

Abstract
Background and Objective
Our goal is to obtain insight into the causes of the pathological lesions in Alzheimer's disease (AD). It is thought that the β-amyloid (Aβ) deposits within the cerebral cortex and hippocampus of AD brains are initiated by a ‘bad seed’ of oligomeric Aβ. The origin of this seed is unknown. Here, we focused on the events that might trigger the formation of neuritic plaques, aiming to explain how these plaques form in cortical and hippocampal regions.
Methods and Results
Using immunocytochemical and biochemical methods, we showed that brainstem-derived, neuronal cells (CAD) – but not cortical or hippocampal neurons – show large amounts of Aβ accumulated at the terminals of their processes. This is similar to what is believed to occur in brain neurons, in the early phases of AD. CAD cells that contain Aβ accumulations also concentrate β-secretase at process terminals. We show that, while the anterograde transport of small vesicles is not significantly affected, the mitochondrial transport is perturbed in CAD cells that contain Aβ accumulations. We further show that intracellular, neuritic Aβ accumulations may become extracellular upon neurite degeneration, thus providing the initial ‘bad seed’ of Aβ oligomers that triggers further aggregation of extracellular proteins.
Conclusion
We propose that brainstem neurons, known to send projections throughout the brain, could provide the ‘bad seed’ of Aβ that nucleates plaques in the cerebral cortex and hippocampus of AD brains.
Key Words: Alzheimer's disease, Neurodegeneration, β-Amyloid precursor protein, β-Amyloid peptide, Neuritic plaques, Autophagy, Mitochondria, Brainstem neurons, CAD cells
Background and Objective
Alzheimer's disease (AD), a complex neurodegenerative disorder, is characterized by two major lesions: the neuritic plaques and the neurofibrillary tangles. Neuritic plaques contain extracellular deposits of β-amyloid (Aβ) peptide, a metabolite of the transmembrane protein, Aβ precursor protein [1].
A characteristic feature of AD neuropathology is the preferential formation of plaques in cortical and hippocampal brain regions. Yet, the initial events that trigger plaque formation in certain brain regions and not in others are not known. According to the seeded polymerization theory [2], the aggregation of soluble Aβ, which leads to plaque formation, is nucleated by ‘bad seeds’ of oligomeric Aβ. The origin and nature of these hypothetical seeds are not known. As described below, our work with the neuronal cell line CAD suggests that Aβ oligomers form at the terminals of projections of brainstem neurons and could act as such seeds.
Methods
CAD cells [a locus coeruleus (LC)-derived cell line] [3] have emerged as an important in vitro experimental system for studying the molecular pathobiology of AD [4,5,6,7,8], and – as highlighted here – may be particularly relevant to the initiation of neuritic plaque formation. Using immunocytochemical and biochemical approaches, we have characterized the CAD cell line with respect to the metabolism of Aβ precursor protein and generation of Aβ.
Results
We discovered that CAD cells are prone to accumulation of large amounts of intracellular Aβ at the terminals of their processes (fig. 1), similar to what may occur in brain neurons, during the initial phases of AD [7]. Using carboxy-terminal-end antibodies to Aβ species, we showed that these Aβ accumulations contain both the Aβ40 and Aβ42 peptides [7]. Cross-reactivity of the accumulations with an antioligomer antibody that preferentially detects species larger than the octamer (A11) [9] indicated that the accumulations include large-molecular-size Aβ oligomers [7].
Fig. 1.
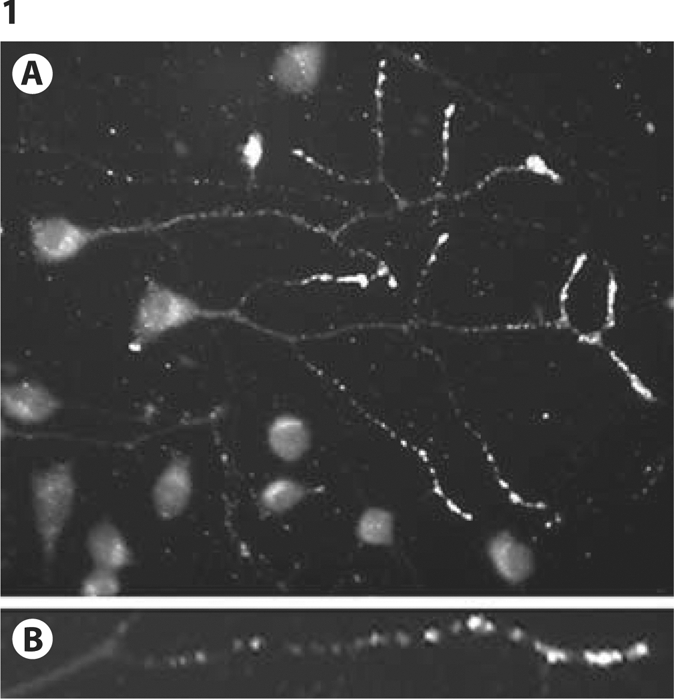
CAD cells immunolabeled with antibody 6E10 (Signet, Dedham, Mass., USA), showing Aβ accumulations at neurite endings (A). B An enlargement of a process, showing localization of Aβ to large particles resembling late endosomes and autophagosomes.
The neuritic accumulation of Aβ in CAD cells is restricted to a small population of cells that show redistribution of β-secretase (BACE1) to the processes, where it colocalizes with Aβ and markers of late endosomes and autophagic vacuoles [7]. These findings suggest that the Aβ accumulations could be generated through endocytosis or macroautophagy, two processes previously implicated in the formation of the neuritically localized Aβ [10, 11]. Importantly, unlike the LC-derived CAD cells, cultured cortical and hippocampal neurons do not show detectable Aβ accumulations at their neuritic terminals (data not shown).
Here, we hypothesize that in AD brains, accumulations of Aβ similar to those observed in CAD cells (fig. 1) could form at the projection terminals of brainstem neurons (which send projections throughout the central nervous system, including the cerebral cortex and hippocampus), and serve as seeds for further Aβ aggregation. This process leads to plaque formation at locations remote from the brainstem, such as the cerebral cortex and hippocampus (fig. 2). The LC is largely affected by cell death early in AD. However, since this brain region mostly lacks neuritic plaques, the prevailing view was that neuronal loss in the LC is caused by the cortical Aβ, which ‘poisons’ the projections of brainstem cells [13]. By contrast, our results suggest that the neuropathology of AD may actually begin in the subcortical regions, and then spread to the cortex and hippocampus. This is in line with earlier reports suggesting that, in AD, pathologic alterations in the LC may spread to cortical and hippocampal brain regions [14,15,16].
Fig. 2.
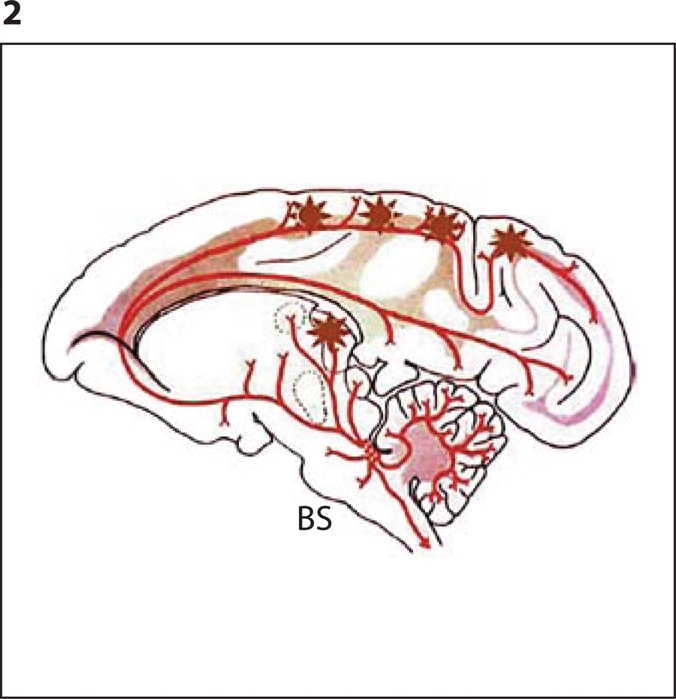
Diagram showing plaques (∗) at the terminals of projections of brainstem (BS) neurons. The drawing was modified from Aston-Jones and Cohen [12], and used with permission from the Annual Review of Neuroscience, vol. 28, 2005, by Annual Reviews (www.annualreviews.org).
How could the intracellular Aβ accumulations (fig. 1) become extracellular and serve as seeds for Aβ deposition? The mechanism could be the fusion of the Aβ-containing autophagosomes and late endosomes with the plasma membrane, or alternatively, neurite degeneration. Indeed, we occasionally found neuritic debris containing Aβ, in the areas enriched in CAD cells having Aβ accumulations (fig. 3). A further question raised by this result is what could trigger neurite degeneration in these cells? We found that, while the anterograde transport of small vesicles is not significantly affected [7], the transport and the neuritic localization of mitochondria is evidently perturbed in CAD cells that contain Aβ accumulations (fig. 4). This could result in diminished supply of ATP in the neurites, which could cause their observed degeneration.
Fig. 3.
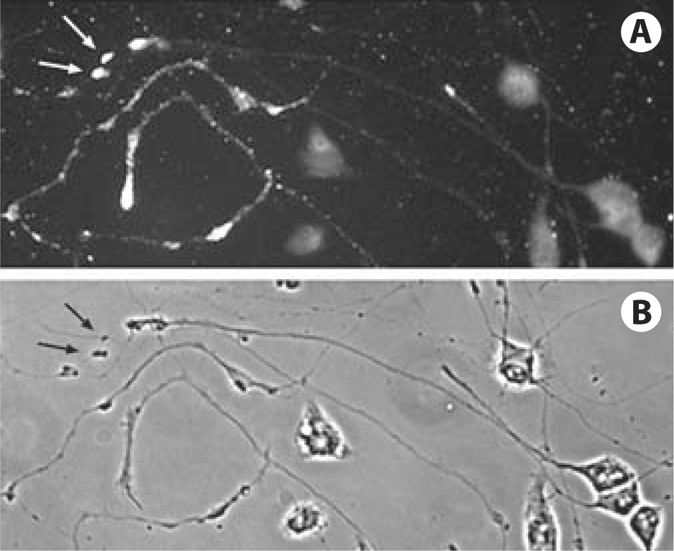
Aβ accumulations in the debris of CAD cell processes (arrows). A CAD cells immunolabeled with antibody 6E10. B Corresponding phase contrast micrograph.
Fig. 4.

Mitochondrial transport into neurites that contain Aβ deposits is disrupted. Fewer mitochondria are present throughout the neurite of a CAD cell that contains Aβ deposits (short arrows). By contrast, neurites that lack Aβ deposits have numerous mitochondria that accumulate at the terminal (long arrows). Mitochondria were detected with an antibody to lipoic acid (A) and Aβ was detected with antibody 6E10 (B).
Conclusions
Taken together, our results point to the brainstem neurons as possible initiators of plaque formation in AD. We propose that the initial seeds of aggregated Aβ are produced at the neurite terminals of the brainstem neurons that project into the brain regions prone to AD pathology (e.g. cerebral cortex, hippocampus). These seeds then trigger further aggregation of soluble, extracellular Aβ into plaques. Thus, our work offers a novel perspective on AD pathogenesis. In addition, the CAD cell system that we established is ideally suited for screening of compounds that could prevent accumulation of Aβ, and thus could be used to treat brainstem neurons at the early stages of AD.
Acknowledgements
This work was supported by National Institutes of Health grant 5R01GM068596 and funds from UMDNJ-NJMS. The work was started at Case Western Reserve University, Cleveland, Ohio, USA.
References
- 1.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 3.Qi Y, Wang JK, McMillian M, Chikaraishi DM. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J Neurosci. 1997;17:1217–1225. doi: 10.1523/JNEUROSCI.17-04-01217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muresan Z, Muresan V. A phosphorylated, carboxy-terminal fragment of β-amyloid precursor protein localizes to the splicing factor compartment. Hum Mol Genet. 2004;13:475–488. doi: 10.1093/hmg/ddh054. [DOI] [PubMed] [Google Scholar]
- 5.Muresan Z, Muresan V. c-Jun NH2-terminal kinase-interacting protein-3 facilitates phosphorylation and controls localization of amyloid-beta precursor protein. J Neurosci. 2005;25:3741–3751. doi: 10.1523/JNEUROSCI.0152-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muresan Z, Muresan V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-Jun NH2-terminal kinase-interacting protein-1. J Cell Biol. 2005;171:615–625. doi: 10.1083/jcb.200502043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muresan Z, Muresan V. Neuritic deposits of amyloid-β peptide in a Subpopulation of central nervous system-derived neuronal cells. Mol Cell Biol. 2006;26:4982–4997. doi: 10.1128/MCB.00371-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muresan Z, Muresan V. The amyloid-β precursor protein is phosphorylated via distinct pathways during differentiation, mitosis, stress, and degeneration. Mol Biol Cell. 2007;18:3835–3844. doi: 10.1091/mbc.E06-07-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy – A novel beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- 13.German DC, Nelson O, Liang F, Liang CL, Games D. The PDAPP mouse model of Alzheimer's disease: locus coeruleus neuronal shrinkage. J Comp Neurol. 2005;492:469–476. doi: 10.1002/cne.20744. [DOI] [PubMed] [Google Scholar]
- 14.Hertz L. Is Alzheimer's disease an anterograde degeneration, originating in the brainstem, and disrupting metabolic and functional interactions between neurons and glial cells? Brain Res Brain Res Rev. 1989;14:335–353. doi: 10.1016/0165-0173(89)90017-9. [DOI] [PubMed] [Google Scholar]
- 15.Mann DM, Yates PO, Hawkes J. The pathology of the human locus ceruleus. Clin Neuropathol. 1983;2:1–7. [PubMed] [Google Scholar]
- 16.Saper CB, Wainer BH, German DC. Axonal and transneuronal transport in the transmission of neurological disease: potential role in system degenerations, including Alzheimer's disease. Neuroscience. 1987;23:389–398. doi: 10.1016/0306-4522(87)90063-7. [DOI] [PubMed] [Google Scholar]