

Summary
Duffy antigen receptor for chemokines (DARC) expressed on red blood cells (RBCs) influences plasma levels of HIV-1-suppressive and proinflammatory chemokines such as CCL5/RANTES. DARC is also the RBC receptor for Plasmodium vivax. Africans with DARC −46C/C genotype, which confers a DARC-negative phenotype, are resistant to vivax malaria. Here, we show that HIV-1 attaches to RBCs via DARC, effecting trans-infection of target cells. In African Americans, DARC −46C/C is associated with 40% increase in the odds of acquiring HIV-1. If extrapolated to Africans, ∼11% of the HIV-1 burden in Africa may be linked to this genotype. After infection occurs, however, DARC-negative RBC status is associated with slower disease progression. Furthermore, the disease-accelerating effect of a previously described CCL5 polymorphism is evident only in DARC-expressing and not in DARC-negative HIV-infected individuals. Thus, DARC influences HIV/AIDS susceptibility by mediating trans-infection of HIV-1 and by affecting both chemokine-HIV interactions and chemokine-driven inflammation.
Introduction
Host genetic polymorphisms contribute to interindividual variation in susceptibility to acquiring HIV-1 infection, the degree of infectiousness to others (as reflected by the viral load), and rates of disease progression. Such polymorphisms might also contribute to significant interpopulation differences in HIV-1 prevalence (Gonzalez et al., 1999, 2005; Schliekelman et al., 2001; Sullivan et al., 2001; Winkler et al., 2004), since sexual behavior and other social factors do not fully explain regional variations of the HIV-1 pandemic (Buve, 2002; Kinoshita-Moleka et al., 2007; Weiss and McMichael, 2004). Thus, identification of transmission and disease modifying polymorphisms that are found more frequently in specific populations may provide insight into the host genetic factors that contribute to the heterogeneity of HIV-AIDS pandemic and has broad public health relevance, as failure to account for such polymorphisms may confound the evaluation of therapeutics, microbicides, and vaccines (Kellam and Weiss, 2006; Pepin, 2005).
Host genetic influence on HIV-AIDS susceptibility by a polymorphism that is differentially distributed between populations is best illustrated by the effects of the European-specific 32 bp deletion (Δ32) in the gene for CC chemokine receptor 5 (CCR5), which is the major coreceptor for R5 strains of HIV-1 that represent the majority of transmissible viruses (Lama and Planelles, 2007). The CCR5-Δ32/Δ32 genotype abrogates cell-surface expression of CCR5 and is associated with profound resistance to HIV infection. Heterozygosity for CCR5-Δ32 is associated with a reduction in risk of acquisition of R5 HIV and with a trend toward a slower rate of progression to AIDS (Gonzalez et al., 1999; Lama and Planelles, 2007; Mangano et al., 2001). It has been proposed (Schliekelman et al., 2001; Sullivan et al., 2001) that the absence of the protective CCR5-Δ32/Δ32 genotype might have contributed to the accelerated spread of HIV in African populations, although this notion is debatable considering the relatively low prevalence of this genotype in European populations (∼1%–3%).
Other as-yet-undefined host factors are expected to have population-specific effects on HIV-AIDS and possibly also contribute to the generally high prevalence of HIV in sub-Saharan Africa. Defining whether there is a genetic basis for the higher incidence of HIV in Africa is especially relevant for two reasons. First, racial differences in incidence and outcome of inflammatory disorders such as sepsis have been observed (Martin et al., 2003). Second, recent studies suggest that compared to individuals of European descent, people of African descent might have a genetic profile that favors a “high-expression profile” of proinflammatory cytokines and a “low-expression profile” of anti-inflammatory cytokines (Mayr et al., 2008; Ness et al., 2004). In light of the aforementioned, and because of the importance of the chemokine system in inflammation and HIV-AIDS pathogenesis (Murphy et al., 2000) and the reasons outlined below, in this study we focused on the impact of variation in the gene encoding Duffy Antigen Receptor for Chemokines (DARC) on risk of acquiring HIV and on disease progression.
DARC is expressed mainly on red blood cells (RBCs) as well as on vascular endothelial and neuronal cells (Hadley and Peiper, 1997). DARC is characterized by its ability to bind a wide array of proinflammatory chemokines (Comerford et al., 2007; Gardner et al., 2004; Rot, 2005) including CCL5 (RANTES), which is highly effective in suppressing HIV-1 replication. However, binding of chemokines to DARC on RBCs does not trigger a signaling cascade because DARC lacks the G protein-signaling domain (Comerford et al., 2007; Rot, 2005). Instead, the promiscuous binding of chemokines to DARC is viewed as a mechanism for regulating plasma levels of chemokines (Fukuma et al., 2003; Jilma-Stohlawetz et al., 2001; Mayr et al., 2008) and, consequently, inflammation (Lee et al., 2006; Mantovani et al., 2006). The observation that DARC substantially influences erythrocyte-bound and plasma chemokine concentrations following administrations of endotoxin (lipopolysaccharide, LPS) (Mayr et al., 2008) is relevant in light of the recent association between LPS and immune activation and HIV pathogenesis (Douek, 2007). Thus, these characteristics of DARC suggest that it could also modulate the levels of HIV-suppressive chemokines and inflammation observed during HIV disease.
It has been proposed that HIV might bind to RBCs by two different mechanisms. First, it was found that HIV-1 can bind to RBCs in vitro and that this binding is mediated through DARC (Lachgar et al., 1998). Supporting this, HIV bound to RBCs has been observed in HIV-infected subjects who have undetectable plasma viral load during highly active antiretroviral therapy (HAART), and this is associated with a poor clinical outcome (Hess et al., 2002). The second is by indirect association of opsonised HIV particles via HIV/anti-HIV immune complexes and complement factor c3b binding to CR1 (CD35) (Banki et al., 2006; Horakova et al., 2004). To further evaluate the impact of DARC on HIV-AIDS pathogenesis, in this study we investigated whether a polymorphism in DARC that has been intensively scrutinized in the context of malaria (Tournamille et al., 1995) also influences HIV-AIDS susceptibility.
The single nucleotide polymorphism (SNP) at the −46 position (T−46C) in the promoter of DARC is widely prevalent in populations of African descent. The −46C allele disrupts the binding site for the erythroid-specific transcription factor GATA-1, and consequently, the −46C/C DARC promoter genotype results in selective loss of expression of DARC on RBCs but not other cell types on which DARC is expressed (Tournamille et al., 1995). Interestingly, this polymorphism influences pathogenesis of another major infectious disease, malaria caused by Plasmodium species. As DARC serves as the receptor on RBCs for merozoites of Plasmodium vivax (Horuk et al., 1993), Africans with the DARC −46C/C genotype are highly resistant to malaria caused by this Plasmodium strain (Tournamille et al., 1995). It is noteworthy that malaria influences the spread of HIV infection, and these two infections together contribute to significant morbidity and mortality in regions devastated by the HIV epidemic (Abu-Raddad et al., 2006).
Here, we show that DARC mediates the binding of HIV to RBCs and the subsequent transfer of virus to HIV-1 target cells. We coupled these in vitro studies with a detailed genetic epidemiological analysis to determine whether the T−46C polymorphism in DARC affects susceptibility to HIV-AIDS of African Americans. Our results affirm that it does, as well as demonstrating a complex interaction between DARC, chemokines, and HIV in vivo.
Results
DARC Binds and Transfers HIV-1 to Target Cells
DARC+ and DARC− RBCs (as in Figure 1A) were incubated with various HIV-1 strains, washed, and then cocultured with HIV-susceptible NP2 cells that express the HIV receptor CD4 as well as either CCR5 or CXCR4 as described in Experimental Procedures. The extent of carriage of HIV by RBCs differed according to coreceptor usage (R5, R5X4, and X4) (Figure 1B). In general, transfer of R5 strains was less than 1% of the infecting inoculum, whereas it was significantly greater for R5X4 and X4 strains, approaching ∼50% for SF2, an X4 strain (Figure 1B). Of these isolates, SL-2 (R5), 2076, 2028 (R5X4), and 2044 (X4) are all primary isolates that have been minimally passaged in peripheral blood mononuclear cells (PBMCs) (Simmons et al., 1996). Similar HIV transfer results were obtained when PBMCs were used as target cells, and HIV reverse transcriptase (RT) activity in the cell supernatants was used as measure of infection (Figure 1C).
Figure 1. DARC Binds and Transfers HIV-1 to Target Cells.

(A) Representative flow-cytometric analysis of RBCs for immunostaining of DARC. RBCs were stained with the DARC monoclonal antibody anti-Fy6 (red) or an isotype control (violet).
(B) Purified human DARC+ RBCs as shown in (A) were incubated with equal amounts of HIV-1 strains with varying coreceptor usages, washed, and added to target NP2 cells bearing CD4 and the appropriate coreceptor (CCR5 or CXCR4). Viral transfer was quantified as the percentage of the input titer recovered from RBCs.
(C) Time course of RT production in human PBMCs cocultured with HIV-loaded RBCs. Using methods similar to those described for (B), HIV-loaded RBCs were cocultured with fresh-activated human PBMCs, and infection was followed by RT output over 9 days.
(D) Relative efficiency of transfer of HIV to donor cells by DARC+ versus DARC− RBCs. RBCs from known DARC+ and DARC− donors as shown in (A) were incubated with PBMCs as in (C), and supernatant RT activity was measured 5 days postinfection. The bars represent the ratio of transfer efficiency of DARC+ RBCs divided by that of DARC− RBCs.
(E) Attachment of the R5X4 strain 89.6 to RBCs was inhibited by rhCCL5 (RANTES; 1 nM), rhCXCL8 (IL-8; 1 nM), and the anti-DARC monoclonal Fy6 (anti-Fy6), but not by rhCCL3 (MIP-1αS; 1 nM). Data are presented as percentage of cell-associated RT activity compared to the untreated RBCs (gray).
(F) rhCXCL8, but not rhCCL3, inhibited transfer of infectious HIV-1 strains to fresh human PBMCs, shown as percent RT output compared to untreated RBCs 5 days postinfection.
(G) Transfer efficiency of SF162 by RBCs to target NP2.CD4.R5 was increased by the addition of 1 and 5 μg/ml of sCD4 compared to media alone (0). Data in (B), (E), and (G) are mean ± SEM, as indicated by error bars, while data in (C), (D), and (F) are from one representative experiment from over three separate experiments conducted.
DARC+ RBCs transferred HIV-1 strains (R5, X4, and R5X4) with 5- to 12-fold more efficiency than DARC− RBCs (Figure 1D), suggesting that DARC provided an HIV-1 binding site to human RBCs. To establish that these effects were mediated specifically by DARC, we investigated whether a DARC antibody or chemokine ligands for DARC can compete for the attachment/binding of HIV-1 to DARC on RBCs. The DARC ligands CXCL8 (IL-8) and CCL5 (RANTES) significantly reduced the attachment of HIV-1 to DARC+ RBCs, whereas CCL3 (MIP-1αS), which is not a ligand for DARC, had minimal effects (Figures 1E and 1F). Interestingly, even R5 strains (SL-2 and BAL) that demonstrated less efficient transfer (Figure 1B) to NP2/CD4/CCR5 cells were sensitive to blockade by CXCL8 (Figure 1F). Monoclonal antibody directed against the DARC epitope Fy6 (anti-Fy6) moderately outcompeted HIV attached to RBCs (Figure 1E). Collectively, these findings demonstrated that HIV-1 virions bind to RBCs in vitro and that RBCs mediate the transfer and infectivity of RBC-bound HIV to target cells predominantly via DARC. The attachment and/or transfer process appeared to be more efficient for R5X4 and X4 HIV strains than R5 strains.
As shown in Figure 1B, R5 strains of HIV-1 were much less efficiently transferred by RBCs than those with X4 tropism or dual tropism, probably because the envelope protein conformation of R5 strains occludes the coreceptor binding site on gp120. Interaction of gp120 with CD4 causes a conformational change that exposes these hidden epitopes and induces binding to chemokine coreceptors. We therefore tested whether soluble CD4 could enhance the binding of the R5 strain SF162 to RBCs and observed a 10-fold increase in attachment and transfer (Figure 1G). This finding implicates specific chemokine receptor binding sites of gp120 for adsorption of virions to DARC-positive RBCs.
As DARC is also expressed on endothelial cell barriers, especially in the brain, we assessed the role of DARC as a functional HIV-1 coreceptor. In NP2/CD4 cells that overexpress DARC, none of the HIV-1 isolates shown in Figure 1B could utilize DARC as an entry coreceptor (data not shown). These results are consistent with previous findings (Doranz et al., 1996).
Distribution of DARC T−46C in Worldwide Populations
To place the results of the genotype-HIV/AIDS phenotype study described below in a broader evolutionary context, we first determined the distribution of the T−46C DARC polymorphism in the well-characterized populations that comprise the Human Genome Diversity Project (HDGP)-CEPH DNA set. In the HGDP-CEPH samples, the presence of the mutated DARC −46C/C genotype was highly specific to subjects of African descent (Figure 2A). The pattern of distribution of DARC genotypes in African Americans from the Wilford Hall Medical Center (WHMC) cohort mimicked those found in the African CEPH DNA samples (Figure 2B).
Figure 2. Distribution of the DARC −46C/C in Worldwide Populations and Its Association with Risk of Acquiring HIV Infection in the African-American Component of the HIV+ WHMC Cohort.
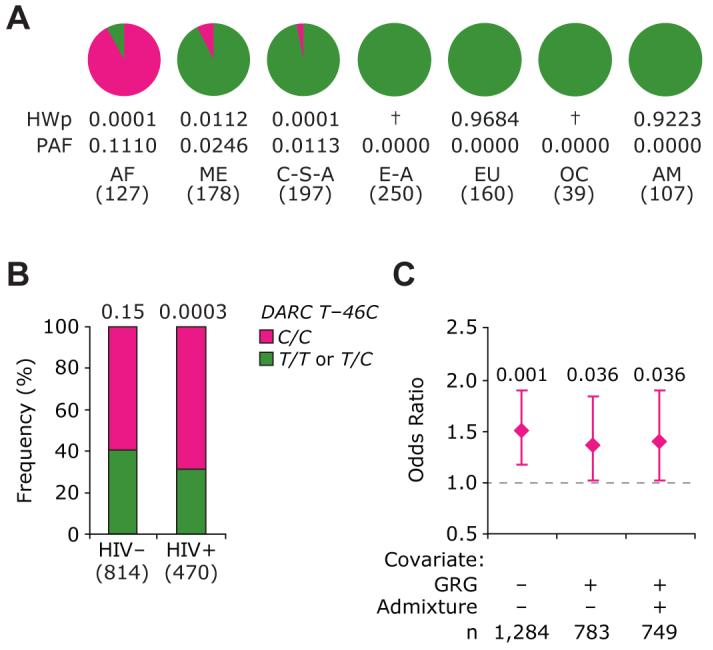
(A) Distribution of and PAF for the DARC −46C/C genotype in different populations represented in the HGDP-CEPH DNA samples. Pie charts show the distribution of the −46C/C genotype in seven populations: AF (Africa), ME (Middle East), C-S-A (Central South Asia), E-A (East Asia), EU (Europe), OC (Pacific Ocean), and AM (America). HWp: Hardy-Weinberg test P value. †, minor allele was absent and HWp not applicable.
(B) Distribution of the color-coded DARC −46C/C genotype in the WHMC HIV-negative (HIV−) and HIV-positive (HIV+) African Americans from the WHMC cohort. The numbers at the top of each bar represent HWp.
(C) Risk of acquiring HIV based on the T−46C genotype in African-American subjects (n = 1284) from the WHMC cohort. The diamonds and error bars show the odds ratio and 95% confidence intervals for risk of acquiring HIV in subjects possessing the DARC −46C/C genotype (DARC− on RBCs) relative to the subjects possessing the DARC −46T/C or T/T genotype (DARC+ on RBCs). Numbers at the top of the error bars are significance values. The results are from multivariate logistic regression analyses. Three models were assessed: first with no covariates, then we added the CCL3L1-CCR5 GRGs as a covariate, and finally we added the degree of population admixture as another covariate. Number of subjects in each genotypic group is shown in parentheses for (A) and (B) and at the bottom of (C), respectively.
However, African Americans are an admixed population and consistent with previous data (Lautenberger et al., 2000), African Americans in the WHMC cohort also have admixture of the −46T allele such that between 30% and 40% of African Americans are phenotypically DARC+ (that is, have at least one −46T allele). Therefore, in the succeeding genotype-phenotype analyses, we accounted for possible confounding effects due to population admixture. Based on 11 genetic markers that are differentially distributed between European and African populations (details provided in Supplemental Data), we predicted the self-reported ethnicity using a multivariate logistic regression model. The model predicted the self-reported ethnicity with an overall accuracy of 98.1% (Supplemental Data). Using results from this model, we generated a probabilistic score that reflects the ethnicity as predicted by these genetic markers. The distribution of this score was similar among HIV+ and HIV− African Americans, indicating that the degree of admixture between these two groups of subjects was similar (Supplemental Data). Thus, wherever appropriate, we adjusted the genotype-phenotype association of the T−46C DARC polymorphism for population admixture by using this score (admixture score) as a covariate in the statistical analyses.
DARC −46C/C Genotype and HIV Susceptibility
The DARC −46C allele was in Hardy-Weinberg (H-W) equilibrium in HIV− African Americans but was in HW disequilibrium in HIV+ African Americans (Figure 2B). This suggested a possible association between the −46C allele and an altered risk of acquiring HIV infection in African Americans. Consistent with this, the prevalence of the −46C/C genotype was greater in HIV+ than HIV− African Americans (Figure 2B), and relative to those possessing a −46T allele (i.e., DARC −46T/T or T/C). African Americans with a DARC −46C/C genotype had ∼50% higher risk of acquiring HIV (Figure 2C). We used this categorization of DARC T −46C genotypes because −46C/C is not associated with DARC expression on RBCs, whereas −46T/T or T/C genotypes are.
In a previous study, we categorized CCR5 genotypes and the copy number of CCL3L1, the most potent HIV-suppressive CCR5-binding chemokine, into three CCL3L1-CCR5 genetic risk groups (GRG) (Dolan et al., 2007). Although the CCL3L1 and CCR5 genotypes are key determinants of HIV acquisition (Gonzalez et al., 2005; Kuhn et al., 2007), even after adjustment for the transmission-modifying effects of the CCL3L1-CCR5 GRGs, the enhanced risk of acquiring HIV associated with DARC −46C/C persisted (Figure 2C). Furthermore, when we adjusted for population admixture, the excess risk of HIV acquisition in subjects possessing the −46C/C genotype was still statistically significant, and the results of the final model suggested that possession of this genotype is associated with ∼40% higher odds of acquiring HIV than those lacking this genotype (Figure 2C). This indicated that DARC −46C/C is an independent determinant of increased risk of HIV acquisition in African Americans, and the excess risk conferred by possession of this genotype is not confounded by population admixture.
We examined how the excess risk of acquiring HIV associated with possession of the DARC −46C/C in subjects of African descent might translate to the increased HIV burden attributable to this genotype in worldwide populations. For this, we used the odds for risk of HIV acquisition and the prevalence of the DARC −46C/C genotype in the populations that comprise the HGDP-CEPH sample to compute the population attributable fractions (PAFs). These data (Figure 2A and Table S1) show the PAFs for excess HIV burden attributable to DARC −46C/C is high (∼11%) in African settings. To place these rather high PAFs in context, the PAF for reduction in risk of transmission by heterozgosity for the CCR5 Δ32 protective mutation in European populations (prevalence between 10%–15%) was only 2.6% (Table S1).
DARC −46C/C and Disease Progression
Although the DARC −46C/C genotype was associated with an increased risk of acquiring HIV infection, within the context of established HIV infection a contrary result was observed: subjects possessing the −46C/C genotype had a slower disease course (Figures 3A and 3B). This relatively slow disease course was statistically more pronounced and significant when the Cox regression models included the following covariates: CCL3L1-CCR5 GRGs, which we have shown previously, have a significant impact on rate of disease progression (Dolan et al., 2007; Gonzalez et al., 2005); the population admixture score; and the three HLA alleles (HLA-A*68, HLA-B*57, and HLA-C*16), which we found previously were independent determinants of disease course in WHMC HIV+ subjects (Ahuja et al., 2008).
Figure 3. Influence of the DARC T−46C Polymorphism on HIV Disease Progression in the African-American Component of HIV + WHMC Cohort.
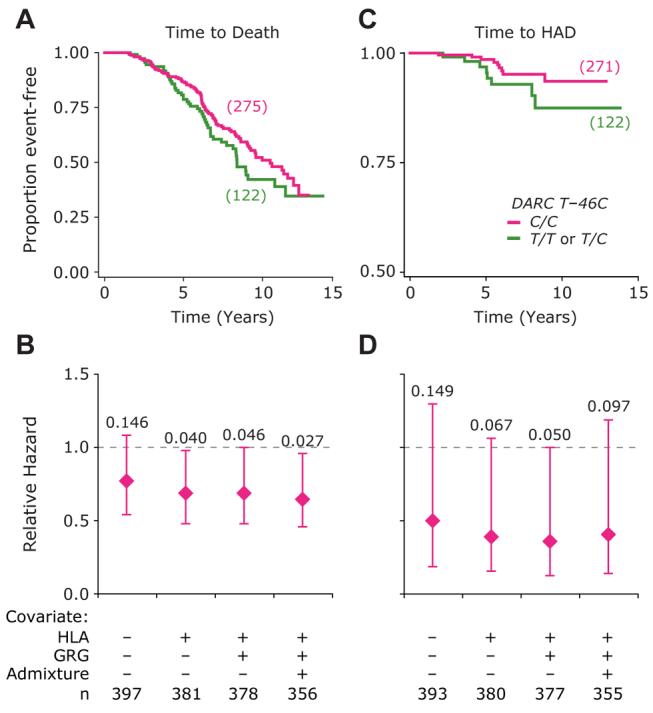
(A) Kaplan-Meier plots for time to death based on possession of −46C/C genotype (pink) or possession of −46T/C or −46T/T genotype (green).
(B) Results of multivariate Cox proportional hazards models for rate of disease progression in subjects possessing the DARC −46C/C genotype (pink) relative to the subjects possessing the DARC −46T/C or T/T genotype (green; relative hazard [RH] = 1). The following three covariates were sequentially included in the model: (1) HLA refers to possession of the following three HLA alleles (HLA-A*68, HLA-B*57, and HLAC*16); (2) CCL3L1-CCR5 GRGs; and (3) degree of population admixture. The diamonds and error bars represent the RHs and 95% confidence intervals (CI) for progression to death. Numbers at the top are significance values. These three HLA alleles associate with a rapid rate of disease progression in the WHMC cohort (Ahuja et al., 2008).
(C) Kaplan-Meier plots for time to HIV-associated dementia (HAD) based on possession of the −46C/C genotype (pink) or possession of −46T/C or −46T/T genotype (green).
(D) The results of multivariate Cox proportional hazards modeling similar to those shown in (B) but for the outcome of HAD. Number of subjects in each genotypic group is shown in parentheses for (A) and (C) and at the bottom of (B) and (D), respectively.
In addition to erythroid cells, DARC is also expressed on subsets of neurons and on endothelial cells at the blood-brain barrier (Hadley and Peiper, 1997). DARC-negative humans display reduced plasma levels of CCL2 (MCP-1) compared to DARC-positive individuals (Jilma-Stohlawetz et al., 2001; Mayr et al., 2008), and in a previous study, we found that a CCL2 genotype associated with increased MCP-1 expression was associated with an increased risk of HIV-associated dementia (HAD) (Gonzalez et al., 2002). We therefore examined whether DARC expression influenced HAD. In accord with its generally protective effect on HIV disease, DARC −46C/C also protected against the rate of progression to HAD (Figure 3C). Furthermore, this protective effect persisted even after adjusting for the GRGs, HLA alleles, and population admixture in multivariate models (Figure 3D). Since the −46C/C genotype affects DARC expression only on RBCs, this finding suggested that the dominant effect of DARC on HIV-AIDS pathogenesis is due to its expression on RBCs and not in the brain.
Population Level Impact of DARC −46C/C on HIV Disease
We compared CD4+ T cell loss and the rate of disease progression in African Americans who possessed or lacked the DARC −46C/C genotype versus the overall European American component of the WHMC HIV+ cohort. Consistent with the protective effects of −46C/C genotype on disease progression, the rate of CD4+ T cell loss was significantly slower in HIV+ African Americans with the DARC −46C/C genotype compared to all other African Americans (Figure 4A). For this reason, we inquired whether the protective effects associated with the African-specific −46C/C genotype might contribute to the finding that HIV+ African Americans have a slight survival advantage compared to European Americans (Figure 4B). A similar survival advantage during the preHAART era was also observed for HIV+ African Americans in the larger Tri-Service AIDS Clinical Consortium (TACC) cohort (Silverberg et al., 2006) of which the WHMC is part. African Americans lacking DARC −46C/C had a disease course that was similar to that of the overall European American component of the cohort, which was significantly faster than that for HIV+ African Americans with the −46C/C genotype (Figure 4C). Concordantly, the rate of CD4+ T cell loss for the overall HIV+ European Americans was similar to that of HIV+ African Americans who lacked the −46C/C genotype (Figure 4A).
Figure 4. Contribution of DARC −46C/C to Ethnicity-Specific Differences in Rates of Disease Progression in the HIV+ WHMC Cohort.

(A) Time course of CD4+ T cell count decline in HIV+ subjects possessing the indicated DARC genotypes. The panel shows Loess curves for HIV+ African Americans with the indicated genotypic groups: −46C/C (pink), −46T/T, or T/C (green),and the overall group of HIV+ European Americans (blue). The average rate of decline in CD4+ T cell count (cells/month) was estimated using GEE, and it is shown at the bottom of the panel. SE stands for standard error and P for significance value for the difference between the rates of CD4+ T cell decline as assessed by Student's t test and using European Americans as the reference group. Data are from subjects who did not receive antiretroviral therapy. n, numbers of subjects; m, numbers of CD4+ T cell count measurements.
(B–D) The Kaplan-Meier plots showing the differences in rate of progression to death (B) between European Americans (blue) and African Americans (gray) in the WHMC HIV+ cohort. (C) shows African Americans with (pink) and without (green) the DARC −46C/C genotype and European Americans (blue); (D) shows African Americans with (pink) and without (green) the −46C/C genotype and European Americans with (brown) and without (blue) the CCR5Δ32 mutation. RH = 1.00 indicates the reference group used in statistical analysis. Numbers of subjects in each genotypic group in (B–D) are shown in parentheses. EAs, European Americans. AAs, African Americans.
To place these results within the perspective of the highly scrutinized European-specific CCR5-Δ32 mutation, we compared the disease-modifying effects associated with the DARC −46C/C genotype in African Americans and CCR5-Δ32 heterozygosity in European Americans. In subjects from the WHMC HIV+ cohort, the survival advantage conferred by DARC −46C/C in African Americans was comparable to that associated with CCR5-Δ32 heterozygosity in European Americans (Figure 4D). Furthermore, African Americans lacking the −46C/C genotype and European Americans lacking the CCR5-Δ32 mutation had similar rates of disease progression (Figure 4D). Taken together, these findings indicate that in the context of HIV disease, the African American-specific DARC −46C/C genotype not only mimicked the disease-retarding effects associated with the European American-specific CCR5-Δ32 mutation, but also contributed to the modest survival advantage observed in the overall group of HIV+ African Americans (Figure 4B).
Interactions between DARC, CCL5, and HIV
To determine whether there was an interaction between DARC, chemokines, and HIV in vivo, we focused on a functional SNP in CCL5 (−471G/A; rs2107538), which is associated with increased RANTES expression levels (Liu et al., 1999). In a previous study (Gonzalez et al., 2001), we found that CCL5 −471A/A-containing genotypes are associated with a faster rate of disease progression to AIDS, but surprisingly, this association was evident only in European Americans but not African Americans. Here, using time to death as a phenotypic endpoint, we confirmed that there were no significant differences in disease progression rates between those African Americans who were CCL5 −471A/A versus African Americans who lack this CCL5 genotype (Figure 5A). Additionally, when we compared the disease-influencing effects associated with the CCL5 −471A/A genotype in European Americans versus African Americans, we found that this genotype was associated with differential effects in these two populations. European Americans with CCL5 −471A/A had a faster rate of disease progression than African Americans with the same genotype (Figure 5B).
Figure 5. Proof-of-Concept Genetic Epidemiologic Studies Illustrating DARC-Chemokine-HIV-1 Interplay.
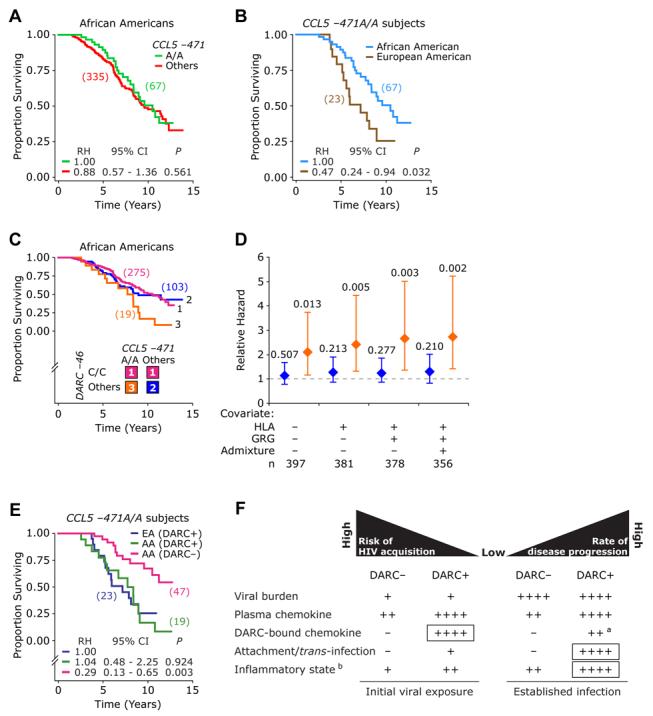
(A) The disease-influencing effect of CCL5 −471G > A SNP in HIV+ African Americans before accounting for DARC genotype. The analysis compares CCL5 −471A/A versus −471A/G or −471G/G (others).
(B) Disease-influencing effects of CCL5 −471A/A genotype in European Americans and African Americans.
(C and D) Disease-influencing effects in African Americans of the three color-coded DARC-CCL5 genotypic groups. (C) shows Kaplan-Meier plots for association with time to death. (D) shows the results of multivariate Cox proportional hazards models that examined the association of the three DARC-CCL5 genotypic groups shown in (C) with time to death. The Cox models used the subjects possessing the DARC −46C/C genotype as the reference group. The covariates used in the models were HLA alleles (A*68, B*57, and C*16), CCL3L1-CCR5 GRGs, and the degree of population admixture. The diamonds and error bars represent the RH and 95% confidence intervals for progression to death was obtained using Cox proportional hazards models. Numbers at the top of the error bars are significance values.
(E) Disease-influencing effects of CCL5 −471A/A genotype in European Americans (EAs) or African Americans (AAs) according to DARC-expression status on RBCs inferred by DARC genotypes (DARC expression on RBCs, i.e., DARC+ in those with −46T/T or −46T/C and DARC nonexpression on RBCs [DARC−] in those with DARC −46C/C). The number of subjects in each genotypic group is shown in parentheses for (A), (B), (E), and at the bottom of (D), respectively.
(F) A hypothetical model of DARC-influencing effects on susceptibility to risk of HIV acquisition (left) and disease progression (right) based on (1) results of the present study, (2) published literature regarding the impact of DARC on plasma and erythrocyte bound-chemokine levels, and (3) the known role of inflammation in HIV pathogenesis. The model is discussed in detail in the text. DARC+ and DARC_are as described in (E). aDARC-bound chemokine levels are anticipated to be lower during established HIV infection in DARC+ individuals than in DARC+ individuals at time of initial viral exposure. This is because during established HIV infection, HIV-1 will compete for DARC-bound chemokines. bChemokine-mediated inflammatory state is shown; boxes designate the dominant effects of DARC at time of initial exposure to virus and during established infection.
How might one explain these differential effects of the CCL5 −471A/A genotype in European Americans and African Americans? Given the interactions between DARC, chemokines, and HIV in vitro, we hypothesized that the differential effects of the CCL5 −471A/A genotype in European Americans and African Americans might relate to differences in DARC-expression status. We postulated that the protective effects of the DARC-negative state (−46C/C genotype) on HIV disease progression in African Americans mask the detrimental effect of the CCL5 −471A/A genotype in DARC-expressing African Americans (−46 T/C or T/T genotype). By contrast, as almost all European Americans possess the DARC-expressing genotype (−46T/C or T/T), the aforementioned masking effect of the −46C/C genotype in African Americans will be absent in European Americans. If true, the CCL5 −471A/A should associate with similar disease-accelerating effects in DARC-expressing individuals, regardless of their ethnic background.
We undertook two approaches to test our hypothesis. In the first approach, we examined the effects of CCL5 −471A/A in African Americans after accounting for DARC-expression status. For this, we categorized the African American component of the WHMC cohort into three DARC-CCL5 genotypic groups (Figure 5C): group 1, African Americans lacking DARC expression (−46C/C); group 2, African Americans expressing DARC (−46T/T or T/C) and lacking the CCL5 −471A/A genotype; and group 3, African Americans expressing DARC and with the CCL5 -471A/A genotype. We found that DARC-CCL5 genotypic groups 1 and 2 associated with similar rates of disease progression, which were significantly slower than those associated with DARC-CCL5 genotypic group 3 (Figure 5C). Of note, these effects were more significant and pronounced after adjusting for HLA alleles, CCL3L1-CCR5 GRGs, and the population admixture score (Figure 5D). As a second approach, we examined the disease-modifying effects of CCL5 −471A/A in European Americans and African Americans after accounting for DARC status. These analyses showed that the CCL5 −471A/A genotype associates with disease acceleration in DARC-expressing subjects regardless of ethnicity (Figure 5E), compared to African Americans who lack expression of DARC and possess CCL5 −471A/A. These findings affirm our hypothesis stated above and provide a genetic-epidemiologic proof of concept for the complex interactions between DARC, chemokines, and HIV in vivo.
Discussion
The association of HIV-1 virions with cell-surface factors on macrophages and dendritic cells (DC) has been shown to protect the virus from degradation and promote viral dissemination (Lekkerkerker et al., 2006; Levy, 2002), but the full repertoire of these factors remains unknown. Here we demonstrate that HIV-1 adsorbs to DARC on the surface of RBCs and transfers virus to target cells, indicating that RBCs might act as carriers of infectious HIV-1 particles to susceptible cells such as CD4+/CCR5+ T lymphocytes. Although this process was most marked for X4-tropic strains, alteration of gp120 conformation of R5 virus by soluble CD4+ significantly increased the binding of HIV to DARC. HIV-1 did not infect CD4+/CCR5− or CD4+/CXCR4− cells transfected with DARC. Collectively, these data suggest that DARC might act like DC-SIGN on DCs (Lekkerkerker et al., 2006) in serving as a tether rather than as a functional entry receptor, although subsequent sequestration and formation of immunological synapses presumably do not occur with RBCs.
To determine whether the in vitro findings have in vivo relevance, we studied the impact of a polymorphism in DARC in a well-characterized cohort of HIV-infected individuals in which confounding factors (e.g., unequal access to health care and unequal socioeconomic status) have been minimized (Dolan et al., 2007; Gonzalez et al., 2005). Contrary to the CCR5-Δ32/Δ32 genotype, which abrogates CCR5 expression and confers protection against HIV transmission in Europeans, we find that the African American-specific DARC −46C/C genotype that abrogates DARC surface expression on RBCs is associated with a 40% increase in the odds of HIV acquisition. Whereas, paradoxically, once infection occurs, the DARC −46C/C genotype confers a survival advantage to African Americans that is comparable in magnitude to the survival advantage conferred by the CCR5-Δ32 genotype to European Americans. Whether the DARC −46C/C genotype also increases the risk of infection and conveys a survival advantage to African populations outside the US remains unknown but would appear likely. If similar effects of the DARC −46C/C genotype on HIV acquisition were to be conveyed in Africans, ∼11% of the HIV burden in these populations may be attributable to this genotype. To validate this hypothesis conclusively, one would need to study subjects in Africa in whom the DARC −46C/C genotype has not attained fixation and in whom other confounding factors, including exposure risk, had been accounted for. Unfortunately, such populations are difficult to identify in Africa because in sub-Saharan black Africans, the −46C/C genotype is almost universal.
The sum of the in vitro and genetic epidemiologic findings demonstrates that HIV might exploit DARC to its advantage: binding of HIV to DARC, a molecule that is expressed on one of the most abundant cell types (RBCs), might afford a unique biological niche that favors viral survival and persistence. Underscoring this possibility is the observation by Hess et al., (2002), who detected RBC-bound HIV in the absence of detectable plasma virus. Thus, it is likely that the net effect of the relationship between DARC and chemokines on HIV disease in vivo is likely to be much more complex, as it seems that DARC regulates the biological effects of chemokines in three distinct ways—either through scavenging, retention, or transportation (Comerford et al., 2007; Rot, 2005). Hence, based on the known functions of DARC as well as the results presented herein, we hypothesize that the ultimate impact of DARC on HIV-AIDS pathogenesis is a complex interplay between levels of DARC, circulating chemokines, and HIV virions.
The few studies that have examined the in vivo impact of DARC genotype on free and erythrocyte-bound chemokine levels paint a very complicated picture. The most definitive work to date is by Jilma and colleagues (Mayr et al., 2008), who determined plasma- and RBC-associated chemokines in those who do (DARC+) or do not (DARC−) express DARC on RBCs, before and after administration of LPS. They found that at baseline (pre-LPS), compared to DARC+ subjects, DARC− individuals had lower plasma- and RBC-associated MCP-1 levels (Mayr et al., 2008). Post-LPS, plasma MCP-1 levels were 2-fold lower in DARC− compared to DARC+ individuals. Consequently, DARC+ individuals—by virtue of having higher chemokine levels during endotoxemia—have a transient proinflammatory state. Additionally, Fukuma et al., (2003) show that DARC genotype influences clearance mechanisms for soluble chemokines as DARC-knockout mice have a more rapid clearance of chemokines injected intravenously with chemokines than their wild-type counterparts. These findings underscore that DARC substantially alters chemokine concentrations in blood with the RBC DARC-phenotype favoring a low chemokine status. These results invoke that one has to consider the complexity of how the amount of plasma chemokine levels and chemokines bound to erythrocyte DARC will impact on competitive binding and displacement of HIV to DARC as well as to CD4+ T cells. Hence, based on our results and these human and murine investigations and knowledge that HIV cell entry invokes a vigorous inflammatory response with release of LPS (Douek, 2007), we propose the following model (Figure 5F), whose confirmation will require additional studies.
Although it is tempting to speculate that akin to its sink function for chemokines (Comerford and Nibbs, 2005), DARC might also serve as a sink for the initial inoculum of HIV, we found that DARC binds X4 virus more efficiently than R5 virus. Due to the low initial inoculum and the preferential primary infection of R5 viruses compared to dual-tropic or X4 viruses, it is less likely that the HIV adsorption/trans-infection function of DARC contributes to the reduced risk of infection in DARC+ subjects. Instead, we suggest this is dominantly attributed by the impact of DARC on chemokine levels. The data of Mayr et al., (2008) indicate that in addition to the DARC ligands CCL2 (MCP-1) and CXCL1 (GROα), DARC+ subjects are also likely to have higher levels of the circulating and RBC-associated HIV-suppressive chemokine CCL5 compared to DARC–individuals (B. Jilma, personal communication). Hence, at the time of initial viral exposure, when a small inoculum of founder R5 virus is attempting to establish infection and disseminate, greater amounts of RBC-associated HIV-suppressive chemokine might provide a relative shield for binding of HIV to RBCs and subsequent transfer to HIV target cells (Figure 5F). Additionally, the higher plasma levels of CCL5 in DARC+ individuals might convey a protective effect against transmission (Figure 5F).
However, once infection occurs, two events might come into play that together confer an accelerated rate of disease progression in DARC+ individuals. First, during established infection, the inflammatory insult conveyed by HIV (Douek, 2007) might be greater in DARC+ than DARC− individuals. This is because DARC binds many more proinflammatory than HIV-suppressive chemokines (Gardner et al., 2004; Rot, 2005), including those that increase viral replication (e.g., CCL2; Monteiro de Almeida et al., 2005). Consequently, the chemokine-driven proinflammatory state (e.g., high levels of CCL2, CCL5, and CXCL1) in DARC+ subjects may far outweigh the HIV-suppressive effects of high CCL5 levels, the only major HIV-suppressive chemokine that binds to DARC. Second, by contrast to what might be observed in the setting of the initial exposure of a host to a small viral inoculum, productive clinical HIV infection is characterized by a high plasma viral burden, which might promote improved virus and target-cell interactions. Consequently, after infection is established, there is a greater probability of HIV displacing DARC-bound chemokines, and in turn, DARC erythrocyte-bound HIV making physical contact with CD4-bearing target cells, improving the efficiency of infection. Accordingly, other studies have found that a virus bound to uninfected cells is more stable and infectious than a similar amount of cell-free virus (Levy, 2002). Hence, after systemic infection has been established, HIV bound to DARC-expressing RBCs might serve as a reservoir for HIV and transport to other cellular targets. The aforementioned two scenarios might explain why HIV-positive subjects who express DARC have a faster rate of disease progression (Figure 5F). Conversely, DARC− HIV-positive subjects might have both a smaller reservoir of HIV-loaded RBCs and a reduced inflammatory state, and this could explain in part the association between the DARC −46C/C genotype and a slower disease course.
Substantiating the aforementioned model, as well the contrasting effects of DARC genotype on HIV acquisition and disease progression, we found that the genotype-phenotype relationships of DARC+ status is remarkably similar to those of a CCL2 genotype (−2578G/G) that we showed previously was associated with increased CCL2 expression (Gonzalez et al., 2002). Thus, it is striking that both DARC+ status as well as the CCL2 −2578G/G genotype track higher expression levels of CCL2. They are also both associated with contrasting effects on HIV acquisition and disease progression, i.e., a reduced risk of HIV acquisition, but increased rate of disease progression to AIDS and HAD (this study and Gonzalez et al., 2002). It is also noteworthy that in our previous work we only detected these associations for the CCL2 −2578G/G genotype in European Americans but not African Americans HIV+ subjects (Gonzalez et al., 2002). However, akin to the genotype-phenotype relationships that we describe for the CCL5 genotype before and after stratification for DARC genotype (Figures 5C–5E), we found that a disease-accelerating effect for the CCL2 genotype was evident only in DARC+ but not DARC− African-American subjects (data not shown).
As there is extensive linkage disequilibrium (LD) around the DARC locus (Lautenberger et al., 2000), we cannot exclude with certainty the possibility that the effects ascribed to the −46C/C genotype might be attributable to some other polymorphism(s)/gene(s) in LD near DARC. However, based on the in vitro (Figure 1) and in vivo (Figure 5) data described herein, as well as previous findings implicating a role for DARC in HIV-AIDS, we suggest that the genotype-phenotype relationships described herein are most likely directly attributable to DARC. Nevertheless, similar studies in other cohorts are warranted to confirm these results. Although the precise mechanisms by which DARC imparts its effects on HIV-AIDS pathogenesis are unclear, our findings indicate that the triangular relationship between this chemokine-binding protein, its natural ligands (chemokines), and pathogen (HIV) is likely to lead to biologically complex and seemingly contrasting outcomes.
In summary, the results of our proof-of-concept genetic-epidemiologic studies support a conceptual model that the interplay between DARC and chemokines might influence the amount of free versus DARC-bound HIV available for eventual transfer from RBCs to target cells and also chemokine levels that promote inflammation (e.g., CCL2, CCL5, CXCL1, and CXCL8) and compete for the binding of HIV (e.g., CCL5) to CD4+ T cells during HIV infection. By extension, the results of our studies indicate that failure to account for the chemokine binding and HIV-adsorption effects of DARC might confound genetic epidemiologic studies that examine the association between chemokine-gene polymorphisms and HIV-AIDS susceptibility in subjects of African descent. Additionally, consistent with our previous contention (Gonzalez et al., 1999, 2005; Moore et al., 2008), the genotype-phenotype vignettes shown here underscore further the possible impact of differentially distributed transmission and/or disease-modifying genotypes on (1) the heterogeneity of HIV disease burden not just at the level of individuals but also populations and (2) evaluation of vaccine efficacy.
The HIV/AIDS-modifying effects of CCR5-Δ32 and DARC −46C, two population-specific alleles, highlight that the impact of ancestral evolutionary factors—including infectious diseases—on the human genome are influencing outcomes of present-day infections. However, these outcomes can be complex. For example, in addition to the DARC −46C allele, polymorphisms such as sickle-cell trait can confer protection against malaria, which can be costly by causing genetic disease in the homozygous state. Thus, it should not be surprising then that what might be beneficial in one context (e.g., protection from malaria by DARC −46C/C genotype) can be detrimental in other settings (e.g., association between DARC −46C/C genotype and increased risk of HIV acquisition).
Of broad interest, our studies underscore that DARC impacts on chemokines, malaria, and HIV, a ménage à trois. However, given the importance of the chemokine system in host defense, immune responses, and inflammation, it will be important to determine the contribution of DARC to the pathogenesis of other infectious disease and inflammatory disease states.
Experimental Procedures
Interactions between DARC and HIV-1 Virions: In Vitro Experiments
Cells
RBCs from donors who were either DARC+ or DARC− were used as virus carriers. Target cells for infection by HIV-loaded RBCs were either purified PBMCs or NP2 cells expressing CD4 and either CXCR4 or CCR5 as target cells for virus-strain binding and infection experiments. The glioma NP2/CD4 cell line has been described in detail previously (Soda et al., 1999). Cells were maintained in Dulbecco's modified Eagle medium (DMEM) with 5% FCS and 1% penicillin/streptomycin at 37°C with 5% CO2.
Transfer of Infectivity
Ten-thousand p24 focus-forming units (ffu) of a variety of primary isolates and laboratory-adapted strains of HIV-1 were incubated at 37°C with 107 purified human RBCs for 1 hour, washed extensively, and overlaid onto target cells. For NP2/CD4 cells, viral transfer of HIV-1 was quantified as the percentage of the input titer (p24 + ve foci by in situ immunostaining) (Neil et al., 2005) recovered from the RBCs by immunostaining target cells for p24 expression. For PBMCs, transferred infectivity was then assessed by RT activity in the cell supernatants at various time points postinfection. Transfer experiments in the presence of soluble CD4 (sCD4) were performed as detailed above, and 1 or 5 μg/ml of sCD4 was/were added to the viral inoculum prior to incubation with the RBCs.
Competitive Infectivity
Recombinant human (rh) CCL5 (RANTES), rhCXCL8 (IL-8), rhCCL3 (MIP-1αS), and monoclonal anti-DARC antibody (anti-Fy6) in the concentrations indicated were incubated with purified RBCs for 1 hour, then the RBCs were washed, and infection and transfer experiments were repeated as described above.
Subjects
We genotyped HIV-1-infected (n = 1266) and noninfected (n = 2218) adult subjects recruited to the WHMC, San Antonio, TX, USA. We also genotyped the HGDP-CEPH (n = 1064) cross-sectional sample of worldwide DNA specimens. Detailed characteristics of the WHMC cohort and the HGDP-CEPH samples are described elsewhere (Dolan et al., 2007; Gonzalez et al., 2005). The protocol and consent for this study were approved by the Institutional Review Boards at the WHMC and the University of Texas Health Science Center at San Antonio.
SNP and Genotyping
The DARC T−46C (rs2814778) SNP was genotyped by TaqMan allelic discrimination assays in the subjects described above. The nomenclature for the DARC polymorphisms was as described by Tournamille et al., 1995 and is based on the reference sequence AC055992. Primer and probe sequences are available upon request from S.K.A. (ahujas@uthscsa.edu).
Statistical Methods
H-W equilibrium was assessed using the χ2 test with one degree of freedom. The risk of HIV acquisition was assessed using multivariate logistic regression analysis, while the association of the DARC genotypes with time to death was assessed using Cox proportional hazard analysis and Kaplan-Meier survival plots after testing for the assumption of proportional hazards using the Schoenfeld residuals. CD4+ T cell trajectories were depicted by Loess curves, and rates of changes in CD4+ T cell counts were assessed using generalized estimatingequations (GEE). PAF was calculated according to Rockhill et al., 1998. The detailed procedure of adjustments for population admixture is provided in the Supplemental Data. STATA 7.0 (Stata Corporation, College Station, TX) software package was used to conduct the statistical analyses.
Supplementary Material
Acknowledgments
We are indebted to the individuals who enrolled in these studies and who made this work possible. We thank S. Wegner and other members of the Infectious Disease Clinical Research Program (IDCRP) for critical support of this work; D. Kazandjian for helpful discussions; R. Sanchez, G. Cortez, V. Ann, B. Smith, and X. He for technical assistance; L. Song for graphic work; and A.S.Ahuja for forbearance. This work was supported by the Veterans Administration Center on AIDS and HIV Infection of the South Texas Veterans Health Care System and by a MERIT award (R37046326) and other awards (AI043279 and MH069270) from the NIH to S.K.A. S.K.A. is a recipient of the Elizabeth Glaser Scientist Award and the Burroughs Wellcome Clinical Scientist Award in Translational Research. Support for the WHMC cohort was provided by the Infectious Disease Clinical Research Program (IDCRP) of the Uniformed Services University of the Health Sciences (USUHS) of which the TriService AIDS Clinical Consortium (TACC) is a component. The IDCRP is a Department of Defense triservice program executed through USUHS and the Henry M. Jackson Foundation for the Advancement of Military Medicine, in collaboration with HHS/NIH/NIAID/DCR through Interagency Agreement HU0001-05-2-0011. This work was also supported by a grant from the UK Medical Research Council to R.A.W. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Departments of the Army, Navy, or Air Force, or the Department of Defense. The authors have no commercial or other association that might pose a conflict of interest.
Footnotes
Supplemental Data include one table, a supplemental note, and supplemental references and can be found online at http://www.cellhostandmicrobe.com/cgi/content/full/4/1/52/DC1/.
Document S1. One Table, Supplemental Note, and Supplemental References
References
- Abu-Raddad LJ, Patnaik P, Kublin JG. Dual infection with HIV and malaria fuels the spread of both diseases in sub-Saharan Africa. Science. 2006;314:1603–1606. doi: 10.1126/science.1132338. [DOI] [PubMed] [Google Scholar]
- Ahuja SK, Kulkarni H, Catano G, Agan BK, Camargo JF, He W, O'Connell RJ, Marconi VC, Delmar J, Eron J, et al. CCL3L1-CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV-1-infected individuals. Nat Med. 2008;14:413–420. doi: 10.1038/nm1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banki Z, Wilflingseder D, Ammann CG, Pruenster M, Mullauer B, Hollander K, Meyer M, Sprinzl GM, van Lunzen J, Stellbrink HJ, et al. Factor I-mediated processing of complement fragments on HIV immune complexes targets HIV to CR2-expressing B cells and facilitates B cell-mediated transmission of opsonized HIV to T cells. J Immunol. 2006;177:3469–3476. doi: 10.4049/jimmunol.177.5.3469. [DOI] [PubMed] [Google Scholar]
- Buve A. HIV epidemics in Africa: what explains the variations in HIV prevalence? IUBMB Life. 2002;53:193–195. doi: 10.1080/15216540212641. [DOI] [PubMed] [Google Scholar]
- Comerford I, Nibbs RJ. Post-translational control of chemokines: a role for decoy receptors? Immunol Lett. 2005;96:163–174. doi: 10.1016/j.imlet.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Comerford I, Litchfield W, Harata-Lee Y, Nibbs RJ, McColl SR. Regulation of chemotactic networks by ‘atypical’ receptors. Bioessays. 2007;29:237–247. doi: 10.1002/bies.20537. [DOI] [PubMed] [Google Scholar]
- Dolan MJ, Kulkarni H, Camargo JF, He W, Smith A, Anaya JM, Miura T, Hecht FM, Mamtani M, Pereyra F, et al. CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat Immunol. 2007;8:1324–1336. doi: 10.1038/ni1521. [DOI] [PubMed] [Google Scholar]
- Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- Douek D. HIV disease progression: immune activation, microbes, and a leaky gut. Top HIV Med. 2007;15:114–117. [PubMed] [Google Scholar]
- Fukuma N, Akimitsu N, Hamamoto H, Kusuhara H, Sugiyama Y, Sekimizu K. A role of the Duffy antigen for the maintenance of plasma chemokine concentrations. Biochem Biophys Res Commun. 2003;303:137–139. doi: 10.1016/s0006-291x(03)00293-6. [DOI] [PubMed] [Google Scholar]
- Gardner L, Patterson AM, Ashton BA, Stone MA, Middleton J. The human Duffy antigen binds selected inflammatory but not homeostatic chemokines. Biochem Biophys Res Commun. 2004;321:306–312. doi: 10.1016/j.bbrc.2004.06.146. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Bamshad M, Sato N, Mummidi S, Dhanda R, Catano G, Cabrera S, McBride M, Cao XH, Merrill G, et al. Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes. Proc Natl Acad Sci U S A. 1999;96:12004–12009. doi: 10.1073/pnas.96.21.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, Dhanda R, Bamshad M, Mummidi S, Geevarghese R, Catano G, Anderson SA, Walter EA, Stephan KT, Hammer MF, et al. Global survey of genetic variation in CCR5, RANTES, and MIP-1alpha: impact on the epidemiology of the HIV-1 pandemic. Proc Natl Acad Sci U S A. 2001;98:5199–5204. doi: 10.1073/pnas.091056898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, et al. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci U S A. 2002;99:13795–13800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- Hadley TJ, Peiper SC. From malaria to chemokine receptor: the emerging physiologic role of the Duffy blood group antigen. Blood. 1997;89:3077–3091. [PubMed] [Google Scholar]
- Hess C, Klimkait T, Schlapbach L, Del Zenero V, Sadallah S, Horakova E, Balestra G, Werder V, Schaefer C, Battegay M, Schifferli JA. Association of a pool of HIV-1 with erythrocytes in vivo: a cohort study. Lancet. 2002;359:2230–2234. doi: 10.1016/S0140-6736(02)09291-7. [DOI] [PubMed] [Google Scholar]
- Horakova E, Gasser O, Sadallah S, Inal JM, Bourgeois G, Ziekau I, Klimkait T, Schifferli JA. Complement mediates the binding of HIV to erythrocytes. J Immunol. 2004;173:4236–4241. doi: 10.4049/jimmunol.173.6.4236. [DOI] [PubMed] [Google Scholar]
- Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, Miller LH. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261:1182–1184. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- Jilma-Stohlawetz P, Homoncik M, Drucker C, Marsik C, Rot A, Mayr WR, Seibold B, Jilma B. Fy phenotype and gender determine plasma levels of monocyte chemotactic protein. Transfusion. 2001;41:378–381. doi: 10.1046/j.1537-2995.2001.41030378.x. [DOI] [PubMed] [Google Scholar]
- Kellam P, Weiss RA. Infectogenomics: insights from the host genome into infectious diseases. Cell. 2006;124:695–697. doi: 10.1016/j.cell.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita-Moleka R, Smith JS, Atibu J, Tshefu A, Hemingway-Foday J, Hobbs M, Bartz J, Koch MA, Rimoin AW, Ryder RW. Low prevalence of HIV and other selected sexually transmitted infections in 2004 in pregnant women from Kinshasa, the Democratic Republic of the Congo. Epidemiol Infect. 2007:1–7. doi: 10.1017/S0950268807009818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn L, Schramm DB, Donninger S, Meddows-Taylor S, Coovadia AH, Sherman GG, Gray GE, Tiemessen CT. African infants' CCL3 gene copies influence perinatal HIV transmission in the absence of maternal nevirapine. Aids. 2007;21:1753–1761. doi: 10.1097/QAD.0b013e3282ba553a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachgar A, Jaureguiberry G, Le Buenac H, Bizzini B, Zagury JF, Rappaport J, Zagury D. Binding of HIV-1 to RBCs involves the Duffy antigen receptors for chemokines (DARC) Biomed Pharmacother. 1998;52:436–439. doi: 10.1016/s0753-3322(99)80021-3. [DOI] [PubMed] [Google Scholar]
- Lama J, Planelles V. Host factors influencing susceptibility to HIV infection and AIDS progression. Retrovirology. 2007;4:52. doi: 10.1186/1742-4690-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenberger JA, Stephens JC, O'Brien SJ, Smith MW. Significant admixture linkage disequilibrium across 30 cM around the FY locus in African Americans. Am J Hum Genet. 2000;66:969–978. doi: 10.1086/302820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Wurfel MM, Matute-Bello G, Frevert CW, Rosengart MR, Ranganathan M, Wong VW, Holden T, Sutlief S, Richmond A, et al. The Duffy antigen modifies systemic and local tissue chemokine responses following lipopolysaccharide stimulation. J Immunol. 2006;177:8086–8094. doi: 10.4049/jimmunol.177.11.8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekkerkerker AN, van Kooyk Y, Geijtenbeek TB. Viral piracy: HIV-1 targets dendritic cells for transmission. Curr HIV Res. 2006;4:169–176. doi: 10.2174/157016206776055020. [DOI] [PubMed] [Google Scholar]
- Levy JA. HIV-1: hitching a ride on erythrocytes. Lancet. 2002;359:2212–2213. doi: 10.1016/S0140-6736(02)09314-5. [DOI] [PubMed] [Google Scholar]
- Liu H, Chao D, Nakayama EE, Taguchi H, Goto M, Xin X, Takamatsu JK, Saito H, Ishikawa Y, Akaza T, et al. Polymorphism in RANTES chemokine promoter affects HIV-1 disease progression. Proc Natl Acad Sci U S A. 1999;96:4581–4585. doi: 10.1073/pnas.96.8.4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangano A, Gonzalez E, Dhanda R, Catano G, Bamshad M, Bock A, Duggirala R, Williams K, Mummidi S, Clark RA, et al. Concordance between the CC chemokine receptor 5 genetic determinants that alter risks of transmission and disease progression in children exposed perinatally to human immunodeficiency virus. J Infect Dis. 2001;183:1574–1585. doi: 10.1086/320705. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. 2006;6:907–918. doi: 10.1038/nri1964. [DOI] [PubMed] [Google Scholar]
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Mayr FB, Spiel AO, Leitner JM, Firbas C, Kliegel T, Jilma-Stohlawetz P, Derendorf H, Jilma B. Duffy antigen modifies the chemokine response in human endotoxemia. Crit Care Med. 2008;36:159–165. doi: 10.1097/01.CCM.0000297875.55969.DB. [DOI] [PubMed] [Google Scholar]
- Monteiro de Almeida S, Letendre S, Zimmerman J, Lazzaretto D, McCutchan A, Ellis R. Dynamics of monocyte chemoattractant protein type one (MCP-1) and HIV viral load in human cerebrospinal fluid and plasma. J Neuroimmunol. 2005;169:144–152. doi: 10.1016/j.jneuroim.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Moore JP, Klasse PJ, Dolan MJ, Ahuja SK. AIDS/HIV. A STEP into darkness or light? Science. 2008;320:753–755. doi: 10.1126/science.1154258. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- Neil SJ, Aasa-Chapman MM, Clapham PR, Nibbs RJ, McKnight A, Weiss RA. The promiscuous CC chemokine receptor D6 is a functional coreceptor for primary isolates of human immunodeficiency virus type 1 (HIV-1) and HIV-2 on astrocytes. J Virol. 2005;79:9618–9624. doi: 10.1128/JVI.79.15.9618-9624.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RB, Haggerty CL, Harger G, Ferrell R. Differential distribution of allelic variants in cytokine genes among African Americans and White Americans. Am J Epidemiol. 2004;160:1033–1038. doi: 10.1093/aje/kwh325. [DOI] [PubMed] [Google Scholar]
- Pepin J. From the Old World to the New World: an ecologic study of population susceptibility to HIV infection. Trop Med Int Health. 2005;10:627–639. doi: 10.1111/j.1365-3156.2005.01441.x. [DOI] [PubMed] [Google Scholar]
- Rockhill B, Newman B, Weinberg C. Use and misuse of population attributable fractions. Am J Public Health. 1998;88:15–19. doi: 10.2105/ajph.88.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot A. Contribution of Duffy antigen to chemokine function. Cytokine Growth Factor Rev. 2005;16:687–694. doi: 10.1016/j.cytogfr.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Schliekelman P, Garner C, Slatkin M. Natural selection and resistance to HIV. Nature. 2001;411:545–546. doi: 10.1038/35079176. [DOI] [PubMed] [Google Scholar]
- Silverberg MJ, Wegner SA, Milazzo MJ, McKaig RG, Williams CF, Agan BK, Armstrong AW, Gange SJ, Hawkes C, O'Connell R J, et al. Effectiveness of highly-active antiretroviral therapy by race/ethnicity. Aids. 2006;20:1531–1538. doi: 10.1097/01.aids.0000237369.41617.0f. [DOI] [PubMed] [Google Scholar]
- Simmons G, Wilkinson D, Reeves JD, Dittmar MT, Beddows S, Weber J, Carnegie G, Desselberger U, Gray PW, Weiss RA, Clapham PR. Primary, syncytium-inducing human immunodeficiency virus type 1 isolates are dual-tropic and most can use either Lestr or CCR5 as coreceptors for virus entry. J Virol. 1996;70:8355–8360. doi: 10.1128/jvi.70.12.8355-8360.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda Y, Shimizu N, Jinno A, Liu HY, Kanbe K, Kitamura T, Hoshino H. Establishment of a new system for determination of coreceptor usages of HIV based on the human glioma NP-2 cell line. Biochem Biophys Res Commun. 1999;258:313–321. doi: 10.1006/bbrc.1999.0633. [DOI] [PubMed] [Google Scholar]
- Sullivan AD, Wigginton J, Kirschner D. The coreceptor mutation CCR5Delta32 influences the dynamics of HIV epidemics and is selected for by HIV. Proc Natl Acad Sci U S A. 2001;98:10214–10219. doi: 10.1073/pnas.181325198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet. 1995;10:224–228. doi: 10.1038/ng0695-224. [DOI] [PubMed] [Google Scholar]
- Weiss RA, McMichael AJ. Social and environmental risk factors in the emergence of infectious diseases. Nat Med. 2004;10:S70–76. doi: 10.1038/nm1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C, An P, O'Brien S. Patterns of ethnic diversity among the genes that influence AIDS. Hum Mol Genet. 2004;13(Spec No 1):R9–19. doi: 10.1093/hmg/ddh075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.