

Abstract
Laboratory mouse plays important role in our understanding of early mammalian development and provides invaluable model for human early embryos, which are difficult to study for ethical and technical reasons. Comprehensive collection of cDNA clones, their sequences, and complete genome sequence information, which have been accumulated over last two decades, have provided even more advantages to mouse models. Here the progress in global gene expression profiling in early mouse embryos and, to some extent, stem cells are reviewed and the future directions and challenges are discussed. The discussions include the restatement of global gene expression profiles as snapshot of cellular status, and subsequent distinction between the differentiation state and physiological state of the cells. The discussions then extend to the biological problems that can be addressed only through global expression profiling, which include: bird’s-eye view of global gene expression changes, molecular index for developmental potency, cell lineage trajectory, microarray-guided cell manipulation, and the possibility of delineating gene regulatory cascades and networks.
Keywords: Expression profiling, Microarray, stem cells, preimplantation embryos, embryogenomics, large-scale analysis
INTRODUCTION
Global gene expression profiling is to measure the expression levels of all genes and is a part of the modern trends towards analyzing biological systems as a whole in a systematic manner. It should therefore be understood from a global framework, including other technologies, such as gene targeting and siRNA technologies. All the functional aspects of mouse embryology are discussed in other articles in this issue, and thus, this review will focus primarily on the gene expression profiling with the emphasis on the problems and challenges that are unique to the analysis of early embryos.
ADVANTAGES OF MOUSE MODEL
Laboratory mouse plays an important role in our understanding of early mammalian embryogenesis. Rather unique developmental feature of mammals, such as the first differentiation into trophectoderm and imprinting, makes it difficult to use other model organisms, such as yeast, worm, fly, frog, and zebrafish in some areas of investigation. Mouse is an especially important model organism for human biology, because human embryos during preimplantation development and implantation are scarce and difficult to obtain for both ethical and technical reasons. The problem has been alleviated by the derivation of human embryonic stem (ES) cells from blastocysts (Thomson et al., 1998), which can provide an excellent in vitro model system to study early differentiation events in human cells. However, the study of in vivo embryos is still difficult and the advantage of mouse system that allow the injection of ES cells into blastocysts to examine further development cannot be achieved in human.
Mouse advantages have been enhanced by the accumulation of molecular reagents and information in this model organism. For example, the majority of cDNA sequences (ESTs) from preimplantation embryos is derived from mouse (reviewed in (Ko, 2005)). The scarcity of materials and small size of early embryos have made the molecular analysis very difficult even for mouse, but genes and their alternatively spliced transcripts, which function primarily in the early embryos, can only be discovered by working on these tissues. Based on the materials and resources generated in this pursuit, global expression profiling became possible. For example, the microarray cannot be built without cDNA clones or cDNA sequences. Similarly, bits of sequences obtained from other expression profiling technologies cannot be identified otherwise. In addition to cDNA/ESTs, the full-length cDNA sequence information (Gerhard et al., 2004; Carninci et al., 2005) and the nearly completed mouse genome sequence (Waterston et al., 2002) have almost completed this particular phase of study, that is, to identify the majority of genes, their structures, and alternative transcripts on the mouse genome. However, the annotation of genes is not complete and the recent discovery of microRNAs (reviewed in (Sevignani et al., 2006)), non-coding RNAs (Ravasi et al., 2006), many pseudogenes, and some truncated form of proteins remains to be understood. The impact of these new discoveries to overall biology is yet to be determined, but definitely one of the areas needs to be studied in the future.
GLOBAL EXPRESSION PROFILING - SNAPSHOT OF CELLULAR STATUS
Ultimate goal of the global expression profiling is to take a snapshot of a cell state and to capture the total activity of a cell at the moment (Ko, 2001). This can be achieved at different levels (Fig. 1). The epigenotype is the first level, which represents the different genome-wide patterns of DNA methylation and histone modifications such as methylation and acetylation according to individual cell types, e.g., ES cells, neurons, and T cells. It has recently become possible to examine epigenomes, but the technology is still labor intensive and not suited to the study of a large number of cell types and conditions (Roh et al., 2005). Specific sets of RNAs are transcribed from cell-specific epigenome and their steady-state levels form a transcript profile, i.e., gene expression profiles at RNA level. This is probably the most accessible, easiest, and cost-effective way to do a global expression profiling. It certainly reflects the status of the whole epigenomes. Protein is, obviously, the molecule that carries out the most function, and thus, is sometimes regarded as the better indicator of the cellular status. It has also been reported that although the expression profiles at RNA levels correlate well with those at protein level, there are some discrepancies between them (Tian et al., 2004). However, the expression profiling of entire protein cohorts in the cells have not been achieved and obviously is not cost-effective at this point. Perhaps the most effective application is to identify proteins expressed differentially among samples by 2D-PAGE and to identify the limited number of proteins by mass spectroscopy (e.g., (Hudelist et al., 2006)). Global protein profiles are also more complex than RNA profiles, because they require not only the measurement of abundance of individual molecules, but also account for a variety of modifications of proteins, such as phosphorylation and glycosylation.
Figure 1. Genome-wide profiling of a cell at different levels.
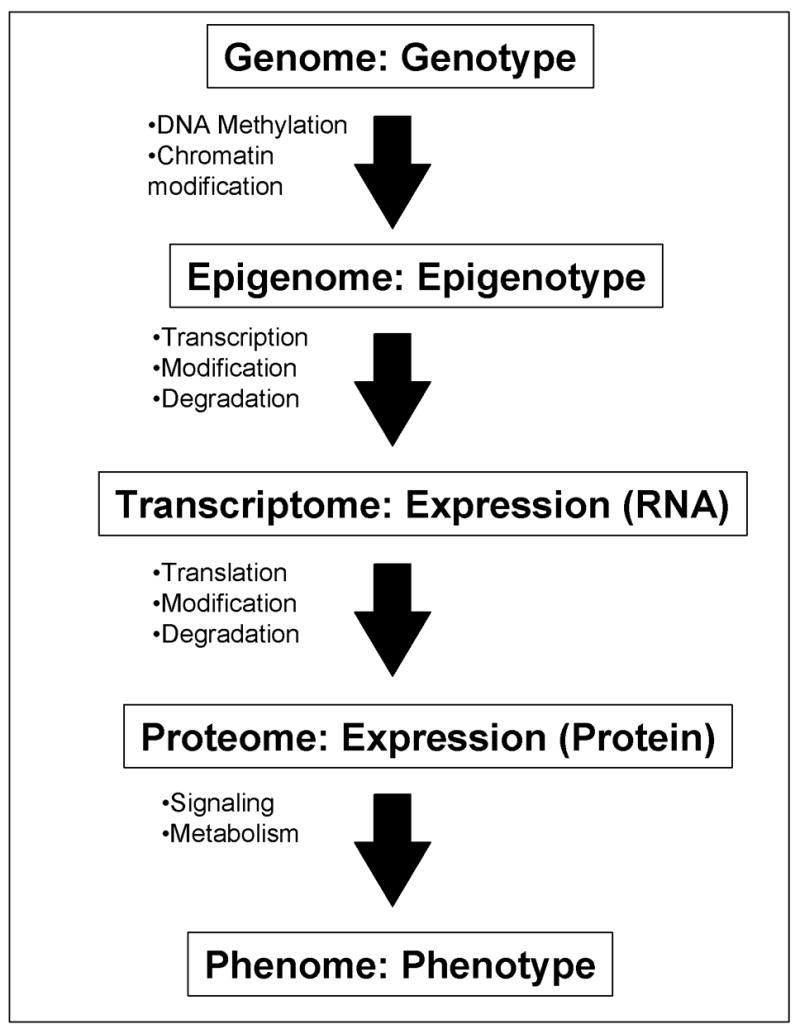
A flow of information from genome/genotype to phenome/phenotype is shown: each step represents one-to-many relationship. All the cells in individual mouse contains the same whole set of DNAs – “genome” (with some exceptions), but individual cells can have different “epigenomes” (Holliday, 2005; Murrell et al., 2005) depending on their state of differentiation after the DNA methylation and the chromatin modifications. Specific sets of genes are transcribed according to the status of “epigenome.” The “transcriptome” of individual cells is the steady-state levels of all the RNA species (after taking the modification and degradation into consideration). Expression profiling at the RNA level can capture the whole or a part of the transcriptome of a cell, tissue, or organ. These RNAs are translated into proteins. The “proteome” of individual cells is the steady-state levels of all the protein species (after taking the modification and degradation into consideration). “Phenome” (Mahner and Kary, 1997; Paigen and Eppig, 2000) is the whole set of phenotypes in a cell, tissue, or organ. Cells with the same proteome can have different phenomes, because they can have different cell state, e.g., different metabolites and signaling molecules. Expression profiling at RNA levels represents the status of epigenome more closely than expression profiling at protein levels. On the other hand, expression profiling at protein levels represents the phenome of a cell more closely than that at RNA level.
It is important to point out that the expressions of RNAs or proteins do not necessary mean that they have functions in the cells. Similarly, the changes of gene expression do not necessary mean significance of these changes. It is conceivable that the future technology advancement will make it possible to use the global profiles of proteins, signaling molecules, and metabolites as a snapshot of cell state. However, the expression profiles at RNA levels will continue to be the method of choice for some time, and thus, the following discussion will focus on RNA profiling.
Methods for gene expression profiling
A variety of methods have been developed and applied to global gene expression profiling in mouse early embryos. Large number of cDNA/EST sequences that have been accumulated in the public database have been used extensively to identify genes specifically expressed in ovary and oocytes (Stanton et al., 2002), preimplantation embryos (Ko et al., 2000; Evsikov et al., 2004), and germ cells (Rajkovic et al., 2001; Yan et al., 2002; Lin and Matzuk, 2005), various stem cells (Bortvin et al., 2003; Mitsui et al., 2003; Sharov et al., 2003). One of the goals for EST project is to discover genes and help the proper annotation of the genome sequences ((Marra et al., 1999)). Although it is a short stretch of sequences, the ESTs can provide important experimental evidences to the coding regions of the genome sequences and their alternative start sites and spliced forms (e.g., (Sharov et al., 2005a)). Recent discovery that many transcripts in mouse oocytes start from the repetitive sequences is another good example of the usage of ESTs (Peaston et al., 2004). EST/cDNA sequences can also be served to identify a potential full-length cDNA clone, which can then be sequenced entirely to produce the full-length cDNA sequence information (Gerhard et al., 2004; Carninci et al., 2005). Physical cDNA clones have also been used as a molecular probe for Northern blotting and in situ hybridization (Lennon et al., 1996). EST/cDNA sequencing project was required to develop the infrastructure that allows the gene expression profiling, including the development of DNA microarray platform (e.g., (Carter et al., 2003)). The existing datasets will continue to provide useful tools to the research community. However, it should be pointed out that EST project can only be justified as a part of initial gene discovery project, because EST projects are not cost-effective as a way to do expression profiling.
PCR-based differential display techniques (Liang and Pardee, 1992) have been applied to the analysis of mouse ES cells (Hollnagel et al., 1999) and parthenogenetic blastocysts (Brown and Kay, 1999). Suppression Subtractive Hybridization (SSH) (Diatchenko et al., 1996) has been used to identify mouse oocyte-specific genes (Zeng and Schultz, 2003). However, these technologies are not suited to do comprehensive profiling. In contrast, serial analysis of gene expression (SAGE) (Velculescu et al., 1995) has been applied to identify genes expressed in mouse ES cells (Anisimov et al., 2002). Another technology for sequence-based expression analysis is massively parallel signature sequencing (MPSS) (Brenner et al., 2000), which has identified a number of genes expressed differentially between human and mouse ES cells (Wei et al., 2005). These sequencing-based technologies are relatively expensive, and thus, SAGE and other methods have been often used to examine samples without biological replications (Ruijter et al., 2002). SAGE’s advantage for greater number of tags compared to ESTs can only add more statistical accuracy and depth within the sample, but the lack of biological replications provide indeed less statistical powers overall. Besides, unlike the cDNA/EST projects, SAGE does not provide any additional resources to justify the costs.
As of today, DNA microarray technologies are the most cost-effective way to obtain a comprehensive genome-wide gene expression profiles. And indeed, a large number of literatures have been published using the microarray technology. Three main platforms for DNA microarrays are currently available. First is a cDNA clone-spotted microarray (mostly homemade microarrays). A notable earlier work with this type of microarray is the expression profiling of three germ layers from gastrulating mouse embryos (Harrison et al., 1995). Second is short oligonucleotide microarray (e.g., Affymetrix). Third is the long oligonucleotide microarray (e.g, Agilent and Illumina).
One promise that still needs to be fulfilled is the accumulation of the available data in the public database so that researchers can do data mining on their own, as it happened to EST database. To facilitate this, the Microarray Gene Expression Data (MGED) Society has proposed the guideline of minimum information required for microarray experiments (MIAME) (Brazma et al., 2001). It has become the journal’s mandatory that any publication reporting the microarray needs to accompany with the submission of the entire data sets to at least one of the MIAME-compliant microarray datasets, such as Gene Expression Omnibus (GEO) (Edgar et al., 2002), ArrayExpress (Brazma et al., 2003) and CIBEX (Ikeo et al., 2003). Although the MIAME ensured the availability of all the detailed information about how samples are collected and how experiments are done, this does not address the issue of platform-to-platform differences. Some have observed concordance between results obtained with different platforms (Yuen et al., 2002; Park et al., 2004; Yauk et al., 2004; Larkin et al., 2005), whereas others have reported disagreement (Kuo et al., 2002; Tan et al., 2003; Mah et al., 2004). Very recent studies have examined variability not only across platforms (e.g,, Affymetrix GeneChips, Agilent 60-mer oligonucleotide arrays, and spotted cDNA arrays), but also across laboratories (Bammler et al., 2005; Irizarry et al., 2005). They have shown that there are large differences between laboratories using the same platform, but the results from the best-performing labs agree well. This indicates that microarray results are reliable and comparable, when performed appropriately.
Differentiation state and physiological state
It is important to point out that the identical cell can have different expression profiles. When one sees that the expression profile of certain cells, e.g., ES cells, one normally assumes that the expression profile is unique and equal to the character of the cells. So the comparison between the different cell types, e.g, ES cells versus trophoblast stem (TS) cells, will be literally interpreted as the difference between ES cells and TS cells (Tanaka et al., 2002). However, what one can actually measure is the global gene expression patterns of a specific state of ES cells (denoted as ES1, ES2, …, ESn in Fig. 2). ES cells at different cell cycle phase will definitely show very different expression patterns. Another example is the stimulation of proliferation or secretion of certain proteins by the stimulation of some factors, e.g., growth hormones. Let’s assume that this does not cause the differentiation of ES cells, which remains undifferentiated ES cells. The expression profiles of undifferentiated ES cells in the presence or absence of this growth factor will probably very different.
Figure 2. Distinction between the differentiation state of a cell and the physiological state of a cell.

Embryonic stem (ES) cells and trophoblast stem (TS) cells are used here as examples. This diagram shows that cells can have different RNA expression profiles/transcriptomes (e.g., ES1 and ES2), while maintaining their cell identity, i.e., differentiation state (e.g., ES).
There seems to be no appropriate term to distinguish these cell types. It is tempting to use the analogy to Plato’s “eidos,” “idea,” or “form,” since the situation is somewhat similar to Plato’s argument that there is a single form (e.g., the horse) representing variety types of the object (e.g., individual horses) in reality. However, it would be perhaps better to call the cell’s identity tentatively “differentiation state of cell,” whereas many different transient forms will be called “physiological state of cell.” In the example discussed above, the expression profile of ES cells that can be obtained experimentally represents one of the physiological states of ES cells (e.g., ES1), but does not directly represent the differentiation state of ES cells. The comparison of the expression profiles between ES and TS in the literature may thus actually mean the comparison of those between ES1 and TS2 or the comparison of those between ES2 and TS3.
This new paradigm provides an explanation to the problem that has been raised about the experiments of “stemness.” Recently, gene expression profiling using several stem cell types, such as ES cells, hematopoietic stem cells, and neural stem cells, was carried out to find a molecular signature of stemness (Ivanova et al., 2002; Ramalho-Santos et al., 2002). Both groups independently identified more than 200 genes involved in stemness, but other groups pointed out that the comparison between their stemness gene lists revealed only six genes in common, even though they used identical microarray chips and the same cell types (Evsikov and Solter, 2003; Fortunel et al., 2003; Pyle et al., 2004; Mikkers and Frisen, 2005). This has often been attributed to the inaccuracy or immaturity of the microarray technology. But at least a part of the problem comes from the misconception that the identical cells or same cell types should have very similar global expression profiles. Distinction of physiological cell state and differentiation cell state will help to understand these discrepancies. These problems are probably more acute in the cultured cells such as ES cells than in the organs.
BIOLOGICAL PROBLEMS THAT CAN BE ADDRESSED ONLY THROUGH GLOBAL EXPRESSION PROFILING
Traditional way of doing profiling is to look at a small number of genes, which are often selected based on the biological relevance, e.g., marker genes involved in apoptosis pathways. Global gene expression profiling has been often seen as an extension of this type of profiling. Indeed, most microarray literatures present and discuss only a small number of specific genes or pathways, even if all the genes are profiled. Therefore, some argues that specialized microarray that contains smaller, but defined sets of genes is more appropriate for most applications, considering a high cost of running a whole genome microarray. However, the power of the comprehensive or the whole-genome analysis lies in the ability to look at global picture so that any important gene expression changes will not be missed. Furthermore, a possibility of retrospective analysis of many genes, which did not receive any attention by the authors at the time of experiments, provides the authors and the research community with strong incentives to do comprehensive genome-wide microarray analysis. Perhaps this will be even more important, if RNA samples from patients or animal models are limited and are not available for later reanalysis. Finally, DNA microarray should not be limited to use for the screening of genes of interest, but should also be used to address biological problems that can be addressed only through global expression profiling. Some of these examples will be discussed below.
Bird’s-eye view of global gene expression changes
Dynamics of gene expression changes can now be reconstructed by linking a series of snapshots of cell’s gene expression activity – global gene expression profiles. A good example is DNA microarray analysis of mouse preimplantation embryos (Hamatani et al., 2004; Wang et al., 2004; Zeng et al., 2004; Wang et al., 2005). Visualization of global expression profiles over time provides for the first time a bird’s-eye view of global gene expression changes during preimplantation embryo development (Fig. 3; (Hamatani et al., 2004)). The view revealed a number of important aspects of preimplantation development. First, the levels of maternal transcripts stored in oocytes are promptly reduced and many of these maternal transcripts are not reactivated during preimplantation development. Second, majority of genes at zygotic gene activation (ZGA) at 2- to 4-cell stage show transient expressions. Third, another wave of gene expression, named mid-preimplantation gene activation (MGA; (Hamatani et al., 2004)), is observed around 8-cell stage. Fourth, the visualization of these transient gene expressions led to the proposal of “waves of activation hypothesis,” which assumes domino effect-like chain reactions (Hamatani et al., 2004). The bird’s-eye view, thus, allows one to see the overall picture of gene expression changes.
Figure 3. An example of bird’s-eye view of gene expression changes: mouse preimplantation development.

Signal intensities (log scale) (which represent relative RNA levels) of individual genes at each developmental stage are shown as color-coded lines. The expression levels of about 22,000 genes were monitored, but only genes that showed statistically significant changes are shown. Color codes are shown in a side bar: from a high expression level (red) to a low expression level (blue). Figure adapted from (Hamatani et al., 2004) and reproduced from (Ko, 2005).
Use of global gene expression patterns to define cell differentiation
Microarray studies comparing two different cell types often address the issue of how similar or how different these two cell types are. People often use correlation coefficient, but similarity or differences is all relative concept. Thus, one needs the third cell type or even more cell types to use as standards. For example, in the case of ES vs. TS comparison, mouse embryo fibroblast (MEF), E12.5 embryo, and E12.5 placenta, was used to satisfy this requirement (Tanaka et al., 2002). Recent work showing the global gene expression difference between the nuclear transfer-derived ES (ntES) cells and the fertilized egg-derived ES (fES) cells used the mouse strain differences of ES cells as a standard (Brambrink et al., 2006). The more the sample numbers are, the more informative the comparison between the cells. However, the analysis and visualization of the data become also a challenge.
The comparison of global gene expression profiles among many samples/cell types will be facilitated by the Principal Component Analysis (PCA), which is a statistical technique to reduce the dimensionality in the dataset and to identify major trends. The PCA can map individual samples/cells in “multidimensional transcript profile space” according to their global gene expression patterns (Fig. 4; (Sharov et al., 2003)). Two different cell types that share similar gene expression patterns will be mapped closer to each other than two different cell types whose gene expression patterns are very different. In this way, the degree of similarity in the gene expression patterns between multiple cell types can be shown in a multidimensional (often represented as three-dimensional) PCA figure. It is conceivable that cells in the same differentiation state, but in the different physiological state will be mapped closer to each other than cells in the different differentiation state in the multidimensional transcript profile space. The PCA may indeed become a tool to distinguish the differences between physiological states and differentiation states.
Figure 4. An example of multi-dimensional transcript profile space, which shows coordinates of cells according to their global gene expression patterns.

The global expression patterns (EST frequencies) of 2812 relatively abundant genes were analyzed by the Principal Component Analysis (PCA). Each cell or embryo (red sphere) was mapped to 3D space along with genes of significance (yellow dot) according to their values of each principal component (PC2, PC3, and PC4). Red arrows indicate the progress of embryos during preimplantation development: the stepwise transition from unfertilized egg to blastocyst. Red circles indicate ES cells and newborn organs (heart and brain). Here the distance between cells represents the degree of similarity in gene expression patterns. Cells or embryos that are mapped closely have more similar overall gene expression patterns than those mapped widely apart. Cells or embryos are labeled as follows (see also Fig. 5): 6.5 EP, E6.5 whole embryo (embryo plus placenta); 7.5 EP, E7.5 whole embryo (embryo plus placenta); 8.5 EP, E8.5 whole embryo (embryo plus placenta); 9.5 EP, E9.5 whole embryo (embryo plus placenta); 7.5 E, E7.5 embryonic part only; 7.5 P, E7.5 extraembryonic part only; NbOvary, newborn ovary; NbBrain, newborn brain; NbHeart, newborn heart; NbKidney, newborn kidney; 13.5 VMB, E13.5 ventral midbrain dopamine cells; 12.5 Gonad (F), E12.5 female gonad/mesonephros; 12.5 Gonad (M), E12.5 male gonad/mesonephros; HS (Kit−, Sca1−), hematopoietic stem/progenitor cells (Lin−, Kit−, Sca1−); HS (Kit−, Sca1+), hematopoietic stem/progenitor cells (Lin−, Kit−, Sca1+); HS (Kit+, Sca1−), hematopoietic stem/progenitor cells (Lin−, Kit+, Sca1−); HS (Kit+, Sca1+), hematopoietic stem/progenitor cells (Lin−, Kit+, Sca1+); and NS-D, differentiated NS cells. Figure adapted from (Sharov et al., 2003).
One difficulty that biologists often face is the interpretation of this PCA figure. The projection of each cell type on PC axis is based on the mathematical transformation of the global gene expression levels, and thus, it is not intuitively clear for many biologists what the location of these cells represents. For any given axis in this transcript profile space, one can derive a list of genes, whose average expression level increases or decreases along the axis. Thus, the relative locations of cells on PC axis can be represented by the average expression levels of these genes. In other words, one can use the average expression level of these selected genes as an “Index” or “Scale,” which represents a major trend of global gene expression changes.
This concept may be easier to understand, if one use the analogy to other indexes used in our society. For example, the Dow Jones Industrial Average is the average value of 30 large, industrial stocks in the US. Individual values of stocks usually do not reflect the global status of the US economy, but average values of selected stocks can provide a global trend of US economy from a certain perspective. The selection of these 30 stocks is made largely arbitrarily. In contrast, the PCA identifies major trends of the global expression data without prior knowledge about what kind of trends they are. The PC axis can then be used to extract sets of genes, whose average expression levels represent the trend. Therefore, the set of genes is not pre-selected arbitrarily to fit to one’s own hypothesis.
Three direct application of this method will be discussed below.
Molecular index for developmental potency
Fig. 4 shows preimplantation embryos, ES cells, adult stem cells, and newborn organs, which are mapped in the 3D transcript profile space according to the global gene expression profiles based on EST frequency (Sharov et al., 2003). The 3D picture is presented as Virtual Reality Modeling Language (VRML), which can be rotated and zoomed in and out (Sharov et al., 2005b). This visualization of the global expression profile data clearly shows the relative distance between embryos and/or cell types in terms of their similarity of gene expression profiles. Furthermore, PC3 axis seems to capture a global trend that the gradual loss of developmental potency from a totipotent fertilized eggs, to ES cells, and to the differentiated cells in newborn organs, although there are a few exceptions. Correlation analysis has identified 88 genes, whose expression levels are significantly associated with the PC3 axis (Sharov et al., 2003). The average expression levels of these 88 genes for each cell type are roughly proportional to the positions of these cells on the PC3 axis and can be used as an index for developmental potency (Fig 5; (Sharov et al., 2003)). Unlike the traditional definition of developmental potency, which requires the experimental manipulation of a cell to see its ability to differentiate into multiple cell types, this index can be obtained by measuring the expression levels of these 88 genes, and thus, may provide a quantitative measure to elusive concept of developmental potency.
Figure 5. A possible molecular index for developmental potency.

Coordinates of each cell or embryo on PC3 axis from Fig. 4 are mapped against the average expression levels of 88 genes. These 88 genes include Birc2, Bmp15, Btg4, Cdc25a, Cyp11a, Dtx2, E2f1, Fmn2, Folr4, Gdf9, Krt2–16, Mitc1, Oas1d, Oas1e, Obox3, Prkab2, Rfpl4, Rgs2, Rnf35, Rnpc1, Slc21a11, Spin, Tcl1, Tclb1, and Tcl1b3. This figure shows that the average expression levels of 88 genes (parameters that can be measured experimentally) can be used as a surrogate for PC3 axis, which seems to capture the trend of losing developmental potency of cells. See Fig. 4 for the labels for cells or embryos. Figure reproduced from (Sharov et al., 2003).
Cell lineage trajectory and the degree of cell commitment/differentiation
Fig. 4 also shows that the progress of embryo development, e.g., unfertilized eggs to blastocysts, can be represented as a trajectory in multi-dimensional transcript profile space (red arrow). Similarly, different cell lineages can be represented by corresponding trajectory, i.e., “cell lineage trajectory,” in the transcript profile space. For example, the global expression profiling of mouse ES cells, embryonal carcinoma (EC) cells, adult neural stem/progenitor (NS) cells, TS cells, and placenta has mapped individual cell types in the specific coordinates in transcript profile space (Fig. 6; (Aiba et al., 2005)). One cell lineage trajectory seems to represent the lineage of trophoblast differentiation, whereas the other seems to represent the lineage of neural differentiation (Fig. 6). When transcript profiles of ES cells during neural differentiation in the monolayer culture for 6 days (Ying et al., 2003) are mapped to this transcript profile space, the differentiating ES cells are positioned along the trajectory of neural lineage and are progressively shifted toward adult neural stem/progenitor cells from undifferentiated ES cells (Fig. 6B, (Aiba et al., 2005)). This supports the notion that the neural lineage trajectory reflects the neural commitment and differentiation from ES cells and a specific direction from ES to neural fate. A set of genes that correspond to this trajectory can also be extracted. For example, the lineage trajectory for neural differentiation represents the average expression of ~4,000 genes whose expression increased with neural commitment/differentiation. This lineage trajectory, thus, not only defines a path to neural fate, but also provides an index for the extent of commitment/differentiation. Thus, these results support the conceptual drawing of well-known “epigenetic landscape” and the concept of “chreod” – the permitted or necessary path taken by cells in development - by Conrad H. Waddington (Waddington, 1957; Slack, 2002).
Figure 6. An example of cell lineage trajectories.

Left Panel: 2D PCA plot of microarray-based global gene expression profiles. The following cells were analyzed and mapped here: undifferentiated mouse ES cells (ES_129a, ES_129b, and ES_R1), undifferentiated embryonal carcinoma cells (EC_P19 and EC_F9), TS, placenta (PL), adult neural stem/progenitor cells (NS1 and NS2), and neural differentiated (DC) cells. Two arrows indicate a trajectory from undifferentiated embryonic cells to the trophoblast/placental lineage, and a trajectory from undifferentiated embryonic cells to the neural lineage. This PC1 seems to define a path to neural fate, providing an index for the degree of commitment/differentiation. Right Panel: 2D PCA plot of microarray-based global gene expression profiles. The following cells were added to the PCA: EC cells induced to neural lineage by all-trans-retinoic acid (EC_P19 RA4) and ES cells differentiated into neural lineage (N2 d0, d1, d2, d3, d4, d5, and d6; ES cells cultured in the N2B27 medium as monolayer culture for 1, 2, 3, 4, 5 or 6 days). These cells differentiating into neural lineage are mapped on the PC1 axis, which supports the notion that PC1 defines a path to neural fate. See the text for more details. Figure reproduced from (Aiba et al., 2005).
Microarray-guided cell manipulation
A single-gene paradigm, which prevails in cell and developmental biology for many years, provides researchers the confidence that if a proper cell assay system can be established, a gene that cause an important function can be found by transfecting a cDNA expression library. In this functional cloning strategy, a cell that received a gene of this particular function by chance will be identified for its phenotype, and the gene will be subsequently isolated. Many genes, including oncogenes and MyoD (Lassar et al., 1986; Tapscott, 2005), have been successfully isolated in this manner. The recent discovery of Nanog gene, which plays a pivotal role in the maintenance of pluripotent ES cells without LIF (Chambers et al., 2003), has provided further assurance about the utility of functional cloning approach.
Although this single-gene paradigm has been and will continue to be successful, it is conceivable that there will be biological problems that require multi-gene paradigm, which requires cooperation of multiple genes for the conversion or alteration of the cell phenotype. In this case, the strategy of transfecting cDNA library into cells will not work, because, as with third-order enzyme kinetics, the chances that proper combination of more than two genes will be co-transfected into the same cell are extremely rare. Identification of such genes may be greatly facilitated by the multi-step cell conversion strategy using the global expression profiling (Ko, 2001). Let’s consider one of the major current interests in the stem cell biology, which is to convert the differentiated cells, such as fibroblast cells, to more potent undifferentiated cells, such as ES cells. First, the global gene expression profiles will map both fibroblast cells and ES cells in multi-dimensional transcript profile space. Fibroblast cells will then be manipulated by either overexpressing or reducing a possible candidate gene. If the position of the manipulated cells is shifted closer to that of ES cells, this cell will be further modified by the overexpression or reduction of another gene. It is conceivable that the cells can be altered to different cell types through such multi-step gene modifications. Similarly, the principle of the microarray-guided cell manipulation can also be applied to the cell conversion by environmental and nutritional manipulation.
TOWARDS THE COMPREHENSIVE UNDERSTANDING OF GENOMIC PROGRAM OF EARLY DEVELOPMENT
As discussed in earlier section, the global gene expression profiling can provide a comprehensive snapshot of cellular state. For example, analysis of different time point or time-course of developing tissues/organs can reveal the developmental program that governs that transition (e.g., (Buttitta et al., 2003)). Analysis of embryos manipulated environmentally or embryos with a specific gene mutated or disrupted can identify candidate downstream genes (e.g., (Zakin et al., 2000; Cui et al., 2002; Okubo et al., 2005; Williams et al., 2005)). Analysis of mouse embryos mutagenized with ENU and other reagents can help to understand the nature of mutation and functions of the affected genes (Seltmann et al., 2005). Despite many such examples of successful application of global gene expression profiles to the analysis of normal and mutant embryos, there are many challenges to be addressed. Three main challenges will be discussed below.
Small size and heterogeneity of embryonic materials
One major technical challenge of the global gene expression profiles of mouse embryos is that their size is very small and cannot provide enough materials to work with. For example, the analysis of early embryonic lethal caused by a gene mutation requires the global gene expression profiling of mouse preimplantation embryos. Unlike the work done previously by pooling embryos from the same state (e.g., (Hamatani et al., 2004)), the analysis of the mutant embryo should ideally be done on individual embryos. When a mutant embryo (e.g., Gene−/−) cannot be distinguished visually from a wild type embryo (e.g., Gene+/+), it is usually impossible to pool embryos according to their genotype. Even for postimplantation embryos or adult organs, the same problem will arise, when the cell heterogeneity in the materials needs to be avoided. Indeed, the global gene expression profiles of the mixed cell population are often problematic, because the critical changes of gene expression patterns in a minor population of the cells would be diluted by the presence of other cells without any changes. This problem can be addressed either by doing microdissection or FACS-sorting the desired cell population, which can be identified morphologically, by immunostaining with a specific antibody, or by cell/tissue-specific promoter-driven GFP (e.g., (Abe et al., 1998; Hubner et al., 2003)).
Performing the global gene expression profiles of small samples, such as a single embryo and a single cell, are still difficult, but the recent improvement of probe-amplification technology seems to provide solutions to this problem (Van Gelder et al., 1990; Ginsberg, 2005; Kurimoto et al., 2006; Nygaard and Hovig, 2006). However, the results are usually noisy with the random fluctuation of individual gene expression levels. Because gene expression levels at a single cell level show intrinsic stochastic variations (Ko, 1992; Kaern et al., 2005; Raser and O’Shea, 2005), such an intrinsic variation of gene expression regulation is difficult to distinguish from the technical noise of the gene expression levels caused by the probe-amplification procedures. Therefore, the global gene expression profiles obtained from a small amount of materials have to be interpreted with extra cautions.
The problem of cell heterogeneity can also be addressed to some extent by using in situ (ISH) or whole-mount in situ (WISH) hybridization technique. Although large-scale ISH/WISH methods have been applied to mouse intestine (Komiya et al., 1997), E9.5 embryos (Neidhardt et al., 2000; Gitton et al., 2002), and E9.5 and E10.5 embryos (Reymond et al., 2002), it has been difficult to apply this method to early embryos, because of their small size (~60 μm diameter in the case of preimplantation embryos) and fragility. This has been recently overcome by a chamber system that utilizes both transwell inserts for parallel processing and capillary action for gentle buffer exchanges (Yoshikawa et al., 2006).
Delineating gene regulatory pathways/cascades
One of the most desirable applications of the global gene expression profiling is to identify downstream genes affected by the mutation, overexpression, or repression of primary gene. Although there are some successful examples (e.g, (Zakin et al., 2000; Cui et al., 2002; Okubo et al., 2005; Williams et al., 2005)), such an application of global gene expression profiles is still difficult for the following reasons. First, it is possible that a defect of primary gene is compensated by other genes, and thus, the alteration of global gene expression patterns is not necessary caused by the primary gene. This problem can be addressed by examining earlier embryos, whose gene expression patterns are not yet fully compensated. Second, genes encoding transcription factors often form a cascade of regulatory chain, where ‘gene 1’ regulates ‘gene 2’, which regulates ‘gene 3’, and so on. In this case, global expression profiling will detect the expression changes of not only ‘gene 2’ – direct downstream of ‘gene 1’, but also ‘gene 3’. In other words, global expression profiling will capture not only the primary changes of gene expressions, but also the secondary and tertiary changes of gene expressions. This problem can be addressed by examining the direct binding of the transcription factor to the regulatory sequences of target genes. As such, chromatin immunoprecipitation (ChIP) microarray (ChIP-on-chip) has been used to identify the downstream target genes by precipitating chromatin-bound DNA with antibody raised against a specific transcription factor and then hybridizing the isolated DNA to microarrays of probes for promoter regions of known genes. Boyer et al. examined the genome-wide distributions of well-known ES cell transcription factors (OCT4, SOX2, and NANOG) in human ES cells (Boyer et al., 2005). Similar technique, ChIP-PET, has been used to identify the downstream of Oct4 and Nanog in mouse ES cells (Loh et al., 2006). Although the validity of individual bindings has not been or cannot be tested, the data narrow down many possible connections based on the analysis of TF-binding sites and possibly provide novel binding sites.
Complexity, stochasticity, and nonlinearity of a gene regulatory network
Early mouse embryos and ES cells will provide an excellent model system to study the structure and dynamics of a gene regulatory network, because the involvement of limited number of cell types makes the complex cell-to-cell interactions less prominent. Nonetheless, even considering a single cell we begin to realize that a gene regulatory network is much more complex than appreciated and the straightforward logic of gene activation cascades may not be relevant. A gene, particularly a transcription factor, regulates a large number of genes, which in turn regulate many other genes. The regulation includes positive- and negative-feedback loops. Here it is important to distinguish the structure, i.e., connections or wiring of the gene regulatory network and the dynamics, i.e., behavior or kinetics of the gene regulatory network. An analysis of this complex network structure is difficult, but an analysis of this nonlinear dynamics will be even more daunting. However, ultimately the embryo development has to be understood and explained as the dynamics of the gene regulatory network (Davidson and Erwin, 2006). To this end, at least the following steps have to be taken as future research directions.
First, genome-wide analysis of all potential regulatory sites has to be carried out. This will be achieved first by mapping computationally the consensus regulatory sequences of all known transcription factors (~3,000 genes). Ideally, binding sites of all these transcription factors should be identified experimentally by using ChIP-on-chip or ChIP-PET assays, as it has been done for Oct3/4, Nanog, and Sox2 (Boyer et al., 2005; Loh et al., 2006). The information will include many false-positives and false-negatives, but these computational and experimental approaches will dramatically reduce the number of possible connection/wiring among genes. The work will elucidate a possible global structure of gene regulatory network.
Second, global expression profiling has to be carried out on a variety of cell types in embryos and ES cells (“differentiation state of a cell”), cells manipulated environmentally, e.g., in different culture conditions (“physiological state of a cell”), cells with the manipulation of individual genes – one gene at a time. Changes of gene expression patterns have to be followed over the course of time. The work will provide the information on both the structure (though indirectly) and dynamics of the gene regulatory network.
Third, a virtual gene regulatory network, which represent both the structure and dynamics information obtained as mentioned above, has to be built in the computer system. The model should incorporate an inherently stochastic nature of gene expression regulation (Ko, 1992; Kaern et al., 2005; Raser and O’Shea, 2005). The model has to be tested and improved by repeating the cycle of computer simulations and experiments on gene- or environmentally-manipulated cells. That is, the changes of global gene expression profiles obtained by computer simulations will be compared to those obtained by the experiments. The discrepancy can be fed into to the global gene regulatory network model so that the results of computer simulation will follow more closely the experimental results. Such an approach has already begun to apply to a small scale network (e.g., (Kastner et al., 2002), and (Kaern et al., 2005; Raser and O’Shea, 2005) for reviews), but the genome-wide analysis and modeling will face enormous complexity of a gene regulatory network.
CONCLUDING REMARKS
Efforts by many researchers for last two decades have established an infrastructure for large-scale genomics methodologies for mouse embryology, including the global gene expression profiling discussed in this review. Global expression profiling will continue to be used to screen genes differentially expressed among different cell types or before and after the cell or gene manipulation. However, its use will gradually shift from the gene discovery phase to the gene expression monitoring phase. Systematic analysis of early embryos and stem cells will not only enhance our understanding of animal development, but also provide means to manipulate the cells, especially stem cells, at will for potential therapeutic applications to dysfunctional and aging organs in regenerative medicine.
Acknowledgments
I would like to thank members of Ko laboratory, especially Drs. Alexei Sharov, Toshio Hamatani, Kazuhiro Aiba, Mark Carter, and Ryo Matoba for the recent original work mentioned in the text. I would like to thank Dr. Linzhao Cheng of the Johns Hopkins University for suggesting the use of stock index as an analogy for the index of developmental potency. I would also like to thank two anonymous reviewers who provided useful and constructive suggestions to improve the manuscript. Finally, I would like to thank Dr. Philippe Soriano for giving me an opportunity to write an article in this special issue of Developmental Dynamics. This work was supported by the Intramural Research Program of the National Institute on Aging, NIH.
References
- Abe K, Ko MSH, MacGregor GR. A systematic molecular genetic approach to study mammalian germline development. Int J Dev Biol. 1998;42:1051–1065. [PMC free article] [PubMed] [Google Scholar]
- Aiba K, Sharov AA, Carter MG, Foroni C, Vescovi AL, Ko MSH. Defining a developmental path to neural fate by global expression profiling of mouse embryonic stem cells and adult neural stem/progenitor cells. Stem Cells. 2005 doi: 10.1634/stemcells.2005-0332. [DOI] [PubMed] [Google Scholar]
- Anisimov SV, Tarasov KV, Tweedie D, Stern MD, Wobus AM, Boheler KR. SAGE identification of gene transcripts with profiles unique to pluripotent mouse R1 embryonic stem cells. Genomics. 2002;79:169–176. doi: 10.1006/geno.2002.6687. [DOI] [PubMed] [Google Scholar]
- Bammler T, Beyer RP, Bhattacharya S, Boorman GA, Boyles A, Bradford BU, Bumgarner RE, Bushel PR, Chaturvedi K, Choi D, et al. Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005;2:351–356. doi: 10.1038/nmeth754. [DOI] [PubMed] [Google Scholar]
- Bortvin A, Eggan K, Skaletsky H, Akutsu H, Berry DL, Yanagimachi R, Page DC, Jaenisch R. Incomplete reactivation of Oct4-related genes in mouse embryos cloned from somatic nuclei. Development. 2003;130:1673–1680. doi: 10.1242/dev.00366. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink T, Hochedlinger K, Bell G, Jaenisch R. ES cells derived from cloned and fertilized blastocysts are transcriptionally and functionally indistinguishable. Proc Natl Acad Sci U S A. 2006;103:933–938. doi: 10.1073/pnas.0510485103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, Abeygunawardena N, Holloway E, Kapushesky M, Kemmeren P, Lara GG, et al. ArrayExpress-a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003;31:68–71. doi: 10.1093/nar/gkg091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 2000;18:630–634. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]
- Brown AL, Kay GF. Bex1, a gene with increased expression in parthenogenetic embryos, is a member of a novel gene family on the mouse X chromosome. Hum Mol Genet. 1999;8:611–619. doi: 10.1093/hmg/8.4.611. [DOI] [PubMed] [Google Scholar]
- Buttitta L, Tanaka TS, Chen AE, Ko MSH, Fan CM. Microarray analysis of somitogenesis reveals novel targets of different WNT signaling pathways in the somitic mesoderm. Dev Biol. 2003;258:91–104. doi: 10.1016/s0012-1606(03)00116-7. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Carter MG, Piao Y, Dudekula DB, Qian Y, VanBuren V, Sharov AA, Tanaka TS, Martin PR, Bassey UC, Stagg CA, et al. The NIA cDNA project in mouse stem cells and early embryos. C R Biol. 2003;326:931–940. doi: 10.1016/j.crvi.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Cui CY, Durmowicz M, Tanaka TS, Hartung AJ, Tezuka T, Hashimoto K, Ko MSH, Srivastava AK, Schlessinger D. EDA targets revealed by skin gene expression profiles of wild-type, Tabby and Tabby EDA-A1 transgenic mice. Hum Mol Genet. 2002;11:1763–1773. doi: 10.1093/hmg/11.15.1763. [DOI] [PubMed] [Google Scholar]
- Davidson EH, Erwin DH. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, et al. Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci U S A. 1996;93:6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evsikov AV, de Vries WN, Peaston AE, Radford EE, Fancher KS, Chen FH, Blake JA, Bult CJ, Latham KE, Solter D, et al. Systems biology of the 2-cell mouse embryo. Cytogenet Genome Res. 2004;105:240–250. doi: 10.1159/000078195. [DOI] [PubMed] [Google Scholar]
- Evsikov AV, Solter D. Comment on “ ‘Stemness’: transcriptional profiling of embryonic and adult stem cells” and “a stem cell molecular signature”. Science. 2003;302:393. doi: 10.1126/science.1082380. author reply 393. [DOI] [PubMed] [Google Scholar]
- Fortunel NO, Otu HH, Ng HH, Chen J, Mu X, Chevassut T, Li X, Joseph M, Bailey C, Hatzfeld JA, et al. Comment on “ ‘Stemness’: transcriptional profiling of embryonic and adult stem cells” and “a stem cell molecular signature”. Science. 2003;302:393. doi: 10.1126/science.1086384. author reply 393. [DOI] [PubMed] [Google Scholar]
- Gerhard DS, Wagner L, Feingold EA, Shenmen CM, Grouse LH, Schuler G, Klein SL, Old S, Rasooly R, Good P, et al. The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC) Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD. RNA amplification strategies for small sample populations. Methods. 2005;37:229–237. doi: 10.1016/j.ymeth.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Gitton Y, Dahmane N, Baik S, Ruiz i Altaba A, Neidhardt L, Scholze M, Herrmann BG, Kahlem P, Benkahla A, Schrinner S, et al. A gene expression map of human chromosome 21 orthologues in the mouse. Nature. 2002;420:586–590. doi: 10.1038/nature01270. [DOI] [PubMed] [Google Scholar]
- Hamatani T, Carter MG, Sharov AA, Ko MSH. Dynamics of global gene expression changes during mouse preimplantation development. Dev Cell. 2004;6:117–131. doi: 10.1016/s1534-5807(03)00373-3. [DOI] [PubMed] [Google Scholar]
- Harrison SM, Dunwoodie SL, Arkell RM, Lehrach H, Beddington RS. Isolation of novel tissue-specific genes from cDNA libraries representing the individual tissue constituents of the gastrulating mouse embryo. Development. 1995;121:2479–2489. doi: 10.1242/dev.121.8.2479. [DOI] [PubMed] [Google Scholar]
- Holliday R. DNA methylation and epigenotypes. Biochemistry (Mosc) 2005;70:500–504. doi: 10.1007/s10541-005-0144-x. [DOI] [PubMed] [Google Scholar]
- Hollnagel A, Oehlmann V, Heymer J, Ruther U, Nordheim A. Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J Biol Chem. 1999;274:19838–19845. doi: 10.1074/jbc.274.28.19838. [DOI] [PubMed] [Google Scholar]
- Hubner K, Fuhrmann G, Christenson LK, Kehler J, Reinbold R, De La Fuente R, Wood J, Strauss JF, 3rd, Boiani M, Scholer HR. Derivation of oocytes from mouse embryonic stem cells. Science. 2003;300:1251–1256. doi: 10.1126/science.1083452. [DOI] [PubMed] [Google Scholar]
- Hudelist G, Singer CF, Pischinger KI, Kaserer K, Manavi M, Kubista E, Czerwenka KF. Proteomic analysis in human breast cancer: identification of a characteristic protein expression profile of malignant breast epithelium. Proteomics. 2006;6:1989–2002. doi: 10.1002/pmic.200500129. [DOI] [PubMed] [Google Scholar]
- Ikeo K, Ishi-i J, Tamura T, Gojobori T, Tateno Y. CIBEX: center for information biology gene expression database. C R Biol. 2003;326:1079–1082. doi: 10.1016/j.crvi.2003.09.034. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Warren D, Spencer F, Kim IF, Biswal S, Frank BC, Gabrielson E, Garcia JG, Geoghegan J, Germino G, et al. Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A Stem Cell Molecular Signature. Science. 2002;12:12. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Kastner J, Solomon J, Fraser S. Modeling a hox gene network in silico using a stochastic simulation algorithm. Dev Biol. 2002;246:122–131. doi: 10.1006/dbio.2002.0664. [DOI] [PubMed] [Google Scholar]
- Ko MSH. Induction mechanism of a single gene molecule: stochastic or deterministic? Bioessays. 1992;14:341–346. doi: 10.1002/bies.950140510. [DOI] [PubMed] [Google Scholar]
- Ko MSH. Embryogenomics: developmental biology meets genomics. Trends Biotechnol. 2001;19:511–518. doi: 10.1016/s0167-7799(01)01806-6. [DOI] [PubMed] [Google Scholar]
- Ko MSH. Molecular biology of preimplantation embryos: primer for philosophical discussions. Reprod Biomed Online. 2005;10(Suppl 1):80–87. doi: 10.1016/s1472-6483(10)62212-2. [DOI] [PubMed] [Google Scholar]
- Ko MSH, Kitchen JR, Wang X, Threat TA, Hasegawa A, Sun T, Grahovac MJ, Kargul GJ, Lim MK, Cui Y, et al. Large-scale cDNA analysis reveals phased gene expression patterns during preimplantation mouse development. Development. 2000;127:1737–1749. doi: 10.1242/dev.127.8.1737. [DOI] [PubMed] [Google Scholar]
- Komiya T, Tanigawa Y, Hirohashi S. A large-scale in situ hybridization system using an equalized cDNA library. Anal Biochem. 1997;254:23–30. doi: 10.1006/abio.1997.2399. [DOI] [PubMed] [Google Scholar]
- Kuo WP, Jenssen TK, Butte AJ, Ohno-Machado L, Kohane IS. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics. 2002;18:405–412. doi: 10.1093/bioinformatics/18.3.405. [DOI] [PubMed] [Google Scholar]
- Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, Ueda HR, Saitou M. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 2006;34:e42. doi: 10.1093/nar/gkl050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin JE, Frank BC, Gavras H, Sultana R, Quackenbush J. Independence and reproducibility across microarray platforms. Nat Methods. 2005;2:337–344. doi: 10.1038/nmeth757. [DOI] [PubMed] [Google Scholar]
- Lassar AB, Paterson BM, Weintraub H. Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell. 1986;47:649–656. doi: 10.1016/0092-8674(86)90507-6. [DOI] [PubMed] [Google Scholar]
- Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics. 1996;33:151–152. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- Lin YN, Matzuk MM. High-throughput discovery of germ-cell-specific genes. Semin Reprod Med. 2005;23:201–212. doi: 10.1055/s-2005-872448. [DOI] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006 doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Mah N, Thelin A, Lu T, Nikolaus S, Kuhbacher T, Gurbuz Y, Eickhoff H, Kloppel G, Lehrach H, Mellgard B, et al. A comparison of oligonucleotide and cDNA-based microarray systems. Physiol Genomics. 2004;16:361–370. doi: 10.1152/physiolgenomics.00080.2003. [DOI] [PubMed] [Google Scholar]
- Mahner M, Kary M. What exactly are genomes, genotypes and phenotypes? And what about phenomes? J Theor Biol. 1997;186:55–63. doi: 10.1006/jtbi.1996.0335. [DOI] [PubMed] [Google Scholar]
- Marra M, Hillier L, Kucaba T, Allen M, Barstead R, Beck C, Blistain A, Bonaldo M, Bowers Y, Bowles L, et al. An encyclopedia of mouse genes. Nat Genet. 1999;21:191–194. doi: 10.1038/5976. [DOI] [PubMed] [Google Scholar]
- Mikkers H, Frisen J. Deconstructing stemness. Embo J. 2005;24:2715–2719. doi: 10.1038/sj.emboj.7600749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- Murrell A, Rakyan VK, Beck S. From genome to epigenome. Hum Mol Genet. 2005;14(Spec No 1):R3–R10. doi: 10.1093/hmg/ddi110. [DOI] [PubMed] [Google Scholar]
- Neidhardt L, Gasca S, Wertz K, Obermayr F, Worpenberg S, Lehrach H, Herrmann BG. Large-scale screen for genes controlling mammalian embryogenesis, using high-throughput gene expression analysis in mouse embryos. Mech Dev. 2000;98:77–94. doi: 10.1016/s0925-4773(00)00453-6. [DOI] [PubMed] [Google Scholar]
- Nygaard V, Hovig E. Options available for profiling small samples: a review of sample amplification technology when combined with microarray profiling. Nucleic Acids Res. 2006;34:996–1014. doi: 10.1093/nar/gkj499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo T, Knoepfler PS, Eisenman RN, Hogan BL. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development. 2005;132:1363–1374. doi: 10.1242/dev.01678. [DOI] [PubMed] [Google Scholar]
- Paigen K, Eppig JT. A mouse phenome project. Mamm Genome. 2000;11:715–717. doi: 10.1007/s003350010152. [DOI] [PubMed] [Google Scholar]
- Park PJ, Cao YA, Lee SY, Kim JW, Chang MS, Hart R, Choi S. Current issues for DNA microarrays: platform comparison, double linear amplification, and universal RNA reference. J Biotechnol. 2004;112:225–245. doi: 10.1016/j.jbiotec.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, Knowles BB. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell. 2004;7:597–606. doi: 10.1016/j.devcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Pyle AD, Donovan PJ, Lock LF. Chipping away at ‘stemness’. Genome Biol. 2004;5:235. doi: 10.1186/gb-2004-5-8-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkovic A, Yan MSC, Klysik M, Matzuk M. Discovery of germ cell-specific transcripts by expressed sequence tag database analysis. Fertil Steril. 2001;76:550–554. doi: 10.1016/s0015-0282(01)01966-5. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. Stemness”: Transcriptional Profiling of Embryonic and Adult Stem Cells. Science. 2002;12:12. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- Raser JM, O’Shea EK. Noise in gene expression: origins, consequences, and control. Science. 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, Fukuda S, Ru K, Frith MC, Gongora MM, et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006;16:11–19. doi: 10.1101/gr.4200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond A, Marigo V, Yaylaoglu MB, Leoni A, Ucla C, Scamuffa N, Caccioppoli C, Dermitzakis ET, Lyle R, Banfi S, et al. Human chromosome 21 gene expression atlas in the mouse. Nature. 2002;420:582–586. doi: 10.1038/nature01178. [DOI] [PubMed] [Google Scholar]
- Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 2005;19:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijter JM, Van Kampen AH, Baas F. Statistical evaluation of SAGE libraries: consequences for experimental design. Physiol Genomics. 2002;11:37–44. doi: 10.1152/physiolgenomics.00042.2002. [DOI] [PubMed] [Google Scholar]
- Seltmann M, Horsch M, Drobyshev A, Chen Y, de Angelis MH, Beckers J. Assessment of a systematic expression profiling approach in ENU-induced mouse mutant lines. Mamm Genome. 2005;16:1–10. doi: 10.1007/s00335-004-3012-x. [DOI] [PubMed] [Google Scholar]
- Sevignani C, Calin GA, Siracusa LD, Croce CM. Mammalian microRNAs: a small world for fine-tuning gene expression. Mamm Genome. 2006;17:189–202. doi: 10.1007/s00335-005-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Dudekula DB, Ko MSH. Genome-wide assembly and analysis of alternative transcripts in mouse. Genome Res. 2005a;15:748–754. doi: 10.1101/gr.3269805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Dudekula DB, Ko MSH. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics. 2005b;21:2548–2549. doi: 10.1093/bioinformatics/bti343. [DOI] [PubMed] [Google Scholar]
- Sharov AA, Piao Y, Matoba R, Dudekula DB, Qian Y, VanBuren V, Falco G, Martin PR, Stagg CA, Bassey UC, et al. Transcriptome analysis of mouse stem cells and early embryos. PLoS Biol. 2003;1:E74. doi: 10.1371/journal.pbio.0000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack JM. Conrad Hal Waddington: the last Renaissance biologist? Nat Rev Genet. 2002;3:889–895. doi: 10.1038/nrg933. [DOI] [PubMed] [Google Scholar]
- Stanton JL, Macgregor AB, Green DP. Using expressed sequence tag databases to identify ovarian genes of interest. Mol Cell Endocrinol. 2002;191:11–14. doi: 10.1016/s0303-7207(02)00046-1. [DOI] [PubMed] [Google Scholar]
- Tan PK, Downey TJ, Spitznagel EL, Jr, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC. Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res. 2003;31:5676–5684. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka TS, Kunath T, Kimber WL, Jaradat SA, Stagg CA, Usuda M, Yokota T, Niwa H, Rossant J, Ko MSH. Gene expression profiling of embryo-derived stem cells reveals candidate genes associated with pluripotency and lineage specificity. Genome Res. 2002;12:1921–1928. doi: 10.1101/gr.670002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ, Yi EC, Dai H, Thorsson V, Eng J, et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics. 2004;3:960–969. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci U S A. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Waddington CH. The strategy of the genes. London: George Allen & Unwin Ltd; 1957. [Google Scholar]
- Wang QT, Piotrowska K, Ciemerych MA, Milenkovic L, Scott MP, Davis RW, Zernicka-Goetz M. A genome-wide study of gene activity reveals developmental signaling pathways in the preimplantation mouse embryo. Dev Cell. 2004;6:133–144. doi: 10.1016/s1534-5807(03)00404-0. [DOI] [PubMed] [Google Scholar]
- Wang S, Cowan CA, Chipperfield H, Powers RD. Gene expression in the preimplantation embryo: in-vitro developmental changes. Reprod Biomed Online. 2005;10:607–616. doi: 10.1016/s1472-6483(10)61668-9. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wei CL, Miura T, Robson P, Lim SK, Xu XQ, Lee MY, Gupta S, Stanton L, Luo Y, Schmitt J, et al. Transcriptome profiling of human and murine ESCs identifies divergent paths required to maintain the stem cell state. Stem Cells. 2005;23:166–185. doi: 10.1634/stemcells.2004-0162. [DOI] [PubMed] [Google Scholar]
- Williams TM, Williams ME, Kuick R, Misek D, McDonagh K, Hanash S, Innis JW. Candidate downstream regulated genes of HOX group 13 transcription factors with and without monomeric DNA binding capability. Dev Biol. 2005;279:462–480. doi: 10.1016/j.ydbio.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Yan W, Rajkovic A, Viveiros MM, Burns KH, Eppig JJ, Matzuk MM. Identification of Gasz, an evolutionarily conserved gene expressed exclusively in germ cells and encoding a protein with four ankyrin repeats, a sterile-alpha motif, and a basic leucine zipper. Mol Endocrinol. 2002;16:1168–1184. doi: 10.1210/mend.16.6.0864. [DOI] [PubMed] [Google Scholar]
- Yauk CL, Berndt ML, Williams A, Douglas GR. Comprehensive comparison of six microarray technologies. Nucleic Acids Res. 2004;32:e124. doi: 10.1093/nar/gnh123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol. 2003;21:183–186. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Piao Y, Zhong J, Matoba R, Carter MG, Wang Y, Goldberg I, Ko MSH. High-throughput screen for genes predominantly expressed in the ICM of mouse blastocysts by whole mount in situ hybridization. Gene Expr Patterns. 2006;6:213–224. doi: 10.1016/j.modgep.2005.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen T, Wurmbach E, Pfeffer RL, Ebersole BJ, Sealfon SC. Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res. 2002;30:e48. doi: 10.1093/nar/30.10.e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakin L, Reversade B, Virlon B, Rusniok C, Glaser P, Elalouf JM, Brulet P. Gene expression profiles in normal and Otx2−/− early gastrulating mouse embryos. Proc Natl Acad Sci U S A. 2000;97:14388–14393. doi: 10.1073/pnas.011513398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng F, Baldwin DA, Schultz RM. Transcript profiling during preimplantation mouse development. Dev Biol. 2004;272:483–496. doi: 10.1016/j.ydbio.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Zeng F, Schultz RM. Gene expression in mouse oocytes and preimplantation embryos: use of suppression subtractive hybridization to identify oocyte- and embryo-specific genes. Biol Reprod. 2003;68:31–39. doi: 10.1095/biolreprod.102.007674. [DOI] [PubMed] [Google Scholar]