

Abstract
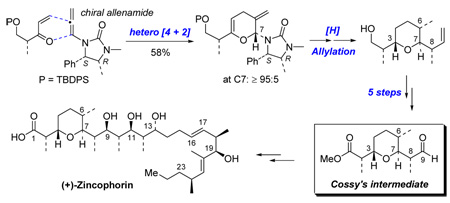
A detailed account on the stereoselective synthesis of the C1–C9 subunit of (+)-zincophorin is described here. This approach features the first application of a stereoselective inverse electron demand hetero [4 + 2] cycloaddition of chiral allenamides in natural product synthesis. The C1–C9 subunit matches Cossy’s intermediate, thereby constituting a formal total synthesis. In addition, details of an unusual urea directed Stork-Crabtree hydrogenation observed during these efforts is also disclosed here.
INTRODUCTION
Polyoxygenated ionophore-containing natural products exhibit potent anti-infectious properties through proton-cation exchange processes across biological membranes. This activity is due to the ability of ionophores to form lipophilic complexes with various cations such as Li+, Na+, and K+, and divalent alkaline earth cations such as Ca2+ and Mg2+.1 In 1984, two new monocarboxylic acid ionophores, griseocholin and antibiotic M144255, were isolated from cultured strains of Streptomyces griseus.2 Extensive NMR experiments of griseocholin and X-ray diffraction of the zinc-magnesium salt of M144255 revealed that these two compounds are actually structurally identical with the same absolute configuration.2b Based on its ability to strongly bind with Zn2+, it was given the trivial name (+)-zincophorin (Figure 1). (+)-Zincophorin possesses strong in vivo activity against Gram-positive bacteria and Clostridium coelchii. Its ammonium and sodium salts display significant anti-coccidal activity against Eimeria tenella in chicken embryos, and its methyl ester was reported in a patent as having strong inhibitory properties against influenza WSN/virus with reduced toxicity for the host cells.2d, 3
Figure 1.

(+)-Zincophorin and Its Methyl Ester.
Over the last twenty plus years, (+)-zincophorin has attracted an impressive array of synthetic efforts including Danishefsky's first total synthesis along with two recent elegant total syntheses reported by Cossy and Miyashita.4 We recently communicated our formal total syntheses of (+)-zincophorin via interception of Miyashita’s advanced intermediate.5 The key strategy features our previously reported stereoselective inverse electron demand hetero-[4 + 2] cycloaddition of chiral allenamides (Scheme 1).6–9 Herein, we report the synthesis of Cossy’s C1–C9 subunit of (+)-zincophorin based on this approach, as well as details of the observation of an unusual urea directed Stork-Crabtree hydrogenation.
Scheme 1.

An Inverse Electron Demand Hetero-[4 + 2] Cycloaddition.
RESULTS AND DISCUSSIONS
1. Retrosynthetic Plan
The construction of Cossy’s intermediate 6, or the C1–C9 subunit of (+)-zincophorin, could be realized from tetrahydropyran 7, which contains all prerequisite stereocenters by several transformations (Scheme 2). In turn, tetrahydropyran 7 could be prepared from pyran 8 via: 1) stereoselective hydrogenation of exo- and endo-cyclic olefins from less hindered top face of the pyranyl ring; and 2) a Lewis acid promoted stereoselective crotylation to remove imidazolidinone auxiliary.10 Finally, the inverse electron demand hetero-[4 + 2] cycloaddition of chiral allenamide 9 with enone 10 could establish the pyranyl skeleton in 8.
Scheme 2.

An Approach to C1–C9 Subunit of (+)-Zincophorin.
2. Synthesis of Chiral Allenamide 9 and Enone 16
Synthesis of chiral allenamide 9 commenced from (+)-ephedrine hydrochloride salt 11 and urea. By condensation of the mixture at 175°C for 0.5 h and then 200–210 °C for 1.5 h, the so-called Close auxiliary 12 could be obtained in 48% yield after recrystallization.11 Subsequently, propargylation followed by base-catalyzed isomerization of propargyl amide 13 furnished chiral allenamide 9 in 93% overall yield (Scheme 3). In the same manner, with the enantiomer of ephedrine, allenamide ent-9 was also prepared.
Scheme 3.

Synthesis of Chiral Allenamide 9.
Chiral enone 16 was prepared from the commercially available chiral hydroxy ester 14 as shown in Scheme 4. Protection of the hydroxyl group as a TBDPS ether followed by reduction of the ester group using DIBALH afforded aldehyde 15 in 76% overall yield.12 Subsequent vinyl Grignard addition and Swern oxidation of the resulting isomeric mixture (1:1) of allylic alcohols gave rise to chiral enone 16 with an overall yield of 66%.
Scheme 4.

Synthesis of Chiral Enone 16.
3. Inverse Demand Hetero-[4 + 2] Cycloaddition
With chiral allenamides 9 and ent-9, in hand, we pursued the key inverse demand hetero-[4 + 2] cycloaddition. Concerned with the fact that the stereochemical outcome could be controlled either through the chiral auxiliary of the allenamide or the chiral enone, leading potentially to matched and/or mismatched scenarios, we examined both reactions of chiral allenamides 9 and ent-9 with enone 16 (Scheme 5). By using our reported cycloaddition protocol with CH3CN as the solvent and the sealed tube as the reaction vessel, after heating at 85°C for 48 h, both reactions of 9 and ent-9 with 16 proceeded readily to provide pyrans 17 and 18 in 58% and 54% yield, respectively, as single isomers. The stereochemistry of 18 was unambiguously confirmed by its single-crystal X-ray diffraction analysis.
Scheme 5.

Hetero-[4 + 2] Cycloadditions with Chiral Enone 16.
Given that the reaction of chiral allenamide 9 should proceed through the matched transition state model shown in Scheme 5 [on the right side], the stereochemical assignment for 18 and the high level of selectivity imply that the cycloaddition of ent-9 also proceeded through the related transition Scheme 5 state, although potentially mismatched [on the left side]. However, any mismatching perceived or real clearly did not impede the reaction or the stereoselectivity. A closer examination reveals that to allow the chiral enone to still approach the open π-face of the internal olefin of allenamide ent-9 as dictated by the chiral Close auxiliary, and to avoid any mismatching as well as allylic strain, the heterodiene 16 would need to position the Me group on the alpha carbon orthogonal to the plane of the chiral enone (Scheme 5).
4. Hydrogenations of Pyran 17 and A Urea Directed Stork-Crabtree Hydrogenation
(a) Hydrogenation of the C6 Exo-Cyclic Olefin
Our efforts then encountered a serious obstacle at what we had believed to be the most trivial stage: sequential hydrogenations of the two olefins at C6 and C3. Given that the exo-cyclic olefin at C6 is more accessible sterically, we intended to accomplish this hydrogenation via mild conditions to ensure good stereoselectivity. As shown in Scheme 6, Lindlar catalyst in MeOH was initially deployed (entry 1), but after stirring for 2 d at 1 atm H2, the desired mono-hydrogenation product 19 was obtained with a best yield of 44% while being contaminated with 23% of the ring-opened byproduct ketone 20 as a 3:2 isomeric mixture with regards to C6. Although increasing the H2 pressure to 60 psi did shorten the reaction time (entry 2), the yield dropped drastically while providing a large amount of ketone 20.
Scheme 6.

Hydrogenation of the C6 Exo-Cyclic Olefin in 17.
We initially believed that the formation of ketone 20 was initiated via hydrolysis of the cyclic aminal through protonation of the electron rich C3 endo-cyclic olefin. Given this consideration, EtOAc and anhydrous hexanes (entries 3 and 4) were employed as solvents, but the formation of 20 still persisted. Changing metal catalyst from Pd to Pt appeared not to be useful either (entry 5). However, intriguingly, we found that without H2, pyran 17 was actually quite stable in the presence of Pt/C catalyst in MeOH (entry 6).
These observations led us to speculate that the formation of ketone 20 is likely promoted by the catalyst-activated H2. As shown in Scheme 7, the same Pt-complex 21 from pyran 17 that could lead to the desired mono-hydrogenation product 19 could also protonate the electron rich endo-cyclic double bond as shown in Pt-complex 22. This would result in ring-opening to give the Pt-π-allyl type complex 23, leading to alkene 24 after reductive elimination. Further hydrogenation of 24 should give 20 as an isomeric mixture as observed. Alternatively, the mono-hydrogenation of pyran 17 could take place predominantly to exclusively afford dihydropyran 19 initially, but 19 could be subsequently converted into 20 via complex 25 through an analogous sequence.
Scheme 7.

A Proposed Mechanism for the Formation of Ketone 20.
Given this proposed process, it seemed that identifying a catalyst with appropriative activity that is effective to hydrogenate the exo-cyclic olefin but slow to protonate is very important. Ultimately, by screening several kinds of base-poisoned catalysts, we were delighted to find that the hydrogenation of this exo-cyclic olefin could be achieved in 64% yield with only 12% of 20 by using Adam’s catalyst along with 3.0 equiv of NaBH4 (entry 8 in Scheme 6).
(b) Hydrogenation of the C3 Endo-Cyclic Olefin
Further hydrogenation of the C3 endo-cyclic olefin, which is more electron rich and sterically less accessible, turned out to be much more difficult by using any heterogeneous catalysts under 15–70 psi of H2. The formation of the ring-opening product 20 was observed as the major product in most cases with no desired product found (Scheme 8).
Scheme 8.

Hydrogenation of Pyran 19.
However, by using Stork-Crabtree's conditions,13 the C3 endo-cyclic olefin was reduced smoothly and provided presumably the desired tetrahydropyran 26 in 74% yield. Unsuspectingly, we proceeded to remove the silyl group in 26, only to be confronted with a product quite different as established by the X-ray structure of 27. While the stereochemistry at C6 is as expected, both stereocenters at C3 and C7 are opposite from what we had expected. These expectations were based on our earlier analyses in which the mono-hydrogenated pyran would assume a unique conformation as shown for 19 (Scheme 8). Hydrogenations of C3 olefin should show a distinct preference at the top face of a flat pyranyl ring away from the urea group, which shields the bottom face while being pseudo-axially situated.
(c) A Urea Directed Stork-Crabtree Hydrogenation.14–16
This unexpected outcome implies a number of possibilities. We considered the following two possibilities as illustrated in Scheme 9. First, the mono-hydrogenation product 19 could epimerize at C7 to give 28 in which the hydrogenation could occur at the bottom face, leading to 27 after desilylation (pathway-a). While this pathway is very likely and provides a sound rationale, we suspected a second scenario involving a urea-directed Stork-Crabtree hydrogenation as shown in 29 to give tetrahydropyran 30 prior to a C7-epimerization that would afford 31 (pathway-b). Tetrahydropyran 30 differs from 27 only in the protection of the primary OH group.
Scheme 9.

A Urea-Directed Hydrogenation vs Epimerization.
Toward this end, we carried out the reaction using 2.0 equiv of Na2CO3 to scavenge protic species or decrease the Lewis acidity of Ir(I) that could promote the C7-epimerization, and found 11% of 30 (a 1:2 inseparable mixture with 19) along with 35% of 31. The relative stereochemistry in 30 was assigned based on the J value between H6 and H7 being in the equatorial-axial range (Scheme 10). Treatment of the mixture of 30 and 19 in CH2Cl2 with a trace amount of p-TsOH quickly give 31 via epimerization at C7 in about 60–70% yield. Meanwhile, the relative stereochemistry in 31 was confirmed via silylation of 27 (Scheme 9). These results collectively imply that the hydrogenation at the hindered bottom face of 19 is possible under the Stork-Crabtree conditions.
Scheme 10.

Experimental Support for Pathway b.
a. Crabtree's catalyst was pre-activated by treatment with 60 psi H2 in CH2Cl2 at rt for 5 min. b. 30 was obtained as an unseparated mixture with 19 (1:2); 31 was isolated in 35% yield.
While pathway-b is supported via the above experiment, we still could not rule out pathway-a by now. Thus, the following reaction to make dihydropyran 28 was examined. When subjecting 19 to pre-activated Crabtree’s catalyst in CH2Cl2, 19 was transformed to 28 in 40% yield via epimerization. The relative stereochemistry in 28 was assigned by the J value between H6 and H7 being in the axial-axial range (Scheme 10). However, under the same directed hydrogenation conditions, dihydropyran 28 could not be converted into 31 and only the ring-opening by-product 20 was formed in about 40% yield.
Based on this result, pathway-a in Scheme 9 can be excluded completely. Semi-empirical calculations on pyranyls 19 and 28 were also performed using the Spartan’02 program (Figure 2). Based on the calculation results, 19 seems thermodynamically more favorable than 28. This is also supported by the acid-catalyzed epimerization experiment in which 2:1 ratio of 19:28 was formed after stirring with PTS in CH2Cl2 for 5 h. The molecular models distinctly reveal that in pyran 19, the urea group occupies the pseudo-axial position allowing the directed hydrogenation to occur, while in pyran 28, the urea group assumes the pseudo-equatorial position, thereby preventing the coordinated metal center to reach the C3 endo-cyclic olefin and thwarting the directed hydrogenation.
Figure 2.

Molecular Modeling of Pyran 19 and 28.
(d) Directed Hydrogenation versus C7-Epimerization
Our efforts also led to another observation involving competition between directed hydrogenation and epimerization at C7. As shown in Scheme 11, it appeared that this competition is dependent upon the pressure of H2, as higher pressure tends to favor the directed hydrogenation product 31. A potential mechanism was proposed as shown in Scheme 12. Upon treatment with H2, the unsaturated Crabtree’s catalyst could complex to the urea group and to the C3 endo-cyclic olefin, leading to the common Ir-complex 32 in which pathway-c and pathway-d could take place depending upon the pressure of H2.
Scheme 11.

Directed Hydrogenation versus Epimerization.
Scheme 12.

Competition Between Directed Hydrogenation and C7-Epimerization.
Under higher pressure (≥60 psi), oxidative addition of H2 could occur to provide 33 en route to alkyl metal complex 34 via migratory insertion. Reductive elimination and epimerization would yield tetrahydropyran 31 and regenerates the active catalyst. However, with lower pressure (≤60 psi), the complexation of H2 to the Ir metal and the subsequent oxidative addition would be slower, thereby allowing 32 to undergo ring-opening by cleavage of C7-O bond and form the Ir-oxo-π-allyl complex 35. An ensuing C6–C7 bond rotation could take place to alleviate steric interactions, leading to the more favored conformer 36, which can afford 28 via ring-closing and release of the Ir-catalyst.
To further explore this urea-direct hydrogenation, we employed the model pyran 37 with the (R)-Close auxiliary because we had unambiguously established the stereochemical outcome of its hydrogenations (Scheme 13).6,10c Specifically, the mono-hydrogenated product 38 obtained from standard hydrogenations contains exclusively a cis relationship for Ha and Hb with a small J value. When hydrogenating pyran 37 with the Crabtree’s catalyst, we isolated 39 with a large J constant, distinctly suggesting a trans relationship between Ha and Hb. Stork-Crabtree hydrogenation of 17 afforded the mono-hydrogenated product 40 also with a large J value in contrast to that of 19 from standard hydrogenations of 17, indicating again a trans relationship between H6 and H7 in 40. Aza-cycle 41, prepared from aza-[4 + 2] cycloaddition of 1-azadiene and chiral allenamide 4,10a is also suitable for this directed hydrogenation, giving the expected stereochemistry of the product. Given the unique conformational preference of 17, 37, and 41, these studies unequivocally support a urea-directed Stork-Crabtree hydrogenation (see the box in Scheme 13).
Scheme 13.

Scope of A Urea-Directed Stork-Crabtree Hydrogenation.
a: 5–10% Heterogeneous catalyst (Pd/C, Pt/C or Lindar), 1 atm H2; b: 10 mol% Crabtree cat, 60 psi H2, CH2Cl2.
(e) High Pressure Hydrogenations
The ultimate solution to this dilemma was using high-pressure hydrogenation. Even at 1500 psi of H2, catalyst and solvent were still crucial to render this reaction feasible leading towards the desired pyran 26 rather than 20. Using the best condition of 20% Pt-Alumina in hexanes for 3 d, 26 was isolated as a single isomer and in the acceptable yield of 50% and its stereochemistry was confirmed by using nOe (see the box in Scheme 14). From molecular modeling of 19 shown in Figure 2, both the urea group and TBDPS group do shield the bottom face of the pyran ring, thereby completely forcing H2 to approach from the top face.
Scheme 14.

High-Pressure Hydrogenations of Pyran 19.
High-pressure hydrogenations of pyrans 28 and 40 were also examined. As shown in Scheme 15, an 89:11 isomeric ratio with respect to C3 stereochemistry was found of 44 while a 67:33 ratio was seen for 45. At this point, we are not sure as to why in these two hydrogenations prefer the same face as the urea group.
Scheme 15.

High-Pressure Hydrogenations of Pyrans 28 and 40.
It is noteworthy that collectively through these hydrogenation and epimerization studies, 6 of 8 possible diastereomers of pyran 26 could be attained from pyran 17 (Scheme 16).
Scheme 16.

Summary: 6 Out of 8 Possible Diastereomers Attainable from Reduction of Pyran 17.
5. Crotylation of Pyran 26 with E- and Z-Crotylsilane
With pyran 26 in hand, crotylation was then investigated. Promoted by SnBr4, 26 was quickly epimerized to 44 with a large J constant meaning a trans relationship between H6 and H7. The following reaction with both E- and Z-crotylsilane proceeded smoothly and gave the crotylation products 50a and 50b in opposing ratios (50a:50b = 4:1 with E-crotylsilane and 1:3 with Z-crotylsilane), and the TBDPS group was also lost in the process. We believe that along with removal of the chiral imidazolidinone, the likely oxocarbenium ion intermediate 49 was generated in which the bulky alkyl group at C3 predominates and assumes an equatorial position.10c,17 Then, addition of E- or Z-crotylsilane to 49 would prefer an anomerically axial trajectory, which is both stereoelectronically and sterically favorable, and provide the expected stereochemical outcome at C7. Based on this analysis, it turned out that 50a and 50b should be diastereomers at C8. Successive oxidation of 50a/50b (as a 4:1 mixture) led to carboxylic acids 51a and 51b, which could be separated. The X-ray structure of 51a unambiguously confirmed all relative stereochemistry. The assignment of C8 in 51a suggests that 51b would be the desired crotylation product for the synthesis.
The rationale for the stereochemical outcome at C8 can be illustrated employing Danishefsky’s C3-crotylation model as shown in Scheme 18.4k In our C7-crotylations, when ruling out transition states from either the anti-periplanar or the synclinal approach with two excessive to severe gauche interactions, and when assuming an anomerically favored axial addition of E- or Z-crotylsilane, what is left behind would be a synclinal approach for the E-crotylation in which ES1 is more favored in leading to 50a, whereas both anti-periplanar and synclinal pathways could be at play for the Z-crotylation with ZA1 playing a more dominant role to favor the formation of 50b.
Scheme 18.

Rationale for the Stereochemical Outcome at C8.
6. Synthesis of the C1–C9 Subunit: Intercepting Cossy's Intermediate
To complete our synthesis of C1–C9 subunit, the crotylated pyran 50b was carried on as a 3:1 isomeric mixture in a sequence of Dess-Martin periodinate oxidation and further oxidation, leading to carboxylic acid 51b still as a 3:1 mixture (Scheme 19). Then, methylation and dihydroxylation of the formed methyl ester furnished the diol mixture 52, in which the major isomer at C8 was readily separated as a 8:1 mixture at C9. Finally, oxidative cleavage of 53 with Pb(OAc)4 provided aldehyde 6, which spectroscopically matched Cossy’s advanced intermediate. 4n
Scheme 19.

Intercepting Cossy’s Intermmediate: C1–C9 Subunit.
a: 1. Dess-Martin [O], 98%; 2. NaH2PO4, NaClO2,t-BuOH/H2O, 2-Me-2-butene, 96%; b: TMSCHN2, MeOH/toluene, 90%; c: NMO, OsO4, acetone/H2O, 79%; separation of the major isomer at C8 completely; d: Pb(OAc)4, CH2Cl2, 53%.
CONCLUSION
We have described here a synthesis of the C1–C9 subunit of (+)-zincophorin that matches Cossy’s advanced intermediate, thereby constituting a formal total synthesis, and details of an unusual urea-directed Stork-Crabtree hydrogenation of the hetero-[4 + 2] cycloadduct derived from a chiral allenamide. This work provides the first application of chiral allenamides in natural product synthesis.
EXPERIMENTAL SECTION
Preparation of pyran 17
To a solution of chiral enone 16 (2.48 g, 7.03 mmol) in anhyd CH3CN (50 mL) was added allenamide 9 (2.22 g, 9.72 mmol). This reaction mixture was sealed under N2 and heated up to 90 °C for 2 d. Concentration under reduced pressure and purification of the crude residue via silica gel flash column chromatography (gradient eluent: 10–20% EtOAc in hexanes) afforded pure cycloadduct 17 (1.96 g, 58% based on recovery of 17% 16) of as a colorless oil. 17: Rf = 0.50 [33% EtOAc/hexanes]; [α] D25 = − 100.4 ° [c 1.00, CH2Cl2]; 1H NMR (500 MHz, CDCl3) δ 0.59 (d, 3H, J = 6.5 Hz), 1.11 (d, 3H, J = 6.5 Hz), 1.13 (s, 9H), 2.23 (dd, 1H, J = 2.5, 20.5 Hz), 2.39 (m, 1H), 2.51 (dd, 1H, J = 4.0, 20.0 Hz), 2.71 (s, 3H), 3.56–3.63 (m, 2H), 3.84 (dd, 1H, J = 6.5, 10.0 Hz), 4.52 (dd, 1H, J = 3.6, 4.0 Hz), 4.78 (s, 1H), 4.86 (d, 1H, J = 8.5 Hz), 5.03 (s, 1H), 6.26 (s, 1H), 7.09–7.10 (m, 2H), 7.24–7.28 (m, 3H), 7.42–7.52 (m, 6H), 7.71–7.74 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 14.7, 14.8, 19.3, 26.9, 26.8, 28.6, 41.3, 56.7, 59.5, 66.5, 80.9, 94.4, 114.7, 127.59, 127.63, 127.68, 127.7, 129.50, 129.57, 133.8, 134.2, 135.46, 135.54, 135.59, 137.78, 137.98, 154.3, 161.4; IR (neat) cm−1 2957s, 1710s, 1456m, 1396s, 1361s; mass spectrum (APCI): m/e (% relative intensity) 581.2 (25) (M+H)+, 439.3 (100), 405.2 (80), 279.2 (50), 191.2 (75), 101.1 (75); HRMS (MALDI) calcd for C36H45N2O3Si (M+H)+ 581.3199, found 581.3761.
Hydrogenation of pyran 17 to 19
To a heterogeneous mixture of Pt/C (5%w/w, 1.40 g, 0.33 mmol) and cycloadduct 17 (3.80 g, 6.55 mmol) in MeOH (140 mL) was added NaBH4 (800.0 mg, 21.0 mmol) in small portions at 0 °C carefully over 2 min. The mixture was hydrogenated with a H2-balloon for 2 h at rt, and after which, the catalyst was filtered and washed with MeOH (50 mL). The MeOH was removed under reduced pressure and the residue was diluted with Et2O (100 mL). This mixture was washed with water (2 × 30 mL), dried over Na2SO4, and concentrated under reduced pressure. Purification of the crude residue via silica gel flash column chromatography (gradient eluent: 10–17% EtOAc in hexanes) afforded pure 19 (2.40 g, 64%) as a colorless oil. 19: Rf = 0.50 [33% EtOAc/hexanes]; [α] D25 = − 29.9 ° [c 1.45, CH2Cl2]; 1H NMR (500 MHz, CDCl3) δ 0.67 (d, 3H, J = 6.5 Hz), 0.76 (d, 3H, J = 6.5 Hz), 1.14–1.16 (m, 12H), 1.22 (m, 1H), 1.94–2.04 (m, 2H), 2.43 (m, 1H), 2.75 (s, 3H), 3.63 (dd, 1H, J = 7.0, 9.5 Hz), 3.73 (dq, 1H, J = 6.5 Hz), 3.88 (dd, 1H, J = 6.5, 10.0 Hz), 4.49 (dd, 1H, J = 3.5, 4.0 Hz), 4.78 (s, 1H, J = 8.5 Hz), 5.83 (d, 1 H, J = 3.5 Hz), 7.33 (m, 4H), 7.43–7.52 (m, 7H), 7.75–7.76 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 14.9, 15.4, 15.6, 19.7, 27.2, 27.3, 29.0, 30.1, 41.5, 57.9, 58.8, 66.9, 83.0, 94.9, 127.89, 127.91, 127.93, 127.98, 129.84, 129.88, 134.26, 134.44, 135.87, 135.93, 139.22, 154.8, 163.5; IR (neat) cm−1 2960s, 1710s, 1426s, 1393s, 1363m; mass spectrum (APCI): m/e (% relative intensity) 583.0 (20) (M+H)+, 439.4 (100), 393.3 (50), 279.2 (30), 191.2 (20), 101.1 (75); HRMS (MALDI) calcd for C36H46N2O3SiNa (M+Na)+ 605.3170, found 605.3202.
Hydrogenation of pyran 19 to 26
A heterogeneous mixture of Pt/Alumina (5%w/w, 80.0 mg, 0.020 mmol) and 19 (60.0 mg, 0.10 mmol) in hexanes (5 mL) was hydrogenated under 1500 psi in a high-pressure bomb at rt for 3 d. After which, the catalyst was filtered and washed with EtOAc (10 mL). Concentration under reduced pressure and purification of the crude residue via silica gel flash column chromatography (gradient eluent: 10–17% EtOAc in hexanes) afforded pure 26 (31.0 mg, 50%) as a colorless oil. 26: Rf = 0.50 [33% EtOAc/hexanes]; [α] D25 = + 49.8 ° [c 1.00, CH2Cl2]; 1H NMR (500 MHz, CDCl3) δ 0.62 (d, 3H, J = 6.5 Hz), 0.69 (d, 3H, J = 6.5 Hz), 0.96 (d, 3H, J = 7.0 Hz), 1.11 (s, 9H), 1.20–1.30 (m, 2H), 1.49 (ddd, 1H, J = 2.0, 6.0, 13.0 Hz), 1.71–1.80 (m, 2H), 1.93 (m, 1H), 2.70 (s, 3H), 3.50–3.60 (m, 3H), 3.75 (dd, 1H, J = 5.5, 10.0 Hz), 4.65 (d, 1H, J = 8.5 Hz), 5.15 (d, 1H, J = 2.0 Hz), 7.26–7.30 (m, 4 H), 7.42–7.48 (m, 7H), 7.26–7.30 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 12.6, 13.1, 15.5, 19.6, 22.3, 27.2, 28.9, 31.5, 31.6, 41.3, 57.8, 58.8, 66.1, 80.1, 86.5, 127.5, 127.8, 127.9, 129.8, 134.40, 134.44, 135.94, 135.95, 135.96, 136.00, 139.8, 168.2; IR (neat) cm−1 2962s, 1708s, 1472m, 1427s, 1390s; mass spectrum (APCI): m/e (% relative intensity) 585.2 (100) (M+H)+, 507.3 (30), 431.4 (20), 329.3 (80), 191.2 (30), 101.1 (90); HRMS (MALDI) calcd for C36H48N2O3SiNa (M+Na)+ 607.3332, found 607.3300.
Crotylation of pyran 26 to 50a/50b
To a solution of 26 (340.0 mg, 0.58 mmol) and Z-crotylsilane (270.0 µL, 1.74 mmol) in CH2Cl2 (30 mL) was added SnBr4 (1.13 g, 2.32 mmol) at −78 °C. The reaction was warmed to −35 °C slowly and stirred at this temperature for 24 h with exposure to the air. Then the mixture was quenched by adding sat aq NaHCO3 (10 mL), and extracted with CH2Cl2 (3 × 50 mL). The combined organic phases were washed with sat aq NaCl (2 × 15 mL), dried over Na2SO4, and concentrated under reduced pressure. Purification of the crude residue via silica gel flash column chromatography (gradient eluent: 7–14% EtOAc in hexanes) afforded a mixture of 50a and 50b (1:3, 80.0 mg, 65%) as a colorless oil. 50b: Rf = 0.30 [20% EtOAc/hexanes]; 1H NMR (500 MHz, CDCl3) δ 0.84 (d, 3H, J = 7.0 Hz), 1.04 (d, 3H, J = 7.0 Hz), 1.06 (d, 3H, J = 7.0 Hz), 1.35–1.40 (m, 1H), 1.48–1.63 (m, 2H), 1.70–1.88 (m, 3H), 2.71 (m, 1H), 3.19 (dd, 1H, J = 3.0, 9.0 Hz), 3.41 (bs, 1H), 3.47 (ddd, 1H, J = 3.5, 9.0, 9.0 Hz), 3.60 (m, 1 H), 5.00 (d, 1H, J = 10.5 Hz), 5.05 (d, 1H, J = 16.5 Hz), 5.64 (ddd, 1H, J = 8.0, 10.0, 17.0 Hz); 13C NMR (100 MHz, CDCl3) δ 13.9, 16.1, 18.6, 24.9, 25.0, 28.4, 38.3, 39.1, 68.2, 76.5, 81.9, 114.9, 142.1. 50a: 1H NMR (500 MHz, CDCl3) δ 0.80 (d, 3H, J = 6.5 Hz), 0.99 (d, 3H, J = 7.0 Hz), 1.03 (d, 3H, J = 7.0 Hz), 1.36–1.41 (m, 1H), 1.50–1.60 (m, 2H), 1.69–1.78 (m, 2H), 1.81–1.87 (m, 1H), 2.65 (m, 1H), 3.06 (dd, 1H, J = 4.0, 8.0 Hz), 3.21 (dd, 1H, J = 4.0, 8.0 Hz), 3.48–3.55 (m, 1H), 3.57 (dd, 1 H, J = 4.0, 10.6 Hz), 3.57–3.64 (m, 1H), 5.05 (d, 1H, J = 10.0 Hz), 5.07 (d, 1H, J = 15.0 Hz), 5.84 (ddd, 1H, J = 8.5, 10.0, 15.0 Hz); 13C NMR (100 MHz, CDCl3) δ 18.47, 18.50, 25.2, 25.7, 29.1, 38.0, 38.9, 67.7, 76.1, 81.7, 114.6, 141.4; IR (neat) cm−1 3439s, 2961s, 1640w, 1459s, 1376m; mass spectrum (APCI): m/e (% relative intensity) 213.3 (100) (M+H)+, 195.2 (85), 177.2 (90), 157.2 (30), 139.2 (45); HRMS (ESI) calcd for C13H24O2Na (M+Na)+ 235.1674, found 235.1673.
Supplementary Material
Experimental procedures, characterization data for all new compounds, 1H NMR, 13C NMR spectra, nOe’s and X-ray structural data are available free of charge via Internet at http://pubs.acs.org.
Scheme 17.

Crotylation of 26 with E and Z-Crotylsilane.
a: 4.0 equiv SnBr4, CH2Cl2, −78 °C to - 35 °C, 24 h, 65%; b: 1) Dess-Martin [O], 96%; 2) NaH2PO4, NaClO2, t-BuOH/H2O, 2-Me-2-butene, 85%; 3) Separation of 51a and 51b.
ACKNOWLEDGEMENT
The authors thank the NIH [GM066055] for funding and Mr. Benjamin E. Kucera and Dr. Vic Young from University of Minnesota for providing X-ray structural analysis.
REFERENCES
- 1.(a) Westley JW, editor. Polyether Antibiotics. Vol. 1. New York: Marcel Dekker; 1982. Vol. 2. 1983. [Google Scholar]; (b) Dobler M, editor. Ionophores and their Structures. New York: Wiley; 1981. [Google Scholar]
- 2.For isolation, see: Grafe U, Schade W, Roth M, Radics L, Incze M, Ujszaszy K. J. Antibiot. 1984;37:836–846. doi: 10.7164/antibiotics.37.836. ; (b) Brooks HA, Gardner D, Poyser JP, King TJ. J. Antibiot. 1984 37;:1501–1504. doi: 10.7164/antibiotics.37.1501. [DOI] [PubMed] [Google Scholar]; (c) Radics L. J. Chem. Soc., Chem. Commun. 1984:599–601. [Google Scholar]; d) Gräfe U. German. [East]: DD 231,793. [Google Scholar]
- 3.Tonew E, Tonew M, Gräfe U, Zopel P. Pharmazie. 1988;43:717–719. [PubMed] [Google Scholar]
- 4.For approaches, see: Cossy J, Blanchard N, Defosseux M, Meyer C. Angew. Chem., Int. Ed. 2002;41:2144–2146. Guindon Y, Murtagh L, Caron V, Landry SR, Jung G, Bencheqroun M, Faucher A-M, Guerin B. J. Org. Chem. 2001;66:5427–5437. doi: 10.1021/jo010310f. Burke SD, Ng RA, Morrison JA, Alberti MJ. J. Org. Chem. 1998;63:3160–3161. Marshall JA, Palovich MR. J. Org. Chem. 1998;63:3701–3705. Chemler SR, Roush WR. J. Org. Chem. 1998;63:3800–3801. Booysen JF, Holzapfel CW. Synth. Commun. 1995;25:1473–1488. Cywin CL, Kallmerten J. Tetrahedron Lett. 1993;34:1103–1106. Balestra M, Wittman MD, Kallmerten J. Tetrahedron Lett. 1988;29:6905–6908. Zelle RE, DeNinno MP, Selnick HG, Danishefsky SJ. J. Org. Chem. 1986;51:5032–5036. The first total synthesis, see: Danishefsky SJ, Selnick HG, DeNinno MP, Zelle RE. J. Am. Chem. Soc. 1987;109:1572–1574. Danishefsky SJ, Selnick HG, Zelle RE, DeNinno MP. J. Am. Chem. Soc. 1988;110:4368–4378. For Cossy’s total synthesis, see: Cossy J, Meyer C, Defosseux M, Blanchard N. Pure Appl. Chem. 2005;77:1131–1137. Defosseux M, Blanchard N, Meyer C, Cossy J. J. Org. Chem. 2004;69:4626–4647. doi: 10.1021/jo0496042. Defosseux M, Blanchard N, Meyer C, Cossy J. Org. Lett. 2003;5:4037–4040. doi: 10.1021/ol035177n. For Miyashita’s total synthesis, see: Komatsu K, Tanino K, Miyashita M. Angew. Chem. Int. Ed. 2004;43:4341–4345. doi: 10.1002/anie.200460434.
- 5.Song Z, Hsung PR. Org. Lett. 2007;9:2199–2202. doi: 10.1021/ol070791a. [DOI] [PubMed] [Google Scholar]
- 6.Wei L-L, Hsung RP, Xiong H, Mulder JA, Nkansah NT. Org. Lett. 1999;1:2145–2148. [Google Scholar]
- 7.For reviews on allenamides, see: Hsung RP, Wei L-L, Xiong H. Acc. Chem. Res. 2003;36:773–782. doi: 10.1021/ar030029i. Tracey MR, Hsung RP, Antoline J, Kurtz KCM, Shen L, Slafer BW, Zhang Y. Chapter 21.4. In: Steve M, Weinreb, editors. Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Stuttagart, Germany: Georg Thieme Verlag KG; 2005.
- 8.For recent reports on the allenamide chemistry, see: Parthasarathy K, Jeganmohan M, Cheng C-H. Org. Lett. 2006;8:621–623. doi: 10.1021/ol0527936. Fenández I, Monterde MI, Plumet J. Tetrahedron Lett. 2005;46:6029–6031. de los Rios C, Hegedus LS. J. Org. Chem. 2005;70:6541–6543. doi: 10.1021/jo050813b. Alouane N, Bernaud F, Marrot J, Vrancken E, Mangeney P. Org. Lett. 2005;7:5797–5800. doi: 10.1021/ol0522743. Antoline JE, Hsung RP, Huang J, Song Z, Li G. Org. Lett. 2007;9:1275–1278. doi: 10.1021/ol070103n. Huang J, Ianni JC, Antoline JE, Hsung RP, Kozlowski MC. Org. Lett. 2006;8:1565–1568. doi: 10.1021/ol0600640. Berry CR, Hsung RP, Antoline JE, Petersen ME, Rameshkumar C, Nielson JA. J. Org. Chem. 2005;70:4038–4042. doi: 10.1021/jo0503411. Shen L, Hsung RP, Zhang Y, Antoline JE, Zhang X. Org. Lett. 2005;7:3081–3084. doi: 10.1021/ol051094q. Huang J, Hsung RP. J. Am. Chem. Soc. 2005;127:50–51. doi: 10.1021/ja044760b.
- 9.For some recent elegant studies on related inverse demand hetero [4 + 2] cycloaddition of chiral enamides, see: Gohier F, Bouhadjera K, Faye D, Gaulon C, Maisonneuve V, Dujardin G, Dhal R. Org. Lett. 2007;9:211–214. doi: 10.1021/ol062626l. Tardy S, Tatibouët A, Rollin P, Dujardin G. Synlett. 2006:1425–1427. and reference cited therein. Also see: Palasz A. Org. Biomol. Chem. 2005;3:3207–3212. doi: 10.1039/b504210k.
- 10.(a) Berry CR, Hsung RP. Tetrahedron. 2004;60:7629–7636. [Google Scholar]; (b) Wei L-L, Xiong H, Douglas CJ, Hsung RP. Tetrahedron Lett. 1999;40:6903–6907. [Google Scholar]; (c) Berry CR, Rameshkumar C, Tracey MR, Wei L-L, Hsung RP. Synlett. 2003:791–796. [Google Scholar]; (d) Rameshkumar C, Hsung RP. Synlett. 2003:1241–1246. [Google Scholar]
- 11.Close WJ. J. Org. Chem. 1950;15:1131–1134. [Google Scholar]
- 12.(a) Fuerstner A, Kattnig E, Lepage O. J. Am. Chem. Soc. 2006;128:9194–9204. doi: 10.1021/ja061918e. [DOI] [PubMed] [Google Scholar]; (b) Bergmeier SC, Stanchina DM. J. Org. Chem. 1997;62:4449–4456. doi: 10.1021/jo970473x. [DOI] [PubMed] [Google Scholar]
- 13.(a) Crabtree RH, Davis MW. J. Org. Chem. 1986;51:2655–2661. [Google Scholar]; (b) Crabtree RH. Acc. Chem. Res. 1979;12:331–337. [Google Scholar]
- 14.For the first application of directed hydrogenations employing Crabtree’s catalyst, see: Stork G, Kahne DE. J. Am. Chem. Soc. 1983;105:1072–1073. For some leading applications, also see: Evans DA, Morrissey MM. J. Am. Chem. Soc. 1984;106:3866–3868. Ginn JD, Padwa A. Org. Lett. 2002;4:1515–1517. doi: 10.1021/ol025746b.
- 15.For an amide directed hydroboration using Crabtree’s catalyst, see: Evans DA, Fu GC. J. Am. Chem. Soc. 1991;113:4042–4043. Evans DA, Fu GC, Hoveyda AH. J. Am. Chem. Soc. 1992;114:6671–6679.
- 16.For a review on directed reactions, see: Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307–1370. For a highlight related to Crabtree’s catalyst, see: Nell PG. Synlett. 2001:160.
- 17.Romero JAC, Tabacco SA, Woerpel KA. J. Am. Chem. Soc. 2000;122:168–169. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, characterization data for all new compounds, 1H NMR, 13C NMR spectra, nOe’s and X-ray structural data are available free of charge via Internet at http://pubs.acs.org.