

Abstract
The S-phase DNA damage checkpoint slows replication when damage occurs during S phase. Cdc25, which activates Cdc2 by dephosphorylating tyrosine-15, has been shown to be a downstream target of the checkpoint in metazoans, but its role is not clear in fission yeast. The dephosphorylation of Cdc2 has been assumed not to play a role in S-phase regulation because cells replicate in the absence of Cdc25, demonstrating that tyrosine-15 phosphorylated Cdc2 is sufficient for S phase. However, it has been reported recently that Cdc25 is required for the slowing of S phase in response to damage in fission yeast, suggesting a modulatory role for Cdc2 dephosphorylation in S phase. We have investigated the role of Cdc25 and the tyrosine phosphorylation of Cdc2 in the S-phase damage checkpoint, and our results show that Cdc2 phosphorylation is not a target of the checkpoint. The checkpoint was not compromised in a Cdc25 overexpressing strain, a strain carrying non-phosphorylatable form of Cdc2, or in a strain lacking Cdc25. Our results are consistent with a strictly Cdc2-Y15 phosphorylation-independent mechanism of the fission yeast S-phase DNA damage checkpoint.
Keywords: S-phase DNA damage checkpoint, inter-S checkpoint, Cdc2, Cdc25, Schizosaccharomyces pombe, fission yeast
Introduction
Cells slow replication in response to DNA damage during S phase.1 This S-phase DNA damage checkpoint, also know as the intra-S checkpoint, does not completely block replication. Instead, it reduces the rate of bulk replication, about 50% in human cells, presumably allowing cells to coordinate replication with repair or bypass of the damage.1,2 Although this checkpoint has been proposed to allow for the repair of damage during S phase, there is not a strong correlation between checkpoint proficiency and damage tolerance. Furthermore, DNA damage induced before or during S phase can persist through the checkpoint and be repaired in G2.3,4 Nonetheless, loss of the checkpoint leads to increased chromosomal rearrangements and profound cancer predisposition in humans.5,6
The checkpoint pathway regulating replication in response to DNA damage is conserved amongst eukaryotes.1 Members of the ATM-family of protein kinases form the center of the checkpoint pathway, serving to recognize DNA damage and initiate checkpoint signaling. ATM itself appears to be the major kinase in the vertebrate S-phase DNA damage checkpoint; the related kinases, Mec1 and Rad3, are required for the checkpoint in budding and fission yeast, respectively. When activated, these kinases phosphorylate a number of downstream effectors including the FHA-containing effector kinases – Rad53 in budding yeast, Cds1 in fission yeast and Chk2/Cds1 in vertebrates. In addition to these checkpoint kinases, an array of accessory damage recognition and checkpoint mediator proteins are also conserved. Although this signaling pathway is well conserved, it is less clear if its targets are also conserved.
A priori, there are two ways that the checkpoint could slow replication. It could reduce the number of replication forks by inhibiting origin firing or arresting a subset of active forks, or it could slow the rate of progression of a majority of forks. Origin firing is inhibited by the checkpoint in vertebrates and in budding yeast.7-10 In vertebrates, Chk2 regulates origin firing by targeting Cdc25A for proteolysis, thus preventing the dephosphorylation and activation of S-phase cyclin-dependent kinases such as Cdk2/Cyclin E, which are required for origin firing throughout S phase.8,11
In addition to the Cdc25-dependent regulation of origin firing, there is a parallel, Cdc25-independent checkpoint mechanism in mammals.12,13 Although the mechanism of this branch of the checkpoint is not well understood, it is known to require ATM phosphorylation of MRN, a heterotrimeric recombinational repair complex consisting of Mre11, Rad50 and Nbs1. MRN is involved in homologous and non-homologous recombinational repair, as well as meiotic recombination, DNA damage signaling and telomere maintenance. The fact that MRN is required only for the Cdc25-independent branch of the checkpoint suggests that it acts downstream in the checkpoint pathway, rather than as an upstream signaling factor.12 The regulation of fork progression has also been shown to require the XRCC3 recombination protein.13 The role of MRN in the regulation of recombination, and the role of XRCC3 in regulating fork progression, has lead to the speculation that the checkpoint may slow replication fork progression through induction of replication-coupled recombinational repair.13,14
The targets of the S-phase DNA damage checkpoint in fission yeast are less well defined. The role of the tyrosine-15 phosphorylation of Cdc2 (the only cyclin-dependent kinase in fission yeast) as a checkpoint target has been well established. In response to DNA damage in G2 or replication blocks during S phase, Cdc25 is inhibited, preventing the dephosphorylation of Cdc2 tyrosine-15 and arresting cells before mitosis.15,16 It has also been reported that inhibition of Cdc25 and phosphorylation of Cdc2 tyrosine-15 are required to slow replication in response to DNA damage.17
The published work notwithstanding, there is reason to suspect that Cdc2 tyrosine phosphorylation is not the target of the S-phase DNA damage checkpoint. Since Cdc2 is the only cyclin-dependent kinase in fission yeast, and since it is required for both replication and mitosis, it has been assumed that there must be different mechanisms of Cdc2 regulation that independently regulate these two events. The model with the most experimental support proposes that different levels of Cdc2 activity trigger the different events: replication is triggered by moderate level of Cdc2 activity, comprised of tyrosine phosphorylated Cdc2/cyclin complexes, and mitosis is triggered by the high level Cdc2 activity achieved when Cdc2/cyclin complexes are dephosphorylated.18 Consistent with this model, tyrosine-15 kinase activity of the Mik1 tyrosine kinase is high in S-phase, while Cdc25 levels are low, favoring Cdc2 tyrosine phosphorylation during S-phase.19,20 Consequently, Cdc2 remains largely phosphorylated during S-phase.21 Furthermore, it is clear that the bulk of Cdc2 cannot be dephosphorylated during S-phase, because such premature dephosphorylation leads to immediate and catastrophic mitosis.22 These observations are inconsistent with general activation of Cdc25 during S-phase. Yet, for inhibition of Cdc25 to be an important target of the S-phase DNA damage target, Cdc25 would have to be active during S-phase, and required for timely replication. Therefore, its activity would have to be limited, either in extent or location, to prevent premature mitosis. Such a subtle regulatory role for Cdc25 seems unlikely, because Cdc25 can be replaced by unrelated tyrosine phosphatases, either human T-cell protein tyrosine phosphatase or over-expression of fission yeast Pyp3.23,24 In both cases, replication appears normal (our unpublished result). This line of reasoning argues against a role for Cdc25 in the S-phase DNA damage checkpoint. Therefore we have revisited the question of whether Cdc25 or the tyrosine phosphorylation of Cdc2 is required for the S-phase DNA damage checkpoint in fission yeast.
Results
We employed flow cytometry to assay the S-phase checkpoint response of fission yeast to DNA damage. To reduce cytoplasmic background and thus increase sensitivity, we performed our analyses on isolated nuclei.25 Initially, we examined the response of asynchronous cultures.17 Fission yeast spend most of their cell cycle in G2, therefore the cytometry profile of an asynchronous culture is largely 2C, with a small 1C and S-phase population (Figure 1A). The alkylating agent methyl methane sulfonate (MMS), which produces DNA damage in the form of base adducts, was used to induce DNA damage and activate the checkpoint. MMS damage is most efficiently recognized during replication, and therefore preferentially activates the S-phase checkpoint, rather than the G2 checkpoint. However, at 0.03% MMS, the standard concentration used in previous synchronous checkpoint experiments26, a significant fraction of cells in an asynchronous culture arrest in G2 (our unpublished observation). Therefore, for these experiments we used 0.015% MMS, a concentration used in previous asynchronous experiments.17 Hydroxyurea (HU), a ribonucleotide reductase inhibitor which arrests cells in the early S-phase by depleting deoxynucleotides, was used as a control for cells containing close to 1C DNA content. As previously reported, wild type cells respond to MMS treatment by accumulating as sub-2C cells, presumably due to slowing of bulk replication (Figure 1A).17,26 In contrast, the rad3Δ strain, which is DNA damage checkpoint defective, showed no significant accumulation of sub-2C cells (Figure 1B).
Figure 1.

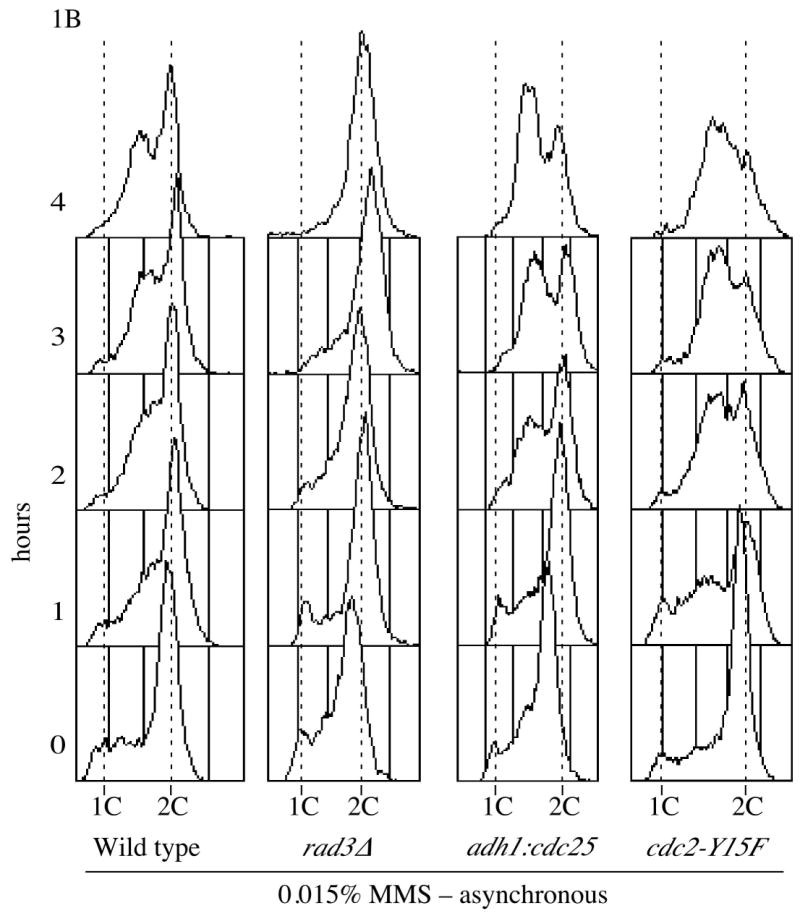
S-phase DNA damage checkpoint analysis in asynchronous cells (A). A mid-log, asynchronous cultures wild type culture (yFS104) was split three ways and incubated in the presence or absence of 0.015% MMS or 10 mM HU; samples were taken for flow cytometry every hour. (B) Asynchronous cultures of wild type (yFS104), rad3Δ (yFS189), adh1:cdc25 (yFS357) and cdc2-Y15F (KGY14) were treated and collected for flow cytometry as in panel A; for clarity, only the MMS treated samples are shown.
Although the asynchronous experiments show a robust checkpoint-dependent accumulation of sub-2C cells, it is difficult to infer cell-cycle kinetics from asynchronous experiments. To more carefully examine the effect of DNA damage on replication, we used synchronous cultures to analyze the progression of cells through S phase in the presence and absence of MMS. We synchronized cells by centrifugal elutriation, which isolates the smallest cells in a culture. Since in fission yeast cytokinesis is coincident with S phase, the smallest, newborn cells are in early G2. Thus, after elutriation we can follow a synchronous G2 population through mitosis into G1 and through S phase back to G2. As in the asynchronous experiment, we used 0.015% MMS because 0.03% MMS causes a significant fraction of the culture to arrest in G2 (our unpublished observation). Most untreated cells replicated between 80 and 120 minutes post elutriation (Figure 2A). The MMS-treated cells begin replicating about the same time as untreated cells but do not complete replication by 180 minutes. This MMS-induced slowing is abrogated in rad3Δ, confirming that it is a checkpoint response (Figure 2B).
Figure 2.


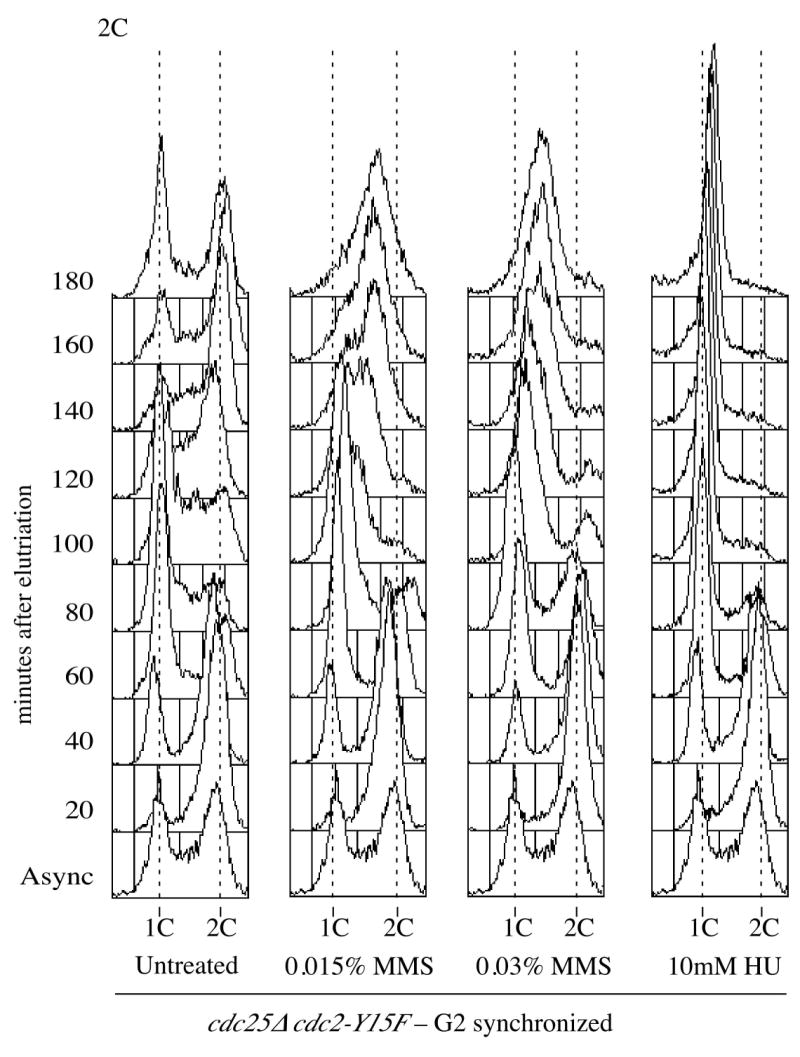
S-phase DNA damage checkpoint analysis in G2 synchronized cells. (A) Wild-type cells (yFS104) were synchronized in G2 by centrifugal elutriation, 0.015% MMS or 10 mM HU were added immediately and samples were collected every 20 minutes for flow cytometry. (B) G2 synchronized cultures of wild type (yFS104), rad3Δ (yFS189), adh1:cdc25 (yFS357) and cdc2-Y15F (KGY14) were treated and collected for flow cytometry as in panel A; for clarity, only the MMS treated samples are shown. (C) cdc25Δ cdc2-Y15F (yFS445) cells synchronized in G2, 0.015% or 0.03% MMS or 10 mM HU was added immediately and samples were collected every 20 minutes for flow cytometry.
As a third approach, we synchronized cells in G1. G1 synchronization has two advantages: the cells are past the G2/M transition, allowing us to use 0.03% MMS without evoking the G2 checkpoint, and the cultures are more synchronous, allowing for meaningful quantitation (Figure 3C). We employed a cdc10-M17ts temperature sensitive allele, which at 35°C inactivates the fission yeast S-phase transcription factor, to block cells in G1. To avoid prolonged G1 arrest, we incubated asynchronous cdc10-M17ts cells at 35°C for 90 minutes, and selected the smallest cells by elutriation. These cells will have just divided, and thus only recently entered G1. We estimate that the cells we isolate spend about 30 minutes arrested in G1 before they are released at the beginning of the time course. After elutriation, we observed a 1C peak showing that cells were arrested in G1. Cells were then released into the cell cycle and S-phase progression was assayed by flow cytometry (Figure 3A). Untreated cells replicated between 40 and 80 minutes after release. MMS-treated cells did not complete replication by 180 minutes, and this slowing was dependent on Rad3 (Figure 3B, C).
Figure 3.


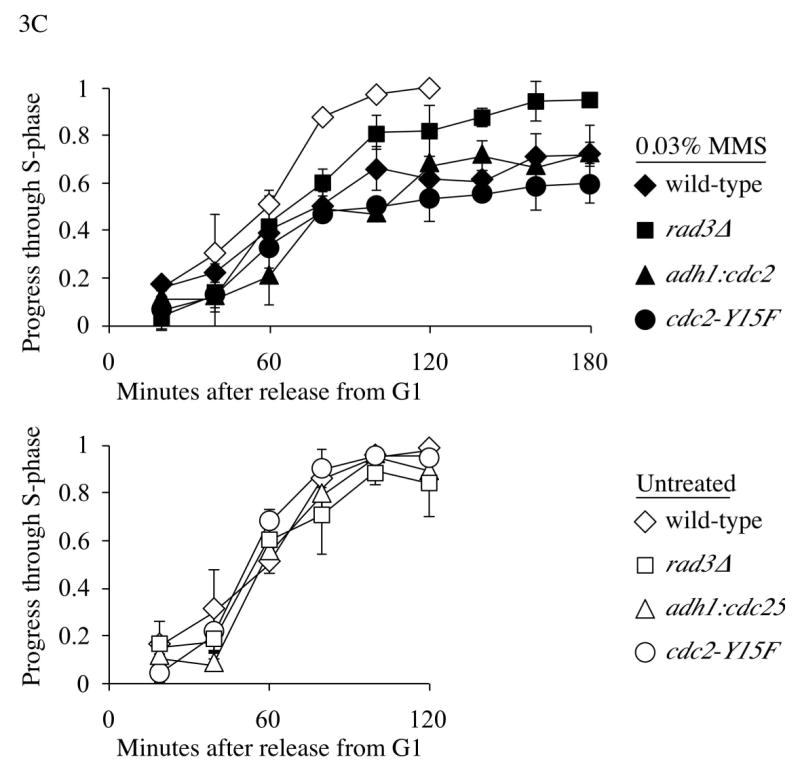
S-phase DNA damage checkpoint analysis in G1 synchronized cells. (A) Flow cytometric analysis of S-phase DNA damage checkpoint in G1 synchronized cdc10-M17ts cells (yFS280). 0.03% MMS or 10 mM HU were added immediately after elutriation and samples collected after every 20 minutes. (B) G1 synchronized cultures of cdc10-M17ts (yFS280), cdc10-M17ts rad3Δ (yFS260), cdc10-M17ts adh1:cdc25 (yFS430) and cdc10-M17ts cdc2-Y15F (yFS437) were treated and collected as in panel A; for clarity, only the MMS treated samples are shown. (C) Quantification of the data of A and B. The previously reported minor, checkpoint independent slowing is evident in the rad3Δ culture.4 Each point is the average of two experiments; the error bars represent the range of the data.
Over-expressing Cdc25 fails to override the S-phase DNA damage checkpoint
As an initial test of the role of Cdc25 in the S-phase DNA damage checkpoint, we examined if we could override the checkpoint by over-expressing Cdc25. Such over-expression efficiently overrides the Cdc25-dependent replication checkpoint arrest in G2.27 We used a strain in which Cdc25 was over-expressed from the strong, constitutive adh1 promoter28. If no difference in the cytometry profiles of cells with or without damage was seen, it would indicate that over-expressing Cdc25 had overcome the S phase DNA damage checkpoint. However, in asynchronous culture, we observed sub-2C DNA content in the presence of damage, indicating that the checkpoint was still active (Figure 1B). In fact, adh1:cdc25 cells accumulate in a sub-2C population to a greater extent than wild-type cells, presumably because some of the wild-type cells arrest in G2, while the adh1:cdc25 cells, lacking the G2 checkpoint, do not.
We also observed an MMS-induced delay of S-phase progression in synchronized adh1:cdc25 cells. In both G2 and G1 synchronous experiments, the wild-type and adh1:cdc25 strains demonstrated a similar degree of MMS-induced slowing of replication (Figures 2B, 3B and 3C). Results from these experiments indicate that Cdc25 over-expression is not sufficient to override the S-phase damage checkpoint.
Inhibitory phosphorylation of Cdc2 is not required for the S-phase DNA damage checkpoint
Although the Cdc25 over-expression results suggest Cdc25 inhibition is not the mechanism for slowing of S phase, it is possible that the S-phase DNA damage checkpoint is able to inhibit even the over-expressed Cdc25. To directly test the role of Cdc2 tyrosine-15 phosphorylation in the checkpoint, we used an allele of cdc2, cdc2-Y15F, in which tyrosine-15 is mutated to phenylalanine, preventing its phosphorylation. Because Cdc2-Y15F cannot be inhibited by tyrosine phosphorylation, it should bypass any Cdc25-dependent S-phase checkpoint, in the same manner that it overrides the G2 checkpoints.15,16 Contrary to that prediction, asynchronous cdc2-Y15F cells treated with 0.015% MMS accumulated in a sub-2C peak, showing no defect in the checkpoint. As with the adh1:cdc25 cells, cdc2-Y15F cells actually accumulate as sub-2C cells to a greater extent than wild-type cells, presumably due to the lack of a G2 checkpoint (Figure 1B).
Synchronous experiments using the cdc2-Y15F strain also showed no defect in S-phase slowing in response to DNA damage. Because the Cdc2-Y15F cannot be inhibited by tyrosine phosphorylation, cdc2-Y15F cells go very quickly through G2. They compensate for this short G2 by expanding G1; this effect can be seen in the large G1 peak in asynchronous cdc2-Y15F cells. Thus cdc2-Y15F cells begin replication later than wild-type cells. Untreated G2 synchronized cdc2-Y15F cells began replicated around 100 minutes and completed replication by 160 minutes (data not shown). In the presence of MMS, cells started replicating at the same time as untreated samples but did not complete replication by 180 minutes (Figure 2B).
G1 synchronized cdc2-Y15F cells begin replication at the same time as wild-type cells, because the arrest is at the end of G1. The MMS induced S-phase slowing is comparable between cdc2-Y15F and wild-type cells, beginning at around 60 minutes and not finishing by 180 minutes (Figures 3B and 3C). These results show that Cdc2 tyrosine-15 phosphorylation is not required for cells to slow replication in response to MMS-induced DNA damage.
Cdc25 is not required for the checkpoint
The previous results show that Cdc2 tyrosine-15 phosphorylation is not required for the S-phase DNA damage checkpoint, but they leave open the possibility that Cdc25 is required to regulate another target besides Cdc2. To test this possibility directly, we wanted to study the S-phase progression in the absence of Cdc25. Since Cdc25 is an essential gene, we created a strain in which the essential function of Cdc25 - the dephosphorylation of Cdc2 - is bypassed by Cdc2-Y15F. cdc25Δ cdc2-Y15F cells were synchronized in the G2 phase by elutriation and their progress through S-phase in the presence or absence of MMS was monitored. Since these cells lack a G2 checkpoint, we were able to use 0.03% MMS without arresting the cells in G2; we also used 0.015% for comparison with the other G2 synchronization experiments. As for cdc2-Y15F cells, untreated cdc25Δ cdc2-Y15F cells replicate later than wild-type, in this case between about 80 and 140 minutes (Figure 2C). cdc25Δ cdc2-Y15F cells treated with 0.015% MMS did not complete replication by 180 minutes; cells treated with 0.03% MMS replicated even more slowly. cdc25Δ cdc2-Y15F cdc10-M17 cells are inviable, precluding G1 synchronization. These results show that S-phase damage checkpoint operates normally in the absence of Cdc25.
Our results are inconsistent with the hypothesis that S-phase damage checkpoint inhibits Cdc25, and are consistent with a strictly Cdc2-Y15 phosphorylation independent mechanism for the S-phase DNA damage checkpoint.
Discussion
We have investigated the role of inhibitory tyrosine-15 phosphorylation of Cdc2 and of the Cdc2 tyrosine-15 phosphatase, Cdc25, in the S-phase DNA damage checkpoint. Inhibition of Cdc25, and thus inhibition of Cdc2 tyrosine-15 dephosphorylation, is the mechanism by which fission yeast arrest in G2 in response to DNA damage or replication blocks.15,16 Recent work has suggested that a similar mechanism may also slow replication in response to DNA damage.17 We have tested this idea and found no evidence for involvement of Cdc25 or Cdc2 tyrosine phosphorylation in the fission yeast S-phase DNA damage checkpoint. Neither the overexpression of Cdc25, nor the mutation of tyrosine-15 to an unphosphorylatable phenylalanine, impairs the S-phase checkpoint; yet both override the G2 checkpoint.15,16,27 Furthermore, cells lacking both Cdc25 and tyrosine-15 of Cdc2 slow replication normally in response to MMS-induced DNA damage. This final result rules out checkpoint mechanisms that involve Cdc2-independent targets of Cdc25, and Cdc25-independent regulation of Cdc2 tyrosine-15 phosphorylation.
These results contradict those of Kumar and Huberman, who, using similar approaches, concluded that adh1:cdc25 and cdc2-Y15F cells lack the S-phase DNA damage checkpoint. There is technical difference between the two studies that may explain the discrepancy. Kumar and Huberman used a whole-cell flow cytometry protocol, in which cytoplasmic background contributes significantly to the total signal, reducing the sensitivity of the assay. We used an isolated-nuclei protocol, which removes the cytoplasm before analysis. This approach greatly increases the resolution of the assay and allows for quantitation of the data. In addition, for their G1 synchrony experiments, cells were arrested in G1 for up to 4 hours, which allows the cells to elongate, further reducing the sensitivity of the whole-cell assay. We find that the combination of the four hour arrest and the whole cell flow-cytometry analysis compromises detection of the checkpoint delay (our unpublished results). We used centrifugal elutriation to isolate cells that had been arrested for only 30 minutes, allowing for a more sensitive analysis of the checkpoint. We believe that these technical differences are responsible for the different conclusion drawn.
Our results implicate a Cdc2 tyrosine phosphorylation independent target of the S-phase DNA damage checkpoint in fission yeast. Precedent for such a target exists. In mammals, the S-phase checkpoint appears to have two branches: one which acts through inhibition of Cdc25A to inhibit origin firing, and one which is Cdc25-independent and requires the MRN complex and XRCC312,13. Since the fission yeast checkpoint requires MRN, it may be mechanistically similar to the MRN-dependent branch of the mammalian checkpoint.29 Little is known about the mechanism or purpose of the MRN-dependent branch of the pathway, except that its loss leads to severe cancer-predisposition in humans.6 The possible role of MRN and XRCC3 in slowing replication fork progression through induction of replication-coupled recombinational repair provides a model that can be tested in fission yeast.14 Whatever the case, fission yeast provides an tractable system for the study of this checkpoint mechanism.
Materials and methods
Yeast methods
Yeast were grown in YES at 30°C and manipulated by standard methods.30 Temperature-sensitive (ts) cells were grown at 25°C unless otherwise stated. Strains used for this study are listed in Table 1.
Table 1. Strain list.
| Strain | Genotype | Source |
|---|---|---|
| yFS104 | h+ leu1-32 ura4-D18 | Lab Stock |
| yFS189 | h- leu1-32 ura4-D18 ade6-704 rad3∷ura4 | Lab Stock |
| yFS260 | h- leu1-32 ura4-D18 cdc10-M17 rad3∷ura4 | Lab Stock |
| yFS280 | h+ leu1-32 ura4-D18 ade6-210 cdc10-M17 | Lab Stock |
| yFS357 | h+ leu1-32 ura4-D18 his3-237 ura4 adh1:cdc25 | Russell Lab28 |
| KGY14 | h- leu1-32 ura4-D18 cdc2∷ura4 cdc2-Y15F LEU2 | Gould Lab21 |
| yFS430 | h- leu1-32 ura4-D18 ade6-210 his3-237 cdc10-M17 ura4 adh1:cdc25 | This study |
| yFS445 | h- leu1-32 ura4-D18 cdc2∷ura4 cdc2-Y15F LEU2 cdc25∷ura4 | This study |
| yFS437 | h+ leu1-32 ura4-D18 cdc2∷ura4 cdc2-Y15F LEU2 cdc10-M17 | This study |
Flow cytometry methods
Isolated nuclei were prepared for flow cytometry by an adaptation of the protocol of Carlson el al.25,30 1.0 OD of cells was fixed in 70% EtOH, washed in 1 ml 0.6M KCl, resuspended in 1 ml 0.6 M KCl, 1 mg/ml Novozym 234 (Sigma L1412), 0.3 mg/ml Zymolyase 20T and incubated for 30 min at 37°C. The cells were pelleted, resuspended in 1 ml 0.1 M KCl 0.1% triton-X100 and incubated for 5 minutes at room temperature. The cells were washed and resuspended in 1 ml 20 mM Tris-HCl, 5 mM EDTA pH 8.0. 10 μl 20 mg/ml RNase A was added and the cells were incubated overnight at 37°C. The spheroplasted cells were disrupted, and isolated nuclei released, by sonication with a Branson Sonifier using a microtip at 0.7 power for 5 seconds. 300 μl of disrupted cells were added to 300 μl of 2 mM Sytox Green (Molecular Probes) in PBS and analyzed on a Becton-Dickinson FACScan flow cytometer. G1 synchronized experiments were quantitated in CellQuest (Becton-Dickinson) by measuring the mean of the S-phase peak as a percentage of the position of between the means of the 1C and 2C controls.
Asynchronous Experiments
Asynchronous experiments were carried out as described,17 except that flow cytometry was carried out using the isolated nuclei protocol described above. Briefly, cells were grown to an O.D. of 1.0, diluted to an O.D. 0.1 and allowed to recover for 1 hour. At this time, the culture was divided and treated as described. Samples were collected after every hour, fixed by 70% ethanol and processed for flow cytometry.
Synchronous Experiments
We used centrifugal elutriation to synchronize cells either in G1 or G2. Since fission yeast spends a short time in G1, experiments were conducted in cdc10-M17 background to synchronize cells in G1. Cultures were grown to O.D. 0.5 arrested at 35°C for 1.5 hours and then synchronized by elutriation. The culture was divided and treated with 0.03% MMS, 10 mM hydroxyurea (HU) or mock treated. The cells were kept at 25°C and samples collected after every 20 minutes for 3 hours.
For G2 synchronization, cultures were grown to O.D. 1.0 and elutriated. The synchronized samples were divided and treated with 0.015% MMS, 10 mM hydroxyurea (HU) or mock treated. Cells were collected after every 20 minutes and processed for flow cytometry.
Acknowledgments
We are grateful to Nicholas Willis for help with the flow cytometry, Alison Bright for help with strain construction, and the rest of the Rhind lab for useful discussions and suggestions. We thank Kathy Gould and Paul Russell for providing strains. This work was supported by NIH grant GM069957 to N.R.
Abbreviations
- MMS
methane methyl sulfonate
- HU
hydroxyurea
References
- 1.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 2.Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:7315–7. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orren DK, Petersen LN, Bohr VA. Persistent DNA damage inhibits S-phase and G2 progression, and results in apoptosis. Mol Biol Cell. 1997;8:1129–42. doi: 10.1091/mbc.8.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhind N, Russell P. The Schizosaccharomyces pombe S-phase checkpoint differentiates between different types of DNA damage. Genetics. 1998;149:1729–37. doi: 10.1093/genetics/149.4.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myung K, Datta A, Kolodner RD. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell. 2001;104:397–408. doi: 10.1016/s0092-8674(01)00227-6. [DOI] [PubMed] [Google Scholar]
- 6.Petrini JH. The Mre11 complex and ATM: collaborating to navigate S phase. Current Opinion in Cell Biology. 2000;12:293–6. doi: 10.1016/s0955-0674(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 7.Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H. Regulation of DNA-replication origins during cell-cycle progression. Nature. 1998;395:618–21. doi: 10.1038/27007. [DOI] [PubMed] [Google Scholar]
- 8.Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, Grieco D, Gautier J. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol Cell. 2000;6:649–59. doi: 10.1016/s1097-2765(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 9.Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–8. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- 10.Larner JM, Lee H, Little RD, Dijkwel PA, Schildkraut CL, Hamlin JL. Radiation down-regulates replication origin activity throughout the S phase in mammalian cells. Nucleic Acids Res. 1999;27:803–9. doi: 10.1093/nar/27.3.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–7. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 12.Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30:290–4. doi: 10.1038/ng845. [DOI] [PubMed] [Google Scholar]
- 13.Henry-Mowatt J, Jackson D, Masson JY, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, Caldecott KW. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell. 2003;11:1109–17. doi: 10.1016/s1097-2765(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 14.Rhind N, Russell P. Checkpoints: It takes more than time to heal some wounds. Current Biology. 2000;10:R908–R911. doi: 10.1016/s0960-9822(00)00849-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhind N, Furnari B, Russell P. Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes & Development. 1997;11:504–11. doi: 10.1101/gad.11.4.504. [DOI] [PubMed] [Google Scholar]
- 16.Rhind N, Russell P. Tyrosine phosphorylation of Cdc2 is required for the replication checkpoint in Schizosaccharomyces pombe. Molecular and Cellular Biology. 1998;18:3782–7. doi: 10.1128/mcb.18.7.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S, Huberman JA. On the slowing of S phase in response to DNA damage in fission yeast. J Biol Chem. 2004;279:43574–80. doi: 10.1074/jbc.M407819200. [DOI] [PubMed] [Google Scholar]
- 18.Stern B, Nurse P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends in Genetics. 1996;12:345–50. [PubMed] [Google Scholar]
- 19.Christensen PU, Bentley NJ, Martinho RG, Nielsen O, Carr AM. Mik1 levels accumulate in S phase and may mediate an intrinsic link between S phase and mitosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:2579–84. doi: 10.1073/pnas.97.6.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moreno S, Nurse P, Russell P. Regulation of mitosis by cyclic accumulation of p80cdc25 mitotic inducer in fission yeast. Nature. 1990;344:549–52. doi: 10.1038/344549a0. [DOI] [PubMed] [Google Scholar]
- 21.Gould KL, Nurse P. Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature. 1989;342:39–45. doi: 10.1038/342039a0. [DOI] [PubMed] [Google Scholar]
- 22.Lundgren K, Walworth N, Booher R, Dembski M, Kirschner M, Beach D. Mik1 and Wee1 cooperate in the inhibitory tyrosine phosphorylation of Cdc2. Cell. 1991;64:1111–22. doi: 10.1016/0092-8674(91)90266-2. [DOI] [PubMed] [Google Scholar]
- 23.Gould KL, Moreno S, Tonks NK, Nurse P. Complementation of the mitotic activator, p80cdc25, by a human protein-tyrosine phosphatase. Science. 1990;250:1573–6. doi: 10.1126/science.1703321. [DOI] [PubMed] [Google Scholar]
- 24.Millar JBA, Lenaers G, Russell P. Pyp3 PTPase acts as a mitotic inducer in fission yeast. EMBO Journal. 1992;11:4933–41. doi: 10.1002/j.1460-2075.1992.tb05600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlson CR, Grallert B, Bernander R, Stokke T, Boye E. Measurement of nuclear DNA content in fission yeast by flow cytometry. Yeast. 1997;13:1329–35. doi: 10.1002/(SICI)1097-0061(199711)13:14<1329::AID-YEA185>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 26.Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes and Development. 1998;12:382–95. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enoch T, Nurse P. Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell. 1990;60:665–73. doi: 10.1016/0092-8674(90)90669-6. [DOI] [PubMed] [Google Scholar]
- 28.Russell P, Nurse P. cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell. 1986;45:145–53. doi: 10.1016/0092-8674(86)90546-5. [DOI] [PubMed] [Google Scholar]
- 29.Chahwan C, Nakamura TM, Sivakumar S, Russell P, Rhind N. The fission yeast Rad32(Mre11)-Rad50-Nbs1 complex is required for the S-phase DNA damage checkpoint. Mol Cell Biol. 2003;23:6564–73. doi: 10.1128/MCB.23.18.6564-6573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forsburg SL, Rhind N. Basic methods for fission yeast. Yeast. 2006;23:173–83. doi: 10.1002/yea.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]