

Abstract
Focal adhesion kinase (FAK) plays key roles in cell adhesion and migration. We now report that the delayed rectifier Kv2.1 potassium channel, through its LD-like motif in N-terminus, may interact with FAK and enhance phosphorylation of FAK397 and FAK576/577. Overlapping distribution of Kv2.1 and FAK was observed on soma and proximal dendrites of cortical neurons. FAK expression promotes a polarized membrane distribution of the Kv2.1 channel. In Kv2.1-transfected CHO cells, formation of the Kv2.1-FAK complex was stimulated by fibronectin/integrin and inhibited by the K+ channel blocker tetraethylammonium (TEA). FAK phosphorylation was minimized by shRNA knockdown of the Kv2.1 channel, point mutations of the N-terminus, and TEA, respectively. Cell migration morphology was altered by Kv2.1 knockdown or TEA, hindering cell migration activity. In wound healing tests in vitro and a traumatic injury animal model, Kv2.1 expression and co-localization of Kv2.1 and FAK significantly enhanced directional cell migration and wound closure. It is suggested that the Kv2.1 channel may function as a promoting signal for FAK activation and cell motility.
Keywords: Adhesion, Migration, FAK, Kv2.1 channel, Phosphorylation, Wound healing
INTRODUCTION
Focal adhesion kinase (FAK) is a major tyrosine kinase activated upon cell attachment with extracellular matrix (Ezratty et al., 2005). It binds to several proteins to regulate cell adhesion and migration (Sieg et al., 1999; Tilghman et al., 2005). FAK phosphorylation induced by integrin-mediated cell adhesion is essential for regulation of focal adhesion turnover, cell spreading, migration, tumor invasion and cell proliferation (Mitra et al., 2005; Westhoff et al., 2004). Conversely, molecular mechanisms underlying FAK activation and functions are poorly understood.
K+ channels are heterogeneous families of membrane proteins that mediate K+ efflux and determine a cell’s intrinsic electrical excitability (Hille, 2001; Misonou et al., 2005; Zhu et al., 1999). Previous studies have shown that some K+ channels may function in migration of non-neuronal cells (Schilling et al., 2004; Schwab et al., 2007). Using hERG1-channel inhibitors, it was shown that tyrosine phosphorylation of FAK and the activity of Rac1, a small GTPase, were dependent on the Kv1.3 activity (Cherubini et al., 2005). The first requirement for a cell to initiate migration is the acquisition of a polarized morphology that enables it to turn intracellularly generated forces into net cell locomotion (Huttenlocher, 2005). Up to now, a putative role for a K+ channel in cell polarization has not been reported.
Voltage-gated outward delayed rectifier Kv channels, specifically the Kv2.1 channel, are the major somatodendritic K+ channels in the brain (Malin and Nerbonne, 2002; Misonou et al., 2005; Pal et al., 2003). Kv2.1 channel often shows clustered distributions on the cell membrane previously linked to regulation of intracellular Ca2+ (Antonucci et al., 2001). The mechanism that controls Kv2.1 channel distribution is still not well defined and whether the Kv2.1 patterning affects cellular functions is not clear. In spite of extensive investigations into Kv2.1 and FAK physiology, a functional or structural link between Kv2.1 and FAK is unknown. We now present initial evidence showing that the cellular distribution of the Kv2.1 channel exhibits a polarized pattern regulated by interaction with FAK and this Kv2.1-FAK interaction in turn promotes a polarized cell morphology and motility in cultured neurons and non-neuronal cells. Further, we provide evidence suggesting a critical role for Kv2.1 expression and possible Kv2.1-FAK interaction in an animal model of traumatic injury.
MATERIAL AND METHODS
Reagents, cell lines and primary cultures
Monoclonal antibodies were purchased from Upstate (Charlottesville, VA; anti-Kv2.1), BD bioscience (San Jose, CA; anti-FAK, anti-pFAK397), and Sigma-Aldrich (St. Louis, MO; anti-Flag, anti-Myc, and β-actin). Rabbit polyclonal antibodies against pFAK576/577 were purchased from Cell Signal (Danvers, MA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) except where specified. The Chinese hamster ovary (CHO) cell line and HEK 293 cells were maintained with Eagle’s MEM medium from ATCC (Manassas, VA) with 10% FBS (Invitrogen, Carlsbad, CA) in a 5% CO2 atmosphere at 37°C. FAK+/+ (CRL-2645) and FAK−/− (CRL-2644) cells were purchased from ATCC and maintained in DMEM (Cellgro, Herndon, VA). Primary cortical neurons and glial cells were cultured as described previously (Yu and Kerchner, 1998; Yu et al., 1997).
Expression vectors and transfection
The cDNA encoding Flag epitope tagged Kv2.1 and Kv2.1 deletion mutants were amplified by PCR using specific primers and subcloned into the mammalian expression vector pCMV-Tag1 (Stratagene, La Jolla, CA). EGFP tagged Kv2.1, Kv1.5, and Kv2.1- Δ1–50 were subcloned into pEGFP-C1 (Clontech, Mountain View, CA) for further experiments. EGFP-Kv2.1-L45S was generated using Quick Change Kit (Strategene La Jolla, CA) from a verified EGFP-Kv2.1 vector. Myc-tagged FAK was amplified from human brain hippocampus Marathon-Ready cDNA (Clontech) with Myc sequence and subcloned into pCMV-Tag1 (Stratagene). FAKK454R vector was generated from verified pCMV-Myc-FAK vector. The authenticity of all vectors was verified by DNA sequencing. All transfection experiments were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to standard protocol. In brief, cell lines were seeded at 2×105 cells per well into six-well plates 24 hr before transfection. Cells were transfected in serum-free medium with 6 μl of Lipofectamine reagent and 1.4 μg total DNA per well. Stable transfection clones were selected by G418 for at least 1 month. Selection of Kv2.1 expressing cell lines was achieved using Western blot and electrophysiological recordings.
Western blot and immunoprecipitation assays
Cells were lysed with modified RIPA buffer (50 mM HEPES, pH 7.3, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, and protease inhibitor cocktail (Roche, Nutley, NJ)) for 30min, followed by centrifugation at 13,000g for 20 min. The protein concentration was subsequently determined using bicinchoninic acid (BCA) protein assay. For immunoblotting, 50 μg protein was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membrane (Amersham, Piscataway, NJ). After blockage with 0.2% Tween in Tris-buffered saline (TBS-T) containing 5% nonfat milk, membranes were probed overnight at 4°C by specific antibodies diluted in TBS-T with 5% nonfat milk. After washing three times with TBS-T, AP-conjugated secondary antibodies were then added for detection. The signal was developed using AP substrate (Promega, Madison, WI). Immunoprecipitation was carried out as previously described (Zeng et al., 2003). Briefly, cell lysates (1 mg) were applied with the indicated antibodies (2–4 μg) overnight at 4°C, followed by incubation with protein G beads (Upstate) for 2 hrs while constantly rocking. The immune complexes were washed three times by modified RIPA buffer without sodium deoxylcholate and SDS at 4°C. The samples were directly applied to immunoblotting after boiling in the NuPAGE LDS sample buffer (Invitrogen).
For control sample of mouse cortex proteins (MBP), the cortex tissue was collected and homogenized in ice-cold lysis buffer (50 mM HEPEs, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, and 1% sodium deoxycholate) supplemented with protease inhibitor cocktail (Roche). The homogenized sample was centrifuged at 13,000 rpm for 30 min and the supernatant containing cortical total proteins was collected. The protein concentration was determined by BCA protein assay (Pierce Biotechnology, Rockford, IL).
Immunocytochemistry
For cells cultured on glass-bottom or plastic dishes, the samples were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.2% Triton X-100 for 5 min. After blocking for 1 hr with 10% horse serum in PBS, the cells were incubated with specific antibodies for 2 hrs. For frozen samples, the slides were fixed with 3% paraformaldehyde/0.1% Triton X-100 for 30 min at room temperature. After blocking with 8% BSA containing 0.03% Triton X-100 for 60 min, the sections were washed with PBS for 15 min. They were then incubated overnight at 4°C with properly diluted antibodies. This was followed by incubation with Cy3–conjugated goat anti–mouse IgG (1:250; Pierce) and Alexa Fluor 488–conjugated goat anti–rabbit antibody (1:200; Invitrogen) for 60 min. Fluorescent images were viewed and analyzed using either a Nikon fluorescence microscope (TE-2000-S; Nikon, Melville, NY) or a Zeiss confocal microscope (LSM PASCAL; Zeiss, Thornwood, NY). Further image analysis was performed using Adobe Photoshop software (San Jose, CA).
Whole-cell patch clamp recording
Outward delayed rectifier K+ currents were recorded using whole-cell voltage clamp techniques as described previously (Yu and Kerchner, 1998; Yu et al., 1997). Briefly, cells in 35-mm dishes were placed on the stage of an inverted microscope (Nikon), whole-cell configuration was obtained, and membrane currents were recorded using an EPC-7 amplifier (List-Electronic, Germany). Series resistance compensation was routinely applied during recordings. Current and voltage signals were collected by the data acquisition/analysis program PULSE (HEKE, Lambrect, Germany). Currents were digitally sampled at 0.33 kHz and filtered at 3 Hz by a 3-pole Bessel filter. The extracellular solution contained (in mM): NaCl 115, KCl 2.5, MnCl2 2.0, HEPES 10, BAPTA 0.1, D-glucose 10 and tetrodotoxin (TTX; 0.1 μM). The electrode solution contained (in mM): KCl 120, MgCl2 1.5, CaCl2 1.0, Na2-ATP 2.0, BAPTA 1.0 and HEPES 10. Solution pH was adjusted to 7.3.
Stimulation of cell adhesion by fibronection
Cells were serum-starved overnight and harvested by 0.25% trypsin-EDTA treatment. Trypsin was inhibited with 0.5 mg/ml soybean trypsin inhibitor (Invitrogen) and the cells were then collected by centrifugation after washed twice with medium containing soybean trypsin inhibitor. Then the cells were resuspended in medium and maintained for 1 hr at 37°C.
To perform cell adhesion experiments, 1×105 EGFP control CHO, EGFP-Kv2.1 CHO cells were plated for the indicated time on 6-well plates pre-coated with 10 μg/ml fibronectin. Random fields were photographed after 90 min, 180 min, or 360 min using 10x phase-contrast microscope (Nikon). Photographs were evaluated for the percentage of cells undergoing spreading. Stationary cells were described as phase-bright and punctual, whereas spreading cells were not phase-bright and contained extensive visible membrane protrusions. >200 cells were counted in each of ten fields for each cell line. The results represent data from four separate experiments.
Kv2.1 targeting shRNA constructs and lentivirus particle packaging
The vectors for shRNA, pLKO.1-TRC cloning vector (10878), pLKO.1-TRC control vector (10879), envelope vector pMD2.G and packaging vector psPAX2were obtained from Addgene (Addgene Inc, Cambridge, MA). The specific Kv2.1 shRNA were designed and generated according to the protocol from Addgene. Briefly, the specific paired oligo DNA targeting specific sequence in Kv2.1 gene (GCCTTGGAGCTAGAACAGAAA, S2; CGCCTTCACCTCTATTCTCAA, S3) was synthesized by Operon (Operon Biotechnologies, Huntsville, AL). Double-strand DNA was subcloned into pLKO.1-TRC cloning vector via AgeI and EcoRI restriction enzyme (New England Biolabs, Hertfordshire, UK) after anealing. The sequences were verified by a DNA sequencer (ABI Prism Model 377; Foster City, CA). The lentivirus particles were packaged according to the manual from Addgene. In brief, the pLKO.1 control vector or vector containing the Kv2.1 shRNA sequence (S2), was co-transfected with envelope vector pMD2.G and packaging vector psPAX2 into HEK293 cells. The medium was changed 24 hrs later and the lentivirus particles in the cell culture supernant were collected after 48 hrs for further investigation.
In vitro wound healing assay
Cell migration was assessed using an in vitro wound healing assay (Zeng et al., 2003). 3×105 cells were grown for 12 hrs on fibronectin-coated 6-well plates. After cell attachment, the monolayer was scratched with a sterile plastic 200 μl micropipette tip. Each well was washed with serum free medium ≥ 5 times, followed by photographs of the initial wound site taken after marking the scratch edge with a permanent marker. At various times up to 24 hrs, the initial wound site was identified and subsequently photographed. The movement speed of the wound edge was determined by the wound size at a given time.
Corneal epithelial wound healing assay
An in vivo assay of epithelial wound healing was performed on two month-old WT (SV129) mice from Jackson Laboratories (Bar Harbor, ME, USA). Experiments were conducted in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The mice were divided into control (mock DNA) and Kv2.1 shRNA treatment groups. Mice were anesthetized with intraperitoneal injection of 4% chloral hydrate at 400 mg/kg. Central corneal epithelium was removed with a dull scalpel from limbus to limbus under a dissecting microscope. Extreme care was taken to minimize injury to the epithelial basement membrane and stroma. While under anesthesia ocular surfaces were protected from drying by topical administration of sterile saline. The shRNA-S2 and mock vector transfection reagents were prepared 30 min before injection. 1.5μl Lipofectamine was added into 50 μl PBS incubated at room temperature for 5 min before 0.5 μg pLKO.1-S2 or pLKO.1 control DNA. The reagent was injected into the subconjunctival region 2 days before and everyday after surgery. Mice were killed by lethal injection (4% chloral hydrate) 3 days after operation. The eyes were then enucleated, frozen, and processed for assessment of wound closure using immunohistology for Kv2.1 and FAK or Hematoxylin and eosin (H&E) staining.
Statistics analysis
Student’s two-tailed t test was used for comparison of two experimental groups; multiple comparisons were done using one-way ANOVA test followed by Dunnett’s post-hoc test and Dunn’s test for comparison to a single control group. Significance was identified if P value was less than 0.05. Mean values were reported together with the standard deviation (SD).
RESULTS
Formation of the Kv2.1-FAK complex
Immunoprecipitation using acutely isolated cortical neuronal lysates from adult mouse brains suggested a possible association between Kv2.1 and FAK (Fig. 1a). Immunofluorescent staining of cultured mouse cortical neurons detected some clustered overlapping distributions of Kv2.1 and FAK on the soma and proximal dendritic membrane (Fig. 1b and 1c).
Figure 1. Interaction and colocalization of Kv2.1 channel and FAK in different cells.

Immunoprecipitation (IP), immunoblotting (IB), and confocal fluorescence imaging were applied to assess Kv2.1 and FAK interaction and colocalization in neuronal and non-neuronal cells. a. The same amount of cell lysate prepared from adult mouse cortical protein was immunoprecipitated with FAK, Kv2.1, or control IgG antibodies. Associated proteins were detected by Western blot with specific antibodies. Mouse brain protein (MBP) from WT mice was used as a positive control for Kv2.1 or FAK expression (see Method for preparation of MBP). b and c. Localized overlapping of Kv2.1 and FAK staining in primary cultured mouse cortical neurons of 4 and 12 days in vitro (b and c, respectively). Fluorescent images were taken after double staining with FAK and Kv2.1 antibodies. Red color corresponds to Kv2.1 and green to FAK. Arrowheads indicate colocalized puncta in the cell membrane and/or proximal dendrites. Orange color in the overlay image signifies colocalization of Kv2.1 and FAK. Scale bar = 10 μm.
In FAK+/+ CRL-2645 fibroblast cells, Kv2.1 channels were transiently expressed. Formation of the migration structure lamellipodia was apparent in Kv2.1-transfected cells (Fig. 2a). Distribution of Kv2.1 channels showed a polarized pattern; plentiful Kv2.1 channels aggregated at the cell’s caudal portion and focal adhesion sites of the leading edge of lamellipodia, and overlapped with FAK staining at these locations (Fig. 2a for fibroblasts and Fig. 3e for CHO cells). On the other hand, in FAK−/− CRL-2644 cells, besides the lack of lamellopodia, Kv2.1 channels expressed in these cells did not show the polarized distribution pattern (Fig. 2b).
Figure 2. Polarized distribution of Kv2.1 channel in FAK+/+ cells.

Kv2.1 channels tagged with GFP probe were transiently expressed in FAK+/+ (CRL-2645, ATCC) and FAK−/− (CRL-2644, ATCC) fibroblast cells. Cellular distributions of Kv2.1 channels (green) and FAK (anti-FAK antibody fluorescent staining, red) were assessed under a confocal microscope 48 hrs after Kv2.1 transfection (24 hrs after planting on glass-bottom dishes). a. FAK+/+ cells developed migratory morphology, showing the formation of lamellipodia (*) on the up portion of the cell. Overlaps of clustered Kv2.1 and FAK staining were observed in both the caudal portion and at the leading edges of lamellipodia (arrowhead) in the cell. The polarized location of Kv2.1 at the caudal part of the cells and overlaps of Kv2.1 and FAK staining are also illustrated in the three-dimensional z-axis side panels of the merged image. The black arrow points to clustered overlapping of Kv2.1 and FAK at the caudal part of the cells. b. Kv2.1 channels were similarly expressed in FAK deficient cells. The majority of these cells did not have lamellipodia morphology. The distribution of Kv2.1 did not show a polarized pattern as in FAK+/+ cells, although Kv2.1 clusters scattered throughout the cell membrane. The z-axis panels reveal the distribution of Kv2.1 channels on the cell membrane, especially the bottom portion of the cell. Note the ruffle like structures but not lamellipodia around the FAK−/− cell. Bar=10 μm.
Figure 3. Kv2.1 current and channel phosphorylation in CHO cells.

Immunoprecipitation and immunoblotting assays were performed in CHO cells transfected with full length Kv2.1. a. Western blot confirmed a lack of expression of Kv2.1 in WT CHO cells and expression in CHO cells stably transfected with Kv2.1 or EGFP-tagged Kv2.1 vector. MBP from WT mice was used as a positive control for Kv2.1 expression. b. Whole-cell recordings of IK currents in EGFP-transfected CHO cells, EGFP-Kv2.1, EGFP-Kv2.1-L45S, and EGFP-Kv2.1-Δ1–50 stably transfected cells. The membrane holding potential was −70 mV, IK current was triggered by stepping the membrane potential to +40 mV for 400 ms. Using this protocol, there was little outward K+ current detectable in control cells. Sizable outward IK current was recorded in EGFP-Kv2.1 cells (representative of 24 cells, see text for the average current amplitude). The currents in cells expressing EGFP-Kv2.1-L45S or EGFP-Kv2.1-Δ1–50 were relatively smaller (n=22 and 10, respectively, per group). The outward K+ currents were sensitive to blockage by the K+ channel blocker TEA (data not shown). c. Regulation of Kv2.1 phosphorylation by FAK kinase activity. pCMV-Kv2.1 vector was co-transfected with Myc-tagged WT FAK gene and the FAK mutant FAKK454R into CHO cells using Lipofectamine. The expression levels of Kv2.1 and FAK were detected with specific antibodies. Binding to WT FAK enhanced phosphorylated Kv2.1, while mutation of the enzymatic site K454 diminished the regulatory effect.
Fibronectin-stimulated Kv2.1-FAK binding and the role of Kv2.1 in FAK phosphorylation
Western blot analysis demonstrated Kv2.1 protein expression in two Kv2.1 stable expression CHO cell lines (Fig. 3a). EGFP-vector control CHO cells had little endogenous K+ currents (Fig. 3b) (Yu and Kerchner, 1998). Transfection of the EGFP-Kv2.1 gene resulted in sizable outward currents (steady-state current = 310±30 pA, `n = 24) sensitive to the K+ channel blocker TEA, while transfection with the mutated gene EGFP-L45S or EGFP-Kv2.1-Δ1–50 led to smaller currents (232±19 pA and 73±27 pA, n=22 and 10, respectively) (Fig. 3b). Interestingly, there was some evidence for a novel regulation of Kv2.1 phosphorylation by FAK. CHO cells were co-transfected with Kv2.1 gene and myc-tagged WT FAK or the FAKK454R mutant, the latter gene lacks an essential site for kinase activity (Sonoda et al., 2000). Forty-eight hours after transfection, phosphorylated Kv2.1 was enhanced by co-transfection with WT FAK but not with FAKK454R (Fig. 3c). These data at least partly explained why the Kv2.1 point mutants at L45, which did not change the p-loop of the K+ channel activity domain, showed attenuated FAK binding as well as reduced K+ currents (Fig. 3c).
Identification of Kv2.1 interaction domain and Kv2.1 membrane distribution
To map the FAK binding domain in the Kv2.1 protein, a flag-tagged Kv2.1 construct and its deletion mutants (1–416, 184–416, 184–857, 416–857, and 712–857) were expressed in CHO cells. Co-immunoprecipitation analysis showed that only mutants retaining the T1 domain in N-terminus (1–857 of full length and 1–416) were able to associate with FAK (Fig. 4a and 4b), suggesting the importance of the N-terminal domain in the binding activity.
Figure 4. The LD-like motif in the N-terminus of Kv2.1 is required for binding to FAK.

Kv2.1 and deletion mutants were tested for essential structures in binding to FAK. a. Schematic representations of flag-tagged full-length and deletion mutants of Kv2.1. b. Full length and deletion mutant genes were introduced into CHO cells with Lipofectamine 2000. The expression level of Kv2.1 and mutants were detected using anti-flag antibodies (top panel) 48 hrs after transfection. The complex was precipitated with the anti-flag antibody and the FAK protein level was determined using the anti-FAK antibody (bottom panel). The asterisk marks the genes that show affinity to FAK at the bottom panel. c. Consensus sequences of the LD motif in paxillin and Kv2.1. d. Site-mutation of LD-like motif in Kv2.1 reduced Kv2.1 binding to FAK. Normal mouse lgG was tested as a binding control. When either the amino acid L45 was replaced with serine or the 1–50 segment was deleted, Kv2.1-FAK binding activity decreased dramatically. Results are representative of three independent experiments. e–g. CHO cells were transiently transfected with EGFP tagged Kv2.1, Kv2.1-L45S, or Kv1.5, respectively, and immunostained against pFAK397 (Red). Overlapping of FAK and Kv2.1 staining was found in cells expressing the full length Kv2.1, especially on the leading edge of the bipolar shaped cells in 3e (the yellow arrow points in the side image point to the clustered overlap of Kv2.1 and FAK at the caudal portion of the cell). Similar overlaps were not seen in cells transfected with Kv2.1-L45S (f), Kv2.1-Δ1–50 (data not shown) and Kv1.5 (g). The confocal side images in 3e to 3g verified the membrane localization of the channels. The similar distribution pattern of FAK and Kv2.1 is additionally demonstrated in the profile line graphs on the right, shown as almost complete overlapped red and green lines across the cell surface (along the white line in merged images). The line graph was generated by the confocal system software LSM Image (Carl Zeiss MicroImaging Inc, San Francisco, CA). Noticeable deviations and separate peaks of the two lines were easily detectable in the line graphs from Kv2.1-L-45S (f) and Kv1.5 transfected (g) cells. Of note that FAK distribution in cells of full length Kv2.1 was markedly different from the other two cells, suggesting the Kv2.1 expression might influence FAK membrane distribution. Bar=10 μm.
Focusing on the N-terminal domain of Kv2.1, we observed an amino acid sequence homologous to the LD motif (45–56, LDRLPRTRLGKL) of paxillin that might bind to FAK (Fig. 4c). When leucine L45 within the motif was site-mutated to serine (Kv2.1-L45S), the Kv2.1-FAK binding activity was reduced in transfected CHO cells (Fig. 4d). Consistent with this, deletion of the 1–50 segment of the N-terminus (Kv2.1-Δ1–50) virtually abolished Kv2.1-FAK binding (Fig. 4d). Confocal imaging was performed on CHO cells transiently transfected with EGFP-tagged Kv2.1, Kv2.1-L45S, and Kv2.1-Δ1–50, respectively. Twenty-four hours after transfection, some clustered distribution of full length Kv2.1 overlapped with FAK staining (Fig. 4e), but much less colocalization was observed in cells transfected with Kv2.1-L45S (Fig. 4f) or Kv2.1-Δ1–50 (data not shown). To elucidate whether the formation of the K+ channel-FAK complex was channel subtype specific, the EGFP-tagged Kv1.5 protein was introduced into WT CHO cells. Immunostaining showed no characteristic overlaps with FAK as seen with Kv2.1 (Fig. 4g).
Next, Kv2.1-FAK interaction was examined using immunoprecipitation with the anti-FAK antibody (Fig. 5 top panel) and anti-Kv2.1 antibody (Fig. 5a bottom panel) in adherent and suspension CHO cells of EGFP control and EGFP-Kv2.1. Ninety minutes after the cells were plated onto fibronectin (FN)-coated dishes, formation of the Kv2.1-FAK complex increased in adherent cells (94±9% and 104±11% increases in the gray intensity of the top and bottom panels, respectively, P<0.05 compared with non FN controls, n=3 independent assays). The K+ channel blocker, TEA (5 mM), significantly attenuated the interaction between Kv2.1 and FAK (20±5% and 40±8% reduction in top and bottom panels, P<0.05 compared with the FN group, Fig. 5).
Figure 5. FN stimulated Kv2.1–FAK interaction.
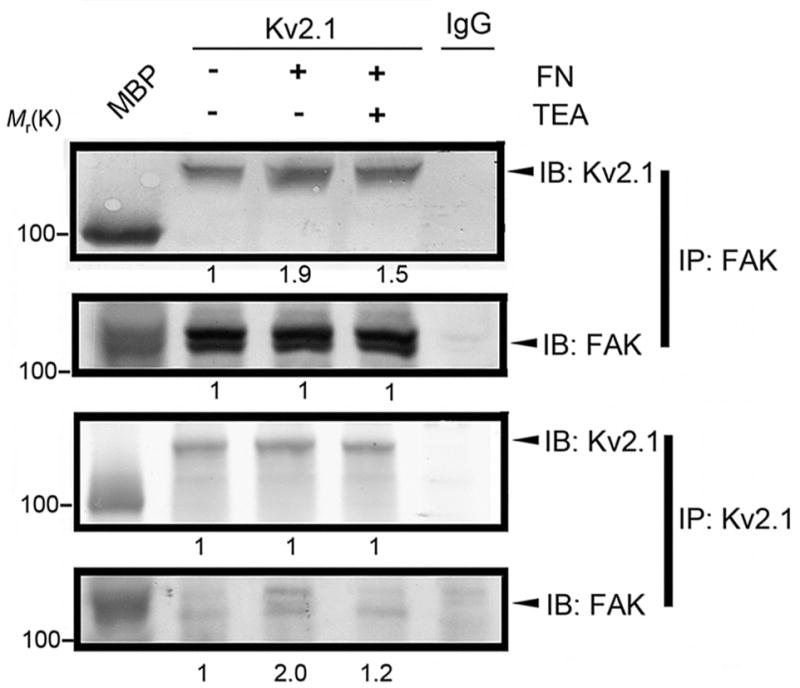
The immunocomplex was pulled down using Kv2.1, FAK, or IgG antibody. Adherent cells stimulated by fibronectin (FN) showed increased Kv2.1-FAK complex formation (top and bottom panels), while the expression of FAK and Kv2.1 proteins was unchanged. The K+ channel blocker TEA (5 mM) showed inhibitory effects on the complex formation. Values under immunoblots represent band intensity ratios normalized to the intensity of suspended cells in the absence of FN. The numbers underneath immunoblots are mean values from 3 independent assays.
We observed that autophosphorylation of FAK tyr-397 increased in EGFP-Kv2.1 cells compared to EGFP blank vector-transfected control CHO cells (91±5% increase in the presence of FN, n=5, P<0.05; Fig. 6a). Phosphorylation of FAK576/577 is subsequent to FAK397 autophosphorylation (Calalb et al., 1995; Owen et al., 1999). Consistent with this notion, the Src kinase inhibitor PP2 (10 μM) showed no effect on FAK397 autophosphorylation, but substantially blocked pFAK576/577 phosphorylation in EGFP-Kv2.1 CHO cells (90±4% increase in Kv2.1/FN group, P<0.05, but no significant difference between vector controls and Kv2.1/FN/PP2 group, n=5 per group; Fig. 6a).
Figure 6. Formation of the Kv2.1–FAK complex and its affect on FAK phosphorylation.

Western blot analysis was applied to examine FAK phosphorylation. a. Kv2.1-FAK interaction increased FAK phosphorylation. Kv2.1 protein levels were verified in EGFP control cells and in EGFP-Kv2.1 transfected CHO cells (top panel). The phosphorylation of pFAK397 and pFAK576/577 was increased in EGFP-Kv2.1 cells compared to EGFP controls. Phosphorylation was further enhanced by FN-mediated cell adhesion (90 min in FN-coated Petri dishes). The Src kinase inhibitor PP2 (10 μM) blocked phosphorylation of pFAK576/577, but not the autophosphorylation of pFAK397. b. Kv2.1-FAK interaction is essential to enhance FAK phosphorylation. The phosphorylation level of FAK was tested in CHO cells stably transfected with EGFP, EGFP-Kv2.1, EGFP-Kv2.1-L45S, and EGFP-Kv2.1Δ1–50, respectively. c. Knockdown of Kv2.1 decreased FAK phosphorylation. EGFP-Kv2.1 CHO cells were transiently transfected with two Kv2.1-targeting sequences shRNA (S2 and S3) or mock DNA for 3 days. Kv2.1 expression was then checked with Western blotting. Kv2.1 manipulation did not alter total FAK expression, while phosphorylation at FAK397 and FAK576/577 was reduced. d. Knockdown of Kv2.1 decreased phosphorylation of exogenously expressed FAK in FAK−/− fibroblast cells. Myc-tagged FAK vector was introduced into FAK−/− cells and a stable expression clone was selected. The expression of Kv2.1 was knocked down using Lipofectamine (Fig. 3h, left 2 lanes) or packaged lentiviral particle (Fig. 3h, right 2 lanes) for 3 days. e. WT CHO cells could adhere to the culture dish in a Kv2.1 or Kv2.1-FAK independent manner. In these cells, neither the K+ channel blocker TEA nor 25 mM K+ showed a significant effect on phosphorylation of FAK397 using pFAK397 specific antibody. In adherent Kv2.1-CHO cells, on the other hand, the above treatments selectively inhibited FAK576/577 phosphorylation. PP2 showed expected inhibitory action on pFAK576/577 in these cells. The phosphorylation levels of FAK397 and FAK576/577 in DNKv2.1-CHO cells were substantially lower than that of Kv2.1-CHO cells. f. In adherent fibroblasts expressing FAK (FAK+/+), TEA, 25 mM K+ or PP2 did not affect FAK397 autophosphorylation while they each attenuated FAK576/577 phosphorylation. g. In FAK+/+fibroblasts, stimulation of K+ efflux by valinomycin (1 μM) selectively augmented phosphorylation of FAK576/577. FAK level was not significantly changes. The phosphorylation level of FAK was detected using indicated antibodies. β-Actin is shown as the loading control. All experiments represent ≥ 3 independent experiments.
To verify whether the putative binding between Kv2.1 and FAK was important for FAK phosphorylation/activation, we used cell lines stably transfected with EGFP-Kv2.1-L45S or EGFP-Kv2.1-Δ1–50 vectors. The phosphorylation of FAK397 and FAK576/577 in EGFP-Kv2.1-L45S and EGFP-Kv2.1-Δ1–50 cells was decreased or even abolished in comparison to EGFP-Kv2.1 CHO cells. The immunoblotting gray intensity was doubled in Kv2.1 cells (P<0.05, n=5) but there was no difference between Kv2.1 mutation groups and vector controls (Fig. 6b).
To further investigate the specificity of Kv2.1 in regulating FAK phosphorylation, the Kv2.1 expression in EGFP-Kv2.1 CHO cells was knocked down using shRNA, targeting the sequence GCCTTGGAGCTAGAACAGAAA (S2) or CGCCTTCACCTCTATTCTCAA (S3) in the Kv2.1 complete DNA sequence (CDS). The Kv2.1 level in these cells was reduced by about 50%, while FAK expression was not affected (Fig. 6c). The down regulation of Kv2.1 significantly attenuated phosphorylation of FAK397 and FAK576/577 (P>0.05 between knockdown and control groups, n=3 per group; Fig. 6c). FAK−/− fibroblast cells stably transfected with FAK were also tested. The phosphorylation of FAK397 and FAK576/577 was reduced when endogenous Kv2.1 expression was suppressed with shRNA through Lipofectamine or Lentivirus particles (Fig. 6d). We verified that the FAK protein level was comparable in cells tested (Fig. 5, 6a, 6b, 6c and 6d), indicating that the observed changes were not due to altered FAK levels.
In the presence of nifedipine (10 μM), elevating extracellular K+ concentration to reduce the K+ gradient across the plasma membrane is an effective way of restraining K+ efflux. A high K+ medium containing 25 mM KCl and the K+ channel blocker TEA were thus tested to understand whether K+ channel activation and K+ efflux were essential for the Kv2.1-induced FAK phosphorylation. In CHO cells transfected with full length Kv2.1, phosphorylation levels of FAK397 and FAK576/577 were higher than that in WT CHO cells. TEA (5 mM) and the high K+ medium had little effect on FAK397 autophosphorylation, but markedly attenuated FAK576/577 phosphorylation (Fig. 6e). As expected, PP2 did not show inhibitory effect on pFAK397, while it blocked FAK576/577 phosphorylation (Fig. 6e). The effects of TEA, high K+ and PP2 on pFAK397 and pFAK576/577 were similarly observed in FAK+/+ fibroblasts (Fig. 6f). Furthermore, stimulation of K+ efflux by the K+ ionophore valinomycin (1 μM) increased phosphorylation of FAK576/577 (Fig. 6g). The phosphorylation levels of FAK proteins were diminished in CHO cells transfected with the dominant-negative Kv2.1 gene (DNKv2.1), further supporting the idea that normal Kv2.1 channel activity either directly or indirectly affected FAK phosphorylation (Fig. 6e).
The role of Kv2.1 in cell polarization, ruffle generation and lamellipodia formation
After the CHO cells were plated for 6 hrs, more EGFP-Kv2.1 cells underwent transformation to a polarized shape than EGFP control cells (75% vs. 40% bipolar shaped cells). To evaluate the morphological changes in a more quantitative manner, wound healing tests were performed, and we counted cells near the wound frontline that displayed orientation towards the wounded area. More such cells were detected in the Kv2.1-CHO group than in two mutated Kv2.1 groups (Fig. 7a and 7b). More specifically, polarized Kv2.1-CHO cells, but not Kv2.1-L45S and Kv2.1-Δ1–50 cells, generated increased numbers of ruffles along the leading edge of orientated cells (Fig. 7a and 7c). Consistent with above observations, the characteristic distribution of β-actin alone the axis of cell body was confirmed in Kv2.1-transfected cells. On the other hand, β-actin staining showed more diffused distribution in cells transfected with EGFP alone, Kv2.1-L45S, or Kv2.1-Δ1–50 (Fig. 7d).
Figure 7. Kv2.1 expression promoted proper morphology for cell migration.

Morphological alterations of migrating cells were examined in Kv2.1-transfected CHO cells. a. Phase contrast images showed enhanced cellular orientation of EGFP-Kv2.1 cells, compared to EGFP control, EGFP-Kv2.1-L45S, or EGFP-Kv2.1-Δ1–50 cells. The second row shows images of enlarged magnification illustrating the ruffle formation along the cellular leading edge. Increased numbers of ruffles were observed in EGFP-Kv2.1 cells in comparison to EGFP control cells and the two Kv2.1 mutated cell lines. The bar graph in b represents data from 6 independent experiments, where orientated cells with body angles of greater than 45° against the wound front edge were identified. The bar graph in c represents 250 cells from 12 experiments. d. Immunostaining with anti-β-actin antibody (red) showed distribution of β-actin alone the axis of cell body in Kv2.1 cells, but not in vector control or cells with mutated Kv2.1 genes where more diffused distribution was seen. Arrow signifies the direction of cell migration. Mean ± SD. *. P < 0.05 compared to EGFP control CHO cells; #. P < 0.05 compared to EGFP-Kv2.1 CHO cells.
Role of Kv2.1 in cell adhesion and migration
CHO cells stably transfected with EGFP blank vector or EGFP-Kv2.1 were tested for integrin-stimulated adhesion to fibronectin-coated Petri dishes. The number of attached EGFP-Kv2.1-CHO cells 90 min after plating was about twice as compared to EGFP control CHO cells (Fig. 8a and 8b). Wound healing test is a widely used assay for directional/directed cell migration. In the in vitro test, EGFP-Kv2.1-CHO cells exhibited significantly higher directional motility than control cells (Fig. 8c and 8d). In line with observed inhibitory effects on Kv2.1-FAK interaction, TEA (5 mM) restricted cell migration (Fig. 8c and 8d). More specifically, shRNA knockdown of the Kv2.1 expression also reduced the directional migration of EGFP-Kv2.1 CHO cells (Fig. 8e and 8f). We compared the wound closure speed using four stable transfection cell lines: EGFP only, EGFP-Kv2.1, EGFP-Kv2.1-L45S, and EGFP-Kv2.1-Δ1–50 cells. Transfection of CHO cells with mutated Kv2.1-L45S or Kv2.1-Δ1–50 resulted in deficient cell motility compared to cells transfected with the full length Kv2.1 (Fig. 8g and 8h). By tracking the passing routes of migrating cells in the wound edge for 6 hrs, it was evident that EGFP vector-transfected control cells exhibited slow and random movements while cells transfected with the full length Kv2.1 displayed polarized shape and faster movements orientated towards the center of the wounded region (Fig. 8i; and see Fig. 7 for morphology changes).
Figure 8. Kv2.1-expression enhanced cell adhesion and directional migration.

CHO cells expressing EGFP blank vector or EGFP-Kv2.1 were tested for their adhesion to FN-coated culture dishes and movement to the scratched region in wound healing tests. a and b. EGFP control cells and EGFP-Kv2.1 cells were used to compare the adhesion activity at various times after plating. The bar graph in b represents the percentage of adherent cells vs. total cells plated. Data were summarized from 3 independent experiments. c. Phase contrast images of wound healing of CHO cells transfected with EGFP control, EGFP-Kv2.1, and in the presence of TEA (5 mM) (blue: initial wound edge; red: 24 hrs later). The phase contrast photos at 0 hr and 24 hrs after scratch show faster and mostly complete wound closure of EGFP-Kv2.1cells compared to EGFP control cells or in the presence of TEA. TEA showed no significant effect on migration of control CHO cells (data not shown). d. The wound closure speed of the leading edge during wound healing tests. The bar graph summarizes 5 assays, showing the accelerated wound closure of EGFP-Kv2.1 CHO cells. e and f. EGFP-Kv2.1 cells were transiently transfected with Kv2.1 specific shRNA or mock DNA for 72 hrs. Using the wound healing test, both knockdown cells (S2 and S3) showed deteriorated directed movement, summarized in the bar graph of f (n = 24, 3 independent experiments). g and h. Cell migration of CHO cells transfected with EGFP blank vector, EGFP-Kv2.1, EGFP-Kv2.1-L45S, or EGFP-Kv2.1-Δ1–50 0 and 24 hrs after the initiation of wound healing. Cells without the Kv2.1 gene or with a mutated Kv2.1 gene exhibited slower movement compared to the EGFP-Kv2.1 cells. The bar graph in h shows pooled data of the migration wound closure speed (n = 24, 3 independent experiments). i. The graph tracks randomly selected cells over 6 hrs for their changes in shape and orientation of movement toward the wound healing edge. Control cells had random movements around the original location, while cells with Kv2.1 underwent to polarized cell shape transformation along a leading-lagging axis within the plane of movement towards the wounded area (upward). Kv2.1 transfected cells, but not control cells, showed directional intrusion and a retraction pattern typical of migrating cells. The bars and values represent the time after wound initiation. Cells were kept in the cell culture incubator between inspections. Mean ± SD. *. P < 0.05 compared to EGFP control CHO cells; #. P < 0.05 compared to EGFP-Kv2.1 CHO cells.
The role of Kv2.1 in corneal wound healing in vivo
In animal studies, we examined epithelial cell migration after corneal injury in a mouse model. Immunostaining with specific antibodies verified that Kv2.1 and FAK were expressed in corneal epithelial cells and their distribution largely overlapped, especially in the basal layers of the epithelium (Fig. 9a). To induce a restricted corneal injury, corneal epithelium was removed from limbus to limbus. TEA (20 mM, 10 μl subconjunctive injection after cornea injury) inhibited the epithelial cell migration and cornea wound closure (Fig. 9b and 9c). To specifically inhibit the Kv2.1 channel, the shRNA (S2) containing lipofectamine transfection reagent or mock DNA containing transfection control was subconjunctivally injected 2 days before and everyday after surgery. Corneas were then harvested 3 days after surgery. Fluorescent imaging showed a distinct reduction of the Kv2.1 expression in shRNA-transfected cells (Fig. 9d). Consistently, the putative Kv2.1-FAK complex in the wound leading edge decreased in Kv2.1 knockdown cells. H&E staining of corneas from mock DNA treated mice revealed the marked repair of epithelium defects by epithelial cell migration towards the corneal center. However, much slower migration and less cornea healing were seen in Kv2.1 knockdown mice (Fig. 9e and 9f).
Figure 9. Kv2.1 channel expression and epithelial cell migration during corneal repair.

Epithelial wound healing in vivo test was performed in a mouse model of corneal repair. Corneal wound was induced by removal of epithelium from limbus to limbus in the central region of the eye under a dissection microscope. a. Expression of Kv2.1 (green) and FAK (red) proteins in the normal mouse cornea. The overlay image shows largely overlapped distribution of Kv2.1 and FAK in epithelial cells of the cornea, especially in the basal region. E and S represent epithelium and stromal, respectively. Bar = 5 μm. b and c. H&E staining shows epithelial migration 3 days after corneal injury. Enlarged images are shown in the lower panel. In figure b, photos show normal (left) and injured epithelium soon after injury (right). In figure c, corneal repair is demonstrated in a control animal with saline injections (left); subconjunctival injection of 10 μl of 20 mM TEA immediately after injury and once a day inhibited epithelial cell migration and wound repair (right). d. Immunostaining of Kv2.1 and FAK at the wound closure edge 3 days after corneal damage in Kv2.1 knockdown experiments. Kv2.1 expression was reduced in corneas receiving Lipofectamine-shRNA-S2 comparing to Lipofectamine-control DNA treatment. Overlay images show Kv2.1/FAK colocalization in epithelial cells at the migrating front (arrowhead). Bar = 5 μm. e. H&E staining shows epithelial migration 3 days after cornea damage. The wound closure speed was markedly reduced in mice treated with Lipofectamine-shRNA-S2. The initial wound is marked with a black arrowhead and the wound leading edge is marked with a red arrowhead. f. The bar graph summarizes data from 3 separate experiments. There were significant less distances of corneal healing in Kv2.1 knockdown mice. *. P < 0.05 compared to control. N=8. Mean ± SD.
DISCUSSION
The present investigation provides novel evidence for a potential role of the Kv2.1 channel in regulating FAK activation, which implicates a new mechanism underlying cell adhesion and migration. We further reveal an essential role for leucine (L45) and the 1–50 fragment in the N-terminus of Kv2.1 in interacting with FAK, and demonstrate an FN/FAK-dependent clustering pattern of the Kv2.1 channel that plays an imperative role in cell adhesion, polarization, and directional migration under normal and pathophysiological conditions. Moreover, the formation of the Kv2.1-FAK complex and FAK576/577 phosphorylation may associate with the Kv2.1 channel activity and its mediated K+ efflux. We recognize from immunohistochemical data that the expression of Kv2.1 on the cell membrane does not entirely overlap or co-localize with FAK expression. The putative protein-protein interaction occurs mainly at the leading edge and caudal portion of migrating cells. Obviously, the channels that do not form the protein complex with FAK should serve their conventional role of controlling the membrane excitability.
According to the present investigation, Kv2.1 may affect migration by regulating FAK phosphorylation. FAK phosphorylation following adhesion is dependent on FAK’s association with Src family kinases, leading to the formation of a multi-molecular signaling complex (Sieg et al., 1999). Residue Y397 of FAK becomes autophosphorylated upon activation, allowing Src family kinases to then associate with Y397 and phosphorylate other tyrosine residues of FAK (Schlaepfer et al., 1994; Sieg et al., 1999). Our investigation shows that the Kv2.1 channel, specifically the N-terminus containing residue L45, can regulate FAK Y397 phosphorylation, and may regulate Y576/577 phosphorylation in a Src dependent manner. Since phosphorylation of Y397 and Y576/Y577 leads to an increased FAK catalytic activity (Calalb et al., 1995), enhanced FAK phosphorylation through interaction with Kv2.1 may provide a unique voltage-dependent mechanism underlying FAK activation in response to membrane activities and micro-environmental changes.
Epithelial cells stimulated with the hepatocyte growth factor revealed an elongated phenotype with process formation and FAK-paxillin complexes that are condensed at the front and back tips of cells (Liu et al., 2002). Phosphorylated FAK is also identified at adhesion sites of front and back of migrating fibroblasts (Rege et al., 2006) and in human astrocytoma cells (Hamadi et al., 2005). In our experiments, the Kv2.1-FAK complex in migrating cells is condensed on the focal adhesion sites of the leading edge and caudal portion of the cells. However, a highly condensed colocalization of FAK and Kv2.1 was identified at non-adhesive caudal portion of fibroblasts and CHO cells (Fig. 2a and Fig. 3e). The functional role of the Kv2.1-FAK complex at this location is unclear. It is likely that the still images revealed a transition state of the moving cells when their caudal portion was detached from the adhesion site. A time-lapse imaging study will be helpful for a better understanding of this issue.
Delayed rectifier IK channels are subjected to Src kinase regulations and have been linked to cellular functions other than regulating the membrane potential. For example, Sobko A et al. (Sobko et al., 1998) showed that the Src family member Fyn kinase is physically associated with and constitutively activated IK channels, including Kv1.5 and Kv2.1, in mouse Schwann cells. Inhibition of Schwann cell proliferation by herbimycin A and by K+-channel blockers suggests that the functional linkage between Src tyrosine kinases and IK channels could be important for Schwann cell proliferation and the onset of myelination. The important role of K+ channels in cell proliferation has been shown in other cell types such as endothelial cells, tumor cells, macrophages, and human myeloblastic Ml-1 cells (Renaudo et al., 2004; Vicente et al., 2003; Wang et al., 1997; Yamazaki et al., 2006). A few previous data support that some K+ channels such as the HERG K+ channels are regulated by the adhesion receptors integrins. In particular, HERG channel activation is dependent upon integrin-mediated cell adhesion (Arcangeli et al., 2004; Hofmann et al., 2001). Upon integrin-mediated cell adhesion to laminin or fibronectin, HERG channels undergo activation, evidenced by the increase in the related current (IHERG). Moreover, this activation drives cell differentiation. An intergrin-mediated activation of the inward recrifier K+ channels (KIR) was linked to phosphyrylation of the pp125FAK and neuritogenesis in neuroblastoma cells (Bianchi et al., 1995). The authors suggest that integrin-mediated activation of KIR channels is a limiting step upstream to the phosphorylation of pp125FAK in the commitment to neuritogenesis. A putative role for the Kv2.1-FAK complex in cell proliferation and differentiation remain to be elucidated, although this function unlikely exists in mature neurons.
Kv2.1 exhibits a clustered distribution in CNS neurons and various cell lines (Antonucci et al., 2001; Muennich and Fyffe, 2004; O’Connell and Tamkun, 2005). The observations in our investigation are consistent with a previous report that Kv2.1 channel is located in high-density clusters on the soma and proximal dendrites, while other Kv channels such as Kv2.2 and Kv1.5 are uniformly distributed throughout the soma and dendrites (Lim et al., 2000). However, mechanisms governing Kv2.1 distribution pattern remain largely unclear. A recent study shows that the clustering and voltage-dependent gating of Kv2.1 in cultured rat hippocampal neurons and human embryonic kidney 293 (HEK293) cells are modulated by cholinergic stimulation (Mohapatra and Trimmer, 2006). The Kv2.1 cytoplasmic C-terminal domain can act as an autonomous domain sufficient to transfer Kv2.1-like clustering, Kv2.1 trafficking, voltage-dependent activation, and cholinergic modulation to diverse Kv channels (Scannevin et al., 1996; Bentley et al., 1999; Levitan, 1999; Leung et al., 2003; Misonou et al., 2005; Mohapatra and Trimmer, 2006). The present investigation suggests that the N-terminus of Kv2.1 may also serve as a molecular structure in determining channel localization and FAK activation that leads to cell directional migration activities. It is likely that more than one mechanism are responsible for regulation of Kv2.1 channel distribution in order to cope with different functions and cellular activities. Of note, the homology between the binding site of the Kv2.1 N-terminus and the LD motif of paxillin imply a possible competition in binding to FAK. Whether Kv2.1 binds to the same paxillin binding site is unknown and requires further examination.
Our data suggest a possible mechanism for Kv2.1 membrane distribution by interacting with FAK. A morphological feature of migrating cells is their polarized cell body along a front-rear axis within the plane of movement (Nabi, 1999). The ability to establish and maintain the polarized morphology and the formation of lamellipodia are essential for directional migration. The Kv1.4 channel was found to be clustered at the leading edge of protruding lamellipodia of migrating MDCK-F cells (Reinhardt et al., 1998). The present study provides evidence that Kv2.1 channel and formation of the Kv2.1-FAK complex play a critical role in lamellipodia formation. How the polarized Kv2.1 channel distribution in migrating cells may affect the Kv2.1 conventional function of regulating excitability of neurons is an intriguing and open question. It is possible that, in accordance to its functional roles during development and under pathological condition, the distribution of Kv2.1 channel undergoes dynamic changes, showing different patterns in migrating cells from the cells that have settled down at their destination.
Previous evidence for a role of K+ channels in cell migration was focused on cultured non-neuronal cells such as tumor cells, epithelial cells, and fibroblasts (Schwab et al., 2007). The human ether-a-go-go-related gene (hERG) channels and Kv1.3 channel was shown to directly interact with β1 integrin to modulate adhesion-dependent signaling in SH-SY5Y neuroblastoma cells, T cells, and HEK293 cells (Cherubini et al., 2005; Levite et al., 2000). Cherubini A et al. (Cherubini et al., 2002) reported that the HERG isoforms could be co-immunoprecipitated with β1 integrin, and strongly modulated upon cell adhesion to appropriate extracellular matrix. Their data indicated the possibility of a complex association between membrane proteins (HERG and integrins) and cytoplasmic components, which could integrate the signaling evoked by cell adhesion to extracellular matrix with the machinery leading to cell differentiation. In the present study, the Kv2.1-FAK complex was detected in cultured neuronal, non-neuronal cells, and gene-transfected cell lines. The wide distribution and high expression of Kv2.1 channel suggest a common mechanism for regulating cell motility. We additionally elucidate a key role for Kv2.1 channel in cell directional migration during wound healing in vitro, as well as in traumatic injury in vivo. Therefore, Kv2.1 channel is not only a key factor in the regulation of membrane excitability, but it may also act as a critical regulator of cell adhesion and migration in both two-dimensional cell cultures and animals.
Some early studies showed that K+ channel blockers such as TEA and 4-amynopuridine (4-AP) prevented cell migration, suggesting that K+ channels such as Kv1.1 might support cell migration (Hendriks et al., 1999; Schwab et al., 1994; Soroceanu et al., 1999; Wang et al., 2000). Precisely how the voltage-gated Kv2.1 channel protein influences FAK and cell migration activities is obscure. Increased intracellular Ca2+ was identified as a mechanism linking K+ channels to migration (Rao et al., 2002; Yacubova and Komuro, 2002). On the other hand, K+ channel-mediated K+ efflux and increased local extracellular K+ concentrations were also proposed as a mechanism stimulating cell migration (Danker et al., 1996; Levite et al., 2000). Specifically, the Ca2+-sensitive K+ channel IK1 mediated the retraction of the trailing edge of migrating MDCK-F cells by inducing localized K+ efflux and shrinkage at the cell’s pole (Schwab et al., 2006). This is consistent with the notion that Kv1.3 mediated K+ efflux stimulates activation of T cell β1 integrin moieties and induces integrin-mediated adhesion and migration (Levite et al., 2000). We observed that increasing extracellular K+ concentration to 25 mM, a general method of antagonizing K+ efflux, reduced Kv2.1-FAK interaction and cell migration (unpublished data). Some data now support that activity of IK1 channel or a charybdotoxin-sensitive, volume/Ca2+-activated K+ channel is required for migration of MDCK-F cells (Jin et al., 2003; Schwab et al., 2006) and microglial cells (Schilling et al., 2004). This supports the importance of K+ efflux and is consistent with our observation that K+ channel blocker TEA and high K+ medium prevent FAK phosphorylation and cell migration. Collectively with the observation that Kv channels of mutated Kv2.1 genes carried smaller K+ currents, it is likely that alterations in Kv2.1 channel activity, K+ efflux, and local K+ concentration are transmembrane signals for Kv2.1-FAK interaction and regulation of cell migration.
Increased cell adhesion has been previously shown to favor cell survival (Reddig and Juliano, 2005). Compelling evidence in recent years has endorsed a pro-apoptotic role for K+ channels, including Kv2.1, in a variety of cell types (Bossy-Wetzel et al., 2004; Lang et al., 2003; Pal et al., 2005; Samoilov et al., 2003; Yu et al., 1997). At first glance, it seems contradictory that the Kv2.1 channel can promote both cell adhesion and apoptosis. A simple and rational explanation is that Kv2.1 mediated effects on cell adhesion and apoptosis are “dose/activity-dependent”. Physiological levels of Kv2.1 channel activity/expression, as in this study, promote adhesion. Alternatively, pathological overactivation or overexpression of the same channel may lead to disruption of cellular K+ homeostasis and cause apoptosis. At the expression levels utilized in this study, the typical outward K+ current observed was within the normal range for delayed rectifier currents in regular cells. We rarely observed changes in cell volume or cell death. Thus, the molecular and cellular activities observed in this investigation do not appear to be influenced by cell death or apoptosis-induced sub-population selection.
Cell adhesion and migration are related to a wide variety of physiological and pathological processes such as embryogenesis, immune defense, wound healing, and the formation of tumor metastases. Understanding the underlying molecular, cellular, and ionic mechanisms, including the adhesion molecules, membrane proteins, and intracellular transduction signaling pathways, will be fundamentally important for improving our basic and clinical knowledge in these fields. We suggest that Kv2.1 and FAK act synergistically to signal downstream targets to promote adhesive and migratory activities of neuronal and non-neuronal cells. This unique regulation may play imperative roles under both normal and pathophysiological conditions.
Acknowledgments
This investigation was supported by research grants from National Institute of Health (NIH NS42236 (S.P.Y.), NS045155 (L.W), NS045810 (S.P.Y.), NS031622 (N.L.B), NS057255 (S.P.Y.) and American Heart Association and Bugher Foundation (AHA-Bugher) Awards (0170064N (S.PY.) and 0170063N (L.W.)). The work was also supported by the NIH grant C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
References
- Antonucci DE, Lim ST, Vassanelli S, Trimmer JS. Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience. 2001;108(1):69–81. doi: 10.1016/s0306-4522(01)00476-6. [DOI] [PubMed] [Google Scholar]
- Arcangeli A, Becchetti A, Cherubini A, Crociani O, Defilippi P, Guasti L, Hofmann G, Pillozzi S, Olivotto M, Wanke E. Physical and functional interaction between integrins and hERG potassium channels. Biochem Soc Trans. 2004;32(Pt 5):826–827. doi: 10.1042/BST0320826. [DOI] [PubMed] [Google Scholar]
- Bentley GN, Brooks MA, O’Neill CA, Findlay JB. Determinants of potassium channel assembly localised within the cytoplasmic C-terminal domain of Kv2.1. Biochim Biophys Acta. 1999;1418(1):176–184. doi: 10.1016/s0005-2736(99)00021-8. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Arcangeli A, Bartolini P, Mugnai G, Wanke E, Olivotto M. An inward rectifier K+ current modulates in neuroblastoma cells the tyrosine phosphorylation of the pp125FAK and associated proteins: role in neuritogenesis. Biochem Biophys Res Commun. 1995;210(3):823–829. doi: 10.1006/bbrc.1995.1732. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41(3):351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15(2):954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini A, Hofmann G, Pillozzi S, Guasti L, Crociani O, Cilia E, Di Stefano P, Degani S, Balzi M, Olivotto M, Wanke E, Becchetti A, Defilippi P, Wymore R, Arcangeli A. Human ether-a-go-go-related gene 1 channels are physically linked to beta1 integrins and modulate adhesion-dependent signaling. Mol Biol Cell. 2005;16(6):2972–2983. doi: 10.1091/mbc.E04-10-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini A, Pillozzi S, Hofmann G, Crociani O, Guasti L, Lastraioli E, Polvani S, Masi A, Becchetti A, Wanke E, Olivotto M, Arcangeli A. HERG K+ channels and beta1 integrins interact through the assembly of a macromolecular complex. Ann N Y Acad Sci. 2002;973:559–561. doi: 10.1111/j.1749-6632.2002.tb04701.x. [DOI] [PubMed] [Google Scholar]
- Danker T, Gassner B, Oberleithner H, Schwab A. Extracellular detection of K+ release during migration of transformed Madin-Darby canine kidney cells. Pflugers Arch. 1996;433(1–2):71–76. doi: 10.1007/s004240050250. [DOI] [PubMed] [Google Scholar]
- Ezratty EJ, Partridge MA, Gundersen GG. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat Cell Biol. 2005;7(6):581–590. doi: 10.1038/ncb1262. [DOI] [PubMed] [Google Scholar]
- Hamadi A, Bouali M, Dontenwill M, Stoeckel H, Takeda K, Ronde P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J Cell Sci. 2005;118(Pt 19):4415–4425. doi: 10.1242/jcs.02565. [DOI] [PubMed] [Google Scholar]
- Hendriks R, Morest DK, Kaczmarek LK. Role in neuronal cell migration for high-threshold potassium currents in the chicken hindbrain. J Neurosci Res. 1999;58(6):805–814. doi: 10.1002/(sici)1097-4547(19991215)58:6<805::aid-jnr7>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Hille H. Potassium channels and chloride channels. In: Hille B, editor. Ion Channels of Excitable Membranes. 3. Sunderland, Massachusetts: Sinauer Associates, Inc.; 2001. pp. 131–162. [Google Scholar]
- Hofmann G, Bernabei PA, Crociani O, Cherubini A, Guasti L, Pillozzi S, Lastraioli E, Polvani S, Bartolozzi B, Solazzo V, Gragnani L, Defilippi P, Rosati B, Wanke E, Olivotto M, Arcangeli A. HERG K+ channels activation during beta(1) integrin-mediated adhesion to fibronectin induces an up-regulation of alpha(v)beta(3) integrin in the preosteoclastic leukemia cell line FLG 29.1. J Biol Chem. 2001;276(7):4923–4931. doi: 10.1074/jbc.M005682200. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A. Cell polarization mechanisms during directed cell migration. Nat Cell Biol. 2005;7(4):336–337. doi: 10.1038/ncb0405-336. [DOI] [PubMed] [Google Scholar]
- Jin M, Defoe DM, Wondergem R. Hepatocyte growth factor/scatter factor stimulates Ca2+-activated membrane K+ current and migration of MDCK II cells. J Membr Biol. 2003;191(1):77–86. doi: 10.1007/s00232-002-1045-1. [DOI] [PubMed] [Google Scholar]
- Lang PA, Kaiser S, Myssina S, Wieder T, Lang F, Huber SM. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am J Physiol Cell Physiol. 2003;285(6):C1553–1560. doi: 10.1152/ajpcell.00186.2003. [DOI] [PubMed] [Google Scholar]
- Leung YM, Kang Y, Gao X, Xia F, Xie H, Sheu L, Tsuk S, Lotan I, Tsushima RG, Gaisano HY. Syntaxin 1A binds to the cytoplasmic C terminus of Kv2.1 to regulate channel gating and trafficking. J Biol Chem. 2003;278(19):17532–17538. doi: 10.1074/jbc.M213088200. [DOI] [PubMed] [Google Scholar]
- Levitan IB. It is calmodulin after all! Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22(4):645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- Levite M, Cahalon L, Peretz A, Hershkoviz R, Sobko A, Ariel A, Desai R, Attali B, Lider O. Extracellular K+ and opening of voltage-gated potassium channels activate T cell integrin function: physical and functional association between Kv1.3 channels and beta1 integrins. J Exp Med. 2000;191(7):1167–1176. doi: 10.1084/jem.191.7.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH, Trimmer JS. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron. 2000;25(2):385–397. doi: 10.1016/s0896-6273(00)80902-2. [DOI] [PubMed] [Google Scholar]
- Liu ZX, Yu CF, Nickel C, Thomas S, Cantley LG. Hepatocyte growth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin-focal adhesion kinase association. J Biol Chem. 2002;277(12):10452–10458. doi: 10.1074/jbc.M107551200. [DOI] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM. Delayed rectifier K+ currents, IK, are encoded by Kv2 alpha-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci. 2002;22(23):10094–10105. doi: 10.1523/JNEUROSCI.22-23-10094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Trimmer JS. Kv2.1: a voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology. 2005;26(5):743–752. doi: 10.1016/j.neuro.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Trimmer JS. The Kv2.1 C terminus can autonomously transfer Kv2.1-like phosphorylation-dependent localization, voltage-dependent gating, and muscarinic modulation to diverse Kv channels. J Neurosci. 2006;26(2):685–695. doi: 10.1523/JNEUROSCI.4620-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muennich EA, Fyffe RE. Focal aggregation of voltage-gated, Kv2.1 subunit-containing, potassium channels at synaptic sites in rat spinal motoneurones. J Physiol. 2004;554(Pt 3):673–685. doi: 10.1113/jphysiol.2003.056192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi IR. The polarization of the motile cell. J Cell Sci. 1999;112(Pt 12):1803–1811. doi: 10.1242/jcs.112.12.1803. [DOI] [PubMed] [Google Scholar]
- O’Connell KM, Tamkun MM. Targeting of voltage-gated potassium channel isoforms to distinct cell surface microdomains. J Cell Sci. 2005;118(Pt 10):2155–2166. doi: 10.1242/jcs.02348. [DOI] [PubMed] [Google Scholar]
- Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19(7):4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23(12):4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal SK, Takimoto K, Aizenman E, Levitan ES. Apoptotic surface delivery of K+ channels. Cell Death Differ. 2005 doi: 10.1038/sj.cdd.4401792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JN, Platoshyn O, Li L, Guo X, Golovina VA, Yuan JX, Wang JY. Activation of K+ channels and increased migration of differentiated intestinal epithelial cells after wounding. Am J Physiol Cell Physiol. 2002;282(4):C885–898. doi: 10.1152/ajpcell.00361.2001. [DOI] [PubMed] [Google Scholar]
- Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005;24(3):425–439. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- Rege TA, Pallero MA, Gomez C, Grenett HE, Murphy-Ullrich JE, Hagood JS. Thy-1, via its GPI anchor, modulates Src family kinase and focal adhesion kinase phosphorylation and subcellular localization, and fibroblast migration, in response to thrombospondin-1/hep I. Exp Cell Res. 2006;312(19):3752–3767. doi: 10.1016/j.yexcr.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Reinhardt J, Golenhofen N, Pongs O, Oberleithner H, Schwab A. Migrating transformed MDCK cells are able to structurally polarize a voltage-activated K+ channel. Proc Natl Acad Sci U S A. 1998;95(9):5378–5382. doi: 10.1073/pnas.95.9.5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaudo A, Watry V, Chassot AA, Ponzio G, Ehrenfeld J, Soriani O. Inhibition of tumor cell proliferation by sigma ligands is associated with K+ Channel inhibition and p27kip1 accumulation. J Pharmacol Exp Ther. 2004;311(3):1105–1114. doi: 10.1124/jpet.104.072413. [DOI] [PubMed] [Google Scholar]
- Samoilov MO, Lazarevich EV, Semenov DG, Mokrushin AA, Tyul’kova EI, Romanovskii DY, Milyakova EA, Dudkin KN. The adaptive effects of hypoxic preconditioning of brain neurons. Neurosci Behav Physiol. 2003;33(1):1–11. doi: 10.1023/a:1021119112927. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Murakoshi H, Rhodes KJ, Trimmer JS. Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+ channel. J Cell Biol. 1996;135(6 Pt 1):1619–1632. doi: 10.1083/jcb.135.6.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T, Stock C, Schwab A, Eder C. Functional importance of Ca2+-activated K+ channels for lysophosphatidic acid-induced microglial migration. Eur J Neurosci. 2004;19(6):1469–1474. doi: 10.1111/j.1460-9568.2004.03265.x. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372(6508):786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schwab A, Nechyporuk-Zloy V, Fabian A, Stock C. Cells move when ions and water flow. Pflugers Arch. 2007;453(4):421–432. doi: 10.1007/s00424-006-0138-6. [DOI] [PubMed] [Google Scholar]
- Schwab A, Wojnowski L, Gabriel K, Oberleithner H. Oscillating activity of a Ca2+-sensitive K+ channel. A prerequisite for migration of transformed Madin-Darby canine kidney focus cells. J Clin Invest. 1994;93(4):1631–1636. doi: 10.1172/JCI117144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab A, Wulf A, Schulz C, Kessler W, Nechyporuk-Zloy V, Romer M, Reinhardt J, Weinhold D, Dieterich P, Stock C, Hebert SC. Subcellular distribution of calcium-sensitive potassium channels (IK1) in migrating cells. J Cell Physiol. 2006;206(1):86–94. doi: 10.1002/jcp.20434. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;112(Pt 16):2677–2691. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- Sobko A, Peretz A, Attali B. Constitutive activation of delayed-rectifier potassium channels by a src family tyrosine kinase in Schwann cells. Embo J. 1998;17(16):4723–4734. doi: 10.1093/emboj/17.16.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda Y, Matsumoto Y, Funakoshi M, Yamamoto D, Hanks SK, Kasahara T. Anti-apoptotic role of focal adhesion kinase (FAK). Induction of inhibitor-of-apoptosis proteins and apoptosis suppression by the overexpression of FAK in a human leukemic cell line, HL-60. J Biol Chem. 2000;275(21):16309–16315. doi: 10.1074/jbc.275.21.16309. [DOI] [PubMed] [Google Scholar]
- Soroceanu L, Manning TJ, Jr, Sontheimer H. Modulation of glioma cell migration and invasion using Cl- and K+ ion channel blockers. J Neurosci. 1999;19(14):5942–5954. doi: 10.1523/JNEUROSCI.19-14-05942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman RW, Slack-Davis JK, Sergina N, Martin KH, Iwanicki M, Hershey ED, Beggs HE, Reichardt LF, Parsons JT. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J Cell Sci. 2005;118(Pt 12):2613–2623. doi: 10.1242/jcs.02380. [DOI] [PubMed] [Google Scholar]
- Vicente R, Escalada A, Coma M, Fuster G, Sanchez-Tillo E, Lopez-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem. 2003;278(47):46307–46320. doi: 10.1074/jbc.M304388200. [DOI] [PubMed] [Google Scholar]
- Wang JY, Wang J, Golovina VA, Li L, Platoshyn O, Yuan JX. Role of K+ channel expression in polyamine-dependent intestinal epithelial cell migration. Am J Physiol Cell Physiol. 2000;278(2):C303–314. doi: 10.1152/ajpcell.2000.278.2.C303. [DOI] [PubMed] [Google Scholar]
- Wang L, Xu B, White RE, Lu L. Growth factor-mediated K+ channel activity associated with human myeloblastic ML-1 cell proliferation. Am J Physiol. 1997;273(5 Pt 1):C1657–1665. doi: 10.1152/ajpcell.1997.273.5.C1657. [DOI] [PubMed] [Google Scholar]
- Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol. 2004;24(18):8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacubova E, Komuro H. Stage-specific control of neuronal migration by somatostatin. Nature. 2002;415(6867):77–81. doi: 10.1038/415077a. [DOI] [PubMed] [Google Scholar]
- Yamazaki D, Aoyama M, Ohya S, Muraki K, Asai K, Imaizumi Y. Novel functions of small conductance Ca2+-activated K+ channel in enhanced cell proliferation by ATP in brain endothelial cells. J Biol Chem. 2006;281(50):38430–38439. doi: 10.1074/jbc.M603917200. [DOI] [PubMed] [Google Scholar]
- Yu SP, Kerchner GA. Endogenous voltage-gated potassium channels in human embryonic kidney (HEK293) cells. J Neurosci Res. 1998;52(5):612–617. doi: 10.1002/(SICI)1097-4547(19980601)52:5<612::AID-JNR13>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278(5335):114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- Zeng L, Si X, Yu WP, Le HT, Ng KP, Teng RM, Ryan K, Wang DZ, Ponniah S, Pallen CJ. PTP alpha regulates integrin-stimulated FAK autophosphorylation and cytoskeletal rearrangement in cell spreading and migration. J Cell Biol. 2003;160(1):137–146. doi: 10.1083/jcb.200206049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XR, Netzer R, Bohlke K, Liu Q, Pongs O. Structural and functional characterization of Kv6.2 a new gamma-subunit of voltage-gated potassium channel. Receptors Channels. 1999;6(5):337–350. [PubMed] [Google Scholar]