications were relatively complete and inclusive, and because the method of identification was not hypothesis driven, we were able to find cell cycle–regulated genes of quite unexpected types. In particular, we found many cell cycle–regulated genes whose functions are essentially not cell cycle specific. These include genes involved in secretion and lipid synthesis, which are probably needed at all times, at least at some level. In these cases, the role of cell cycle regulation may be to provide extra transcript when there is extra demand, i.e., at the time of budding. The best single example of such a gene may be PMA1, encoding the major plasma membrane proton pump, a stable protein. The PMA1 function is essential, and although its function is required throughout the cell cycle (Serrano et al., 1986), its transcription is strongly periodic. Peak expression probably coincides with the time of fastest growth of the plasma membrane in the bud; presumably periodic expression is needed to provide the PMA1 for the daughter cell. In this case, it is impractical to make a store of PMA1 beforehand, because excess PMA1 is toxic, causing accumulation of intracellular membranous structures (Espinet et al., 1995). The cell cycle regulation of PMA1 might be considered partly an answer to a problem of stoichiometry. Stoichiometric considerations are important for other cell cycle–regulated genes as well, notably the histones, SPB components, and microtubules.
Conclusions
To summarize, we found 800 yeast genes whose transcripts oscillate through one peak per cell cycle. We defined these 800 genes by using an objective, empirical model of cell cycle regulation, whose threshold was somewhat arbitrary. Below this threshold there may well be genes whose expression is truly periodic and whose periodicity might even have biological significance. Unfortunately we cannot reliably detect such genes, but it is likely that they are relatively few in number, because very few of the genes known beforehand to be periodically expressed during the cell cycle fall below our threshold.
With respect to mechanism, we could account for the periodicity of expression of about half. We found relevant DNA motifs upstream of these genes, and we observed independently that their expression was affected by induction of Cln3p and Clb2p. The basis of the regulation of the remaining genes remains to be elucidated, and some of the detailed behavior of some of the cyclin-dependent gene expression also remains to be explained.
Finally, we hope that our colleagues in the scientific community will find this paper to be valuable not as only a description of our results but also as a resource for data for some time to come. We made measurements for virtually every gene or open reading frame in the yeast genome but are in a position to interpret explicitly only a tiny fraction of these measurements. We make our data available, as did Cho et al. (1998), in the expectation that there will be increasing value in genomic data sets as more of them accumulate and that together these will fully realize the promise of the genome sequencing projects.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jon Mulholland, Holly Sundberg, Trisha Davis, Anita Sil, and Lee Hartwell for their assistance and helpful conversations. We also thank those who assisted in the tedious task of microarray manufacture (Cammy Kao, Audrey Gasch, Tracy Ferea, Joseph DeRisi, Craig Cummings, and Barbara Dunn). Herman Wijnen, Max Diehn, and Doug Ross deserve thanks for critical reading of the manuscript. Our efforts were greatly aided by the genome databases, particularly the Saccharomyces Genome Database (http://genome-www.stanford.edu/Saccharomyces/) and the Yeast Protein Database (http://quest7.proteome.com/YPDhome.html). This work was supported by grants from the National Institutes of Health to D.B. (GM46406 and CA77097), to B.F. (GM45410), to P.O.B. (HG00450), and to M.Q.Z. (HG01696). This work was also supported by the Howard Hughes Medical Institute. P.O.B. is an associate investigator of the Howard Hughes Medical Institute. M.Q.Z. is also supported by a Cold Spring Harbor Laboratory Association Award, and G.S. was supported by a postdoctoral fellowship from the Department of the Army (DAMD17-97-1-7316).
Footnotes
A complete data set for this article is available at www.molbiolcell.org.
REFERENCES
- Acton TB, Zhong H, Vershon AK. DNA-binding specificity of Mcm1: operator mutations that alter DNA- bending and transcriptional activities by a MADS box protein. Mol Cell Biol. 1997;17:1881–1889. doi: 10.1128/mcb.17.4.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althoefer H, Schleiffer A, Wassmann K, Nordheim A, Ammerer G. Mcm1 is required to coordinate G2-specific transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:5917–5928. doi: 10.1128/mcb.15.11.5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon A, Tyers M, Futcher B, Nasmyth K. Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell. 1993;74:993–1007. doi: 10.1016/0092-8674(93)90722-3. [DOI] [PubMed] [Google Scholar]
- Andrews B, Measday V. The cyclin family of budding yeast: abundant use of a good idea. Trends Genet. 1998;14:66–72. doi: 10.1016/s0168-9525(97)01322-x. [DOI] [PubMed] [Google Scholar]
- Andrews BJ, Herskowitz I. Identification of a DNA binding factor involved in cell-cycle control of the yeast HO gene. Cell. 1989;57:21–29. doi: 10.1016/0092-8674(89)90168-2. [DOI] [PubMed] [Google Scholar]
- Araki H, Hamatake RK, Morrison A, Johnson AL, Johnston LH, Sugino A. Cloning DPB3, the gene encoding the third subunit of DNA polymerase II of Saccharomyces cerevisiae. Nucleic Acids Res. 1991;19:4867–4872. doi: 10.1093/nar/19.18.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaiseau PL, Isnard AD, Surdin-Kerjan Y, Thomas D. Met31p and Met32p, two related zinc finger proteins, are involved in transcriptional regulation of yeast sulfur amino acid metabolism. Mol Cell Biol. 1997;17:3640–3648. doi: 10.1128/mcb.17.7.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobola N, Jansen RP, Shin TH, Nasmyth K. Asymmetric accumulation of Ash1p in postanaphase nuclei depends on a myosin and restricts yeast mating-type switching to mother cells. Cell. 1996;84:699–709. doi: 10.1016/s0092-8674(00)81048-x. [DOI] [PubMed] [Google Scholar]
- Botstein D, Amberg D, Mulholland J, Huffaker T, Adams A, Drubin D, Stearns T. The yeast cytoskeleton. In: Pringle JR, Broach JR, Jones EW, editors. The Molecular and Cellular Biology of the Yeast Saccharomyces: Cell Cycle and Cell Biology. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1997. pp. 1–90. [Google Scholar]
- Caro LH, Smits GJ, van Egmond P, Chapman JW, Klis FM. Transcription of multiple cell wall protein-encoding genes in Saccharomyces cerevisiae is differentially regulated during the cell cycle. FEMS (Fed Eur Microbiol Soc) Microbiol Lett. 1998;161:345–349. doi: 10.1111/j.1574-6968.1998.tb12967.x. [DOI] [PubMed] [Google Scholar]
- Chapman JW, Johnston LH. The yeast gene, DBF4, essential for entry into S phase is cell cycle regulated. Exp Cell Res. 1989;180:419–428. doi: 10.1016/0014-4827(89)90068-2. [DOI] [PubMed] [Google Scholar]
- Chevalier S, Blow JJ. Cell cycle control of replication initiation in eukaryotes. Curr Opin Cell Biol. 1996;8:815–821. doi: 10.1016/s0955-0674(96)80082-2. [DOI] [PubMed] [Google Scholar]
- Cho RJ, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Cross FR. Starting the cell cycle: what’s the point? Curr Opin Cell Biol. 1995;7:790–797. doi: 10.1016/0955-0674(95)80062-x. [DOI] [PubMed] [Google Scholar]
- Cross FR, Hoek M, McKinney JD, Tinkelenberg AH. Role of SWI4 in cell cycle regulation of CLN2 expression. Mol Cell Biol. 1994;14:4779–4787. doi: 10.1128/mcb.14.7.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Dohrmann PR, Butler G, Tamai K, Dorland S, Greene JR, Thiele DJ, Stillman DJ. Parallel pathways of gene regulation: homologous regulators SWI5 and ACE2 differentially control transcription of HO and chitinase. Genes & Dev. 1992;6:93–104. doi: 10.1101/gad.6.1.93. [DOI] [PubMed] [Google Scholar]
- Dohrmann PR, Voth WP, Stillman DJ. Role of negative regulation in promoter specificity of the homologous transcriptional activators Ace2p and Swi5p. Mol Cell Biol. 1996;16:1746–1758. doi: 10.1128/mcb.16.4.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson AD, Kilmartin JV. Spc42p: a phosphorylated component of the S. cerevisiae spindle pole body (SPB) with an essential function during SPB duplication. J Cell Biol. 1996;132:887–901. doi: 10.1083/jcb.132.5.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan JD, Toyn JH, Johnson AL, Johnston LH. P40SDB25, a putative CDK inhibitor, has a role in the M/G1 transition in Saccharomyces cerevisiae. Genes & Dev. 1994;8:1640–1653. doi: 10.1101/gad.8.14.1640. [DOI] [PubMed] [Google Scholar]
- Eisen, M.B., and Brown, P.O. (1999). DNA arrays for analysis of gene expression. Methods Enzymol. (in press). [DOI] [PubMed]
- Eisen, M.B., Spellman, P.T., Brown, P.O., and Botstein, D. (1999). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA (in press). [DOI] [PMC free article] [PubMed]
- Espinet C, de la Torre MA, Aldea M, Herrero E. An efficient method to isolate yeast genes causing overexpression-mediated growth arrest. Yeast. 1995;11:25–32. doi: 10.1002/yea.320110104. [DOI] [PubMed] [Google Scholar]
- Fitch I, Dahmann C, Surana U, Amon A, Nasmyth K, Goetsch L, Byers B, Futcher B. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol Biol Cell. 1992;3:805–818. doi: 10.1091/mbc.3.7.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman KB, Karns LR, Lutz KA, Smith MM. Histone H3 transcription in Saccharomyces cerevisiae is controlled by multiple cell cycle activation sites and a constitutive negative regulatory element. Mol Cell Biol. 1992;12:5455–5463. doi: 10.1128/mcb.12.12.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futcher AB. Analysis of the cell cycle in S. cerevisiae. In: Fantes P, Brooks R, editors. The Cell Cycle: A Practical Approach. Oxford: IRL Press; 1993. pp. 69–92. [Google Scholar]
- Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Hereford LM, Osley MA, Ludwig TRd, McLaughlin CS. Cell-cycle regulation of yeast histone mRNA. Cell. 1981;24:367–375. doi: 10.1016/0092-8674(81)90326-3. [DOI] [PubMed] [Google Scholar]
- Igual JC, Johnson AL, Johnston LH. Coordinated regulation of gene expression by the cell cycle transcription factor Swi4 and the protein kinase C MAP kinase pathway for yeast cell integrity. EMBO J. 1996;15:5001–5013. [PMC free article] [PubMed] [Google Scholar]
- Jarvis EE, Hagen DC, Sprague GF., Jr Identification of a DNA segment that is necessary and sufficient for alpha-specific gene control in Saccharomyces cerevisiae: implications for regulation of alpha-specific and a-specific genes. Mol Cell Biol. 1988;8:309–320. doi: 10.1128/mcb.8.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LH, Eberly SL, Chapman JW, Araki H, Sugino A. The product of the Saccharomyces cerevisiae cell cycle gene DBF2 has homology with protein kinases and is periodically expressed in the cell cycle. Mol Cell Biol. 1990a;10:1358–1366. doi: 10.1128/mcb.10.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LH, White JH, Johnson AL, Lucchini G, Plevani P. Expression of the yeast DNA primase gene, PRI1, is regulated within the mitotic cell cycle and in meiosis. Mol Gen Genet. 1990b;221:44–48. doi: 10.1007/BF00280366. [DOI] [PubMed] [Google Scholar]
- Jones EW, Fink GR. The regulation of amino acid and nucleotide biosynthesis in yeast. In: Strathern JN, Jones EW, Broach JR, editors. The Molecular Biology of the Yeast Saccharomyces: Metabolism and Gene Expression. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1982. [Google Scholar]
- Kaiser CA, Gimeno RE, Shaywitz DA. Protein secretion, membrane biogenesis, and endocytosis. In: Pringle JR, Broach JR, Jones EW, editors. The Molecular and Cellular Biology of the Yeast Saccharomyces: Cell Cycle and Cell Biology. Cold Spring Harbor NY: Cold Spring Harbor Press; 1997. pp. 91–227. [Google Scholar]
- Kaiser P, Sia RAL, Bardes EGS, Lew DJ, Reed SI. Cdc34 and the F-box protein Met30 are required for degradation of the Cdk-inhibitory kinase Swe1. Genes & Dev. 1998;12:2587–2597. doi: 10.1101/gad.12.16.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, Dyos SL, Kershaw D, Finch JT. A spacer protein in the Saccharomyces cerevisiae spindle poly body whose transcript is cell cycle-regulated. J Cell Biol. 1993;123:1175–1184. doi: 10.1083/jcb.123.5.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapholz S, Esposito RE. Isolation of SPO12-1 and SPO13-1 from a natural variant of yeast that undergoes a single meiotic division. Genetics. 1980;96:567–588. doi: 10.1093/genetics/96.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klis FM. Review: cell wall assembly in yeast. Yeast. 1994;10:851–869. doi: 10.1002/yea.320100702. [DOI] [PubMed] [Google Scholar]
- Knapp D, Bhoite L, Stillman DJ, Nasmyth K. The transcription factor Swi5 regulates expression of the cyclin kinase inhibitor p40SIC1. Mol Cell Biol. 1996;16:5701–5707. doi: 10.1128/mcb.16.10.5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C, Moll T, Neuberg M, Ahorn H, Nasmyth K. A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science. 1993;261:1551–1557. doi: 10.1126/science.8372350. [DOI] [PubMed] [Google Scholar]
- Koch C, Nasmyth K. Cell cycle regulated transcription in yeast. Curr Opin Cell Biol. 1994;6:451–459. doi: 10.1016/0955-0674(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Kovacech B, Nasmyth K, Schuster T. EGT2 gene transcription is induced predominantly by Swi5 in early G1. Mol Cell Biol. 1996;16:3264–3274. doi: 10.1128/mcb.16.7.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Weinert T, Pringle JR. Cell cycle control in Saccharomyces cerevisiae. In: Pringle JR, Broach JR, Jones EW, editors. The Molecular and Cellular Biology of the Yeast Saccharomyces: Cell Cycle and Cell Biology. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1997. pp. 607–695. [Google Scholar]
- Louis EJ. The chromosome ends of Saccharomyces cerevisiae. Yeast. 1995;11:1553–1573. doi: 10.1002/yea.320111604. [DOI] [PubMed] [Google Scholar]
- Louis EJ, Haber JE. The structure and evolution of subtelomeric Y′ repeats in Saccharomyces cerevisiae. Genetics. 1992;131:559–574. doi: 10.1093/genetics/131.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes NF, Johnson AL, Breeden L, Johnston LH. Swi6 protein is required for transcription of the periodically expressed DNA synthesis genes in budding yeast. Nature. 1992;357:505–508. doi: 10.1038/357505a0. [DOI] [PubMed] [Google Scholar]
- Lowndes NF, Johnson AL, Johnston LH. Coordination of expression of DNA synthesis genes in budding yeast by a cell-cycle regulated trans factor. Nature. 1991;350:247–250. doi: 10.1038/350247a0. [DOI] [PubMed] [Google Scholar]
- Lydall D, Ammerer G, Nasmyth K. A new role for MCM1 in yeast: cell cycle regulation of SWI5 transcription. Genes & Dev. 1991;5:2405–2419. doi: 10.1101/gad.5.12b.2405. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Lu Q, Grunstein M. A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in Saccharomyces cerevisiae. Genes & Dev. 1996;10:1327–1340. doi: 10.1101/gad.10.11.1327. [DOI] [PubMed] [Google Scholar]
- McInerny CJ, Partridge JF, Mikesell GE, Creemer DP, Breeden LL. A novel Mcm1-dependent element in the SWI4, CLN3, CDC6, and CDC47 promoters activates M/G1-specific transcription. Genes & Dev. 1997;11:1277–1288. doi: 10.1101/gad.11.10.1277. [DOI] [PubMed] [Google Scholar]
- McIntosh EM, Atkinson T, Storms RK, Smith M. Characterization of a short, cis-acting DNA sequence which conveys cell cycle stage-dependent transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:329–337. doi: 10.1128/mcb.11.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney JD, Chang F, Heintz N, Cross FR. Negative regulation of FAR1 at the Start of the yeast cell cycle. Genes & Dev. 1993;7:833–843. doi: 10.1101/gad.7.5.833. [DOI] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- Moll T, Tebb G, Surana U, Robitsch H, Nasmyth K. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 1991;66:743–758. doi: 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. At least 1400 base pairs of 5′-flanking DNA is required for the correct expression of the HO gene in yeast. Cell. 1985;42:213–223. doi: 10.1016/s0092-8674(85)80117-3. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol. 1993;5:166–179. doi: 10.1016/0955-0674(93)90099-c. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Liu JS, Lawrence CE. Gibbs motif sampling: detection of bacterial outer membrane protein repeats. Protein Sci. 1995;4:1618–1632. doi: 10.1002/pro.5560040820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlen LJ, McKinney JD, Cross FR. Ste12 and Mcm1 regulate cell cycle-dependent transcription of FAR1. Mol Cell Biol. 1996;16:2830–2837. doi: 10.1128/mcb.16.6.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlean P. Biogenesis of yeast wall and surface components. In: Pringle JR, Broach JR, Jones EW, editors. The Molecular and Cellular Biology of the Yeast Saccharomyces: Cell Cycle and Cell Biology. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1997. pp. 229–362. [Google Scholar]
- Partridge JF, Mikesell GE, Breeden LL. Cell cycle-dependent transcription of CLN1 involves swi4 binding to MCB- like elements. J Biol Chem. 1997;272:9071–9077. doi: 10.1074/jbc.272.14.9071. [DOI] [PubMed] [Google Scholar]
- Parviz F, Hall DD, Markwardt DD, Heideman W. Transcriptional regulation of CLN3 expression by glucose in Saccharomyces cerevisiae. J Bacteriol. 1998;180:4508–4515. doi: 10.1128/jb.180.17.4508-4515.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Willems AR, Sa D, Kuras L, Thomas D, Craig KL, Tyers M. Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box proteincomplexes that regulate cell division and methionine biosynthesis in yeast. Genes & Dev. 1998;12:692–705. doi: 10.1101/gad.12.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellman D, Bagget M, Tu YH, Fink GR, Tu H. Two microtubule-associated proteins required for anaphase spindle movement in Saccharomyces cerevisiae. J Cell Biol. 1995;130:1373–1385. doi: 10.1083/jcb.130.6.1373. (erratum 131, 561) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price C, Nasmyth K, Schuster T. A general approach to the isolation of cell cycle-regulated genes in the budding yeast, Saccharomyces cerevisiae. J Mol Biol. 1991;218:543–556. doi: 10.1016/0022-2836(91)90700-g. [DOI] [PubMed] [Google Scholar]
- Sanders SL, Herskowitz I. The BUD4 protein of yeast, required for axial budding, is localized to the mother/BUD neck in a cell cycle-dependent manner. J Cell Biol. 1996;134:413–427. doi: 10.1083/jcb.134.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BL, Yang QH, Futcher AB. Linkage of replication to start by the Cdk inhibitor Sic1. Science. 1996;272:560–562. doi: 10.1126/science.272.5261.560. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Cochran BH. MAT alpha 1 can mediate gene activation by a-mating factor. Genes & Dev. 1991;5:1924–1934. doi: 10.1101/gad.5.10.1924. [DOI] [PubMed] [Google Scholar]
- Serrano R, Kielland-Brandt MC, Fink GR. Yeast plasma membrane ATPase is essential for growth and has homology with (Na+ + K+), K+- and Ca2+-ATPases. Nature. 1986;319:689–693. doi: 10.1038/319689a0. [DOI] [PubMed] [Google Scholar]
- Shalon D, Smith SJ, Brown PO. A DNA microarray system for analyzing complex DNA samples using two- color fluorescent probe hybridization. Genome Res. 1996;6:639–645. doi: 10.1101/gr.6.7.639. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. In: Fink CGaGR., editor. Guide to Yeast Genetics and Molecular Biology. San Diego: Academic Press; 1991. pp. 3–21. [Google Scholar]
- Siede W, Robinson GW, Kalainov D, Malley T, Friedberg EC. Regulation of the RAD2 gene of Saccharomyces cerevisiae. Mol Microbiol. 1989;3:1697–7707. doi: 10.1111/j.1365-2958.1989.tb00155.x. [DOI] [PubMed] [Google Scholar]
- Strunnikov AV, Larionov VL, Koshland D. SMC1: an essential yeast gene encoding a putative head-rod-tail protein is required for nuclear division and defines a new ubiquitous protein family. J Cell Biol. 1993;123:1635–1648. doi: 10.1083/jcb.123.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Cherest H, Surdin-Kerjan Y. Elements involved in S-adenosylmethionine-mediated regulation of the Saccharomyces cerevisiae MET25 gene. Mol Cell Biol. 1989;9:3292–3298. doi: 10.1128/mcb.9.8.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyn JH, Araki H, Sugino A, Johnston LH. The cell-cycle-regulated budding yeast gene DBF2, encoding a putative protein kinase, has a homologue that is not under cell-cycle control. Gene. 1991;104:63–70. doi: 10.1016/0378-1119(91)90465-n. [DOI] [PubMed] [Google Scholar]
- Toyn JH, Johnston LH. Spo12 is a limiting factor that interacts with the cell cycle protein kinases Dbf2 and Dbf20, which are involved in mitotic chromatid disjunction. Genetics. 1993;135:963–971. doi: 10.1093/genetics/135.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger MW, Hartwell LH. Control of cell division in Saccharomyces cerevisiae by methionyl-tRNA. Proc Natl Acad Sci USA. 1976;73:1664–1668. doi: 10.1073/pnas.73.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Xu H, Grunstein M. CDC14 of Saccharomyces cerevisiae. Cloning, sequence analysis, and transcription during the cell cycle. J Biol Chem. 1992;267:11274–11280. [PubMed] [Google Scholar]
- Wang ZF, Whitfield ML, Ingledue TC, III, Dominski Z, Marzluff WF. The protein that binds the 3′ end of histone mRNA: a novel RNA-binding protein required for histone pre-mRNA processing. Genes & Dev. 1996;10:3028–3040. doi: 10.1101/gad.10.23.3028. [DOI] [PubMed] [Google Scholar]
- White JH, Green SR, Barker DG, Dumas LB, Johnston LH. The CDC8 transcript is cell cycle regulated in yeast and is expressed coordinately with CDC9 and CDC21 at a point preceding histone transcription. Exp Cell Res. 1987;171:223–231. doi: 10.1016/0014-4827(87)90265-5. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Guacci V, Koshland D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s) J Cell Biol. 1996;133:99–110. doi: 10.1083/jcb.133.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.