

Abstract
Autophagy, an evolutionally conserved homeostatic process for catabolizing cytoplasmic components, has been implicated in the elimination of intracellular pathogens during mammalian innate immune responses. However, the mechanisms underlying cytoplasmic infection-induced autophagy, and the role of autophagy in host survival against intracellular pathogens are unknown. Here we report that in drosophila, recognition of diaminopimelic acid-type peptidoglycans by the pattern recognition receptor PGRP-LE is crucial for the induction of autophagy, and that autophagy prevents the intracellular growth of Listeria monocytogenes and promotes host survival against this infection. Autophagy induction occurs independently of the Toll and IMD innate signaling pathways. These findings define a clear pathway leading from the intracellular pattern recognition receptors to the induction of autophagy to host defense.
INTRODUCTION
The innate immune system is a powerful and evolutionally well-conserved barrier to infectious pathogens. In drosophila, which rely almost entirely on innate immunity to fight microbial infection, members of the peptidoglycan recognition protein (PGRP) family act as microbe sensors1, 2. These receptors are found in the hemolymph, on immune cell surfaces, and within the immune cells, and they recognize bacterial peptidoglycans and activate immune signaling pathways such as the Toll and IMD pathways. The Toll and IMD pathways control the production of antimicrobial peptides (AMPs) through NF-κB transcription factors1,2. PGRP-LE, a drosophila PGRP family member present both in the hemolymph and inside immune cells3, binds to diaminopimelic acid (DAP)-type peptidoglycans (PGN)4 and is sufficient for the induction of AMP genes following immune stimulation with a monomeric DAP-type PGN, tracheal cytotoxin (TCT)5. AMPs are quite effective against the growth of bacteria or fungi in the hemolymph6 and perhaps in phagosomal compartments as well, but cytosolic pathogens escape these humoral defenses. Mammalian cells also express intracellular receptors that detect intracellular pathogens or their products7. For example, NOD1 and NOD2 sense substructures from bacterial PGN in the cell cytoplasm and activate innate immune signaling pathways8, 9. CARD9 is an adaptor protein that associates with NOD2 and elicits innate immune responses critical for protecting mice from Listeria monocytogenes infection10. The immune responses activated by these receptors to defend against intracellular bacterial growth are not clear, however, in either mammals or insects.
Autophagy is a highly conserved cellular mechanism in which cytoplasmic components are sequestered into double-membrane structures called autophagosomes and eventually degraded in lysosomes11. Autophagy is involved in diverse functions, including the removal of damaged organelles, protein turnover, the supply of nutrients under nutrient-deprived conditions, and in cell survival and death12. In mammalian cells, autophagy also plays a role in innate immune defenses against invading pathogens, such as group A Streptococcus, Shigella flexneri, Mycobacterium tuberculosis, and Toxoplasma gondii13–17. Group A Streptococcus is an extracellular bacteria that can also invade the host cell cytoplasm where it is rapidly sequestered into autophagosomes associated with microtubule-associated protein 1 light chain 3 (LC3, also called Atg8), resulting in its degradation in the vacuole after fusion with lysosomes13. M. tuberculosis resides in phagosomes where it interferes with phagosomal maturation and reduces the acidification of the bacteria-containing vacuole. The activation of M. tuberculosis-infected macrophages by interferon-γ induces autophagy; this autophagy is induced in an IRGM (immunity-related GTPase family, M; p47 GTPase)-dependent manner, and inhibits bacterial survival14, 18. Although these reports suggest that some intracellular pathogens induce autophagy and are controlled by autophagy in cultured cells, these studies show only limited effects on pathogen survival. Moreover, the mechanism(s) and sensor(s) mediating the induction of these autophagic antibacterial responses remain to be elucidated.
Recent studies also suggest that Toll-like receptor (TLR) signaling can promote autophagy. TLR4 stimulation promotes the colocalization of mycobacterial phagosomes with autophagosomes19, but this is unlikely to be relevant in an infection because M. tuberculosis does not express any TLR4 ligands. A number of other TLR ligands also induce autophagy in cultured macrophage cell lines or murine primary macrophages20. It is not yet clear, however, if and how M. tuberculosis infections induce autophagy. Moreover, it is not yet known if autophagy plays an important role in protecting the whole animal from lethal infection by M. tuberculosis, Group A Streptococcus, or other intracellular infections.
A link between the autophagy pathway and TLR signaling is further supported by a recent report that phagocytosis of TLR agonist-coated beads promotes phagosome maturation by recruiting elements of the autophagy pathway to the phagosome 21. Localized TLR signaling within the phagosome is therefore likely to be important for the recruitment of autophagic components.
The cytoplasmic sensor(s) that recognize bacteria invading the cytoplasm and trigger autophagy remain to be identified. Here, we demonstrate that in drosophila, PGRP-LE recognizes cytosolic L. monocytogenes, and is essential for inducing autophagy, which inhibits intracellular growth of the bacteria and is necessary for host survival after L. monocytogenes infection. In addition, the induction of autophagy after detection of the bacteria via PGRP-LE is independent of the Toll pathway and IMD pathways, suggesting the existence of a distinct innate immune pathway responsible for the induction of autophagy by a cytoplasmic sensor.
RESULTS
PGRP-LE in resistance to L. monocytogenes infection
We previously demonstrated that in drosophila, PGRP-LE functions as an intracellular receptor for a monomeric DAP-type PGN, TCT5. This finding led us to examine whether PGRP-LE is essential for the recognition of intracellular pathogens. We first analyzed the resistance of the PGRP-LE null mutant PGRP-LE112 to infection by L. monocytogenes, an intracellular Gram-positive bacteria that is pathogenic to mammals and drosophila and that expresses DAP-containing PGN, which is potentially recognized by PGRP-LE4. As indicated by survival experiments, the PGRP-LE112 mutant was hyper-susceptible to low-dose L. monocytogenes infection compared with wild-type (Oregon R) or yw flies (flies with the same genetic background as the PGRP-LE112 mutant) (Fig. 1a). Consistent with a previous report demonstrating that IMD pathway mutants are susceptible to L. monocytogenes infection22, PGRP-LC7454 mutants, in which PGRP-LC, a cell surface sensor required for the activation of the IMD pathway, is hardly expressed, were also susceptible to L. monocytogenes infection. The survival rate of PGRP-LE112,PGRP-LC7454 double mutants following L. monocytogenes infection was similar to that of the PGRP-LE112 and PGRP-LC7454 mutants, suggesting that PGRP-LE and PGRP-LC do not have redundant functions in enabling resistance to L. monocytogenes. L. monocytogenes requires listeriolysin O to lyse the host phagocytic vacuole and access the cytosol23. The Δhly strain of L. monocytogenes, which harbors a deletion of the gene encoding listeriolysin O, can enter host cells by phagocytosis, but is incapable of entering the cytoplasm24. PGRP-LE112 mutants were not susceptible to this strain, consistent with the notion that cytosolic recognition requires PGRP-LE (Fig. 1b).
Figure 1.
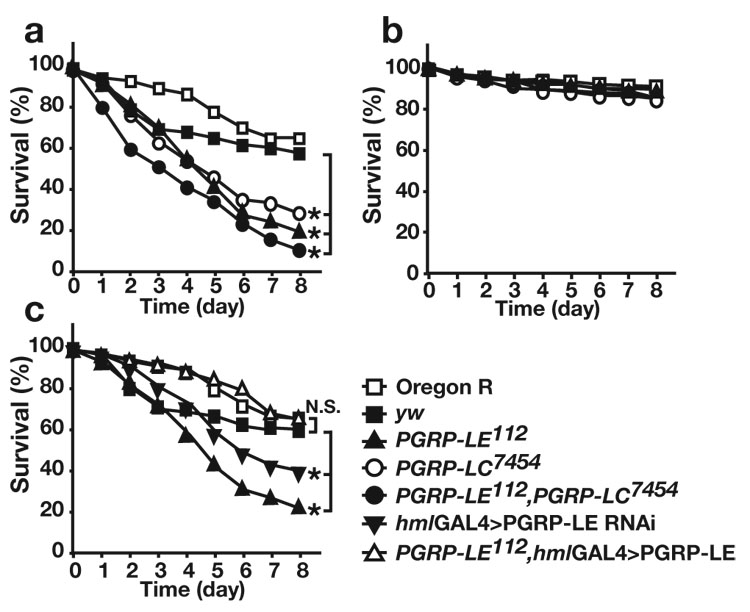
PGRP-LE in hemocytes is essential for resistance against L. monocytogenes infection in vivo. (a,b) Survival rate of wild-type (Oregon R) flies, yw flies, ywPGRP-LE112 (PGRP-LE112), w;;PGRP-LC7454 (PGRP-LC7454), and ywPGRP-LE112,PGRP-LC7454 double-mutant flies (PGRP-LE112,PGRP-LC7454), after (a) wild-type or (b) Δhly L. monocytogenes injections. (c) Survival rate of Oregon R, yw, ywPGRP-LE112 (PGRP-LE112), UAS-PGRP-LE IR/+; hml-GAL4/+, (hml-GAL4>PGRP-LE RNAi) and ywPGRP-LE112; UAS-PGRP-LE/+;hml-GAL4/+ (PGRP-LE112, hml-GAL4>PGRP-LE) flies after wild-type L. monocytogenes injections. All experiments were performed at 28°C. The average survival rate of four independent experiments is shown. In each experiment, >30 flies of the indicated genotypes were examined at the same time. * P<0.01 (Wilcoxon-Mann-Whitney test) (P=0.0006, 0.0043, 0.0043, yw versus PGRP-LE112, PGRP-LC7454, PGRP-LE112,PGRP-LC7454 flies, respectively in (a) P=0.0095 yw versus hml-GAL4>PGRP-LE RNAi flies in (c))
L. monocytogenes infects phagocytic cells such as macrophages in mammals and hemocytes in Drosophila22, 23. PGRP-LE is expressed in hemocytes3. Susceptibility to L. monocytogenes in PGRP-LE112 mutants was rescued by expressing PGRP-LE specifically in hemocytes using the hemocyte-specific driver hemolectin-GAL4 (hml-GAL4)25, demonstrating that PGRP-LE expression in hemocytes is important for resistance against L. monocytogenes infection (Fig. 1c). Consistent with this finding, of the expression of RNA interference (RNAi) targeting PGRP-LE using the hml-GAL4 driver induced susceptibility to L. monocytogenes infection (Fig. 1c). In these conditions, hml-GAL4-drived RNAi efficiently reduced PGRP-LE expression in the hemocytes (Supplementary Fig. 1, online) but did not reduce the PGRP-LE expression in the fat body or gut (data not shown).
Next, we examined if PGRP-LE is involved in suppressing bacterial growth in vivo. In wild-type flies, the growth of L. monocytogenes was relatively suppressed in both the humoral and the cellular fractions, whereas in the PGRP-LE112 mutant flies, L. monocytogenes growth in the cellular fraction began immediately after infection, and bacterial growth in the humoral fraction started to increase 6 h later (Supplementary Fig. 2, online). In contrast to the PGRP-LE112 mutant, bacterial growth in the humoral fraction of the PGRP-LC7454 mutant began to increase immediately after infection, but bacterial growth in the cellular fraction of the PGRP-LC7454 mutant was indistinguishable from that in wild-type flies (Supplementary Fig. 2, online). These data suggest that PGRP-LE is essential for suppressing the growth of L. monocytogenes within cells in vivo, while PGRP-LC protects the extracellular environment from these microbes.
PGRP-LE and autophagy suppress L. monocytogenes growth
To directly demonstrate the requirement for PGRP-LE in limiting the intracellular growth of L. monocytogenes, ex vivo-cultured hemocytes from drosophila larvae were infected with L. monocytogenes for 1 h and then incubated for 5 h in gentamicin-containing medium to kill the extracellular bacteria. L. monocytogenes infection of hemocytes was visualized by 4,6-diamido-2-phenylinodole (DAPI) staining (Fig. 2a) and the number of bacteria per cell was quantified (Fig. 2b). Wild-type but not Δhly L. monocytogenes produced an actin comet in hemocytes (Supplementary Fig. 3, online), suggesting that wild-type L. monocytogenes invaded the cytoplasm of the ex vivo-cultured hemocytes, which is consistent with a previous report that L. monocytogenes associates with actin in hemocytes derived from infected larvae22. The number of wild-type L. monocytogenes contained in the PGRP-LE112 mutant hemocytes was significantly greater than that in wild-type hemocytes (Fig. 2a, b). The number of Δhly strain L. monocytogenes growing in PGRP-LE112 hemocytes was similar to that in wild-type hemocytes (Fig. 2b). These results suggest that PGRP-LE recognizes L. monocytogenes only after it enters the hemocyte cytoplasm and that phagocytic activity required for entry of the bacteria was not affected in the PGRP-LE112 mutant. PGRP-LE expression induced by the hml-GAL4 driver in PGRP-LE112 hemocytes rescued the growth suppression of the wild-type bacteria (Fig. 2b). These results indicate that PGRP-LE acts cell-autonomously in suppressing the cytosolic growth of the bacteria. In contrast to PGRP-LE hemocytes, those from mutant strains of Imd, the critical IMD pathway adaptor, or Relish, the key NF-κB transcription factor of the pathway, did not exhibit impaired control of intracellular Listeria growth (Fig. 2a,b). The control of intracellular bacterial growth was also not affected in hemocytes from larvae of PGRP-LC7454, MyD88kra1, a mutant of an adaptor of the Toll pathway, or J4 homozygous mutants, which lack both Dif and dorsal (dl), key transcription factors in the Toll pathway (Fig. 2b). These results demonstrated that neither the two classical innate immune signaling pathways, nor the cell surface receptor for DAP-type PGN, are required to control the intracellular growth of L. monocytogenes; in contrast, the intracellular PGN receptor PGRP-LE is crucial. These results argue that some mechanism other then AMP production is essential for defending against cytosolic L. monocytogenes.
Figure 2.
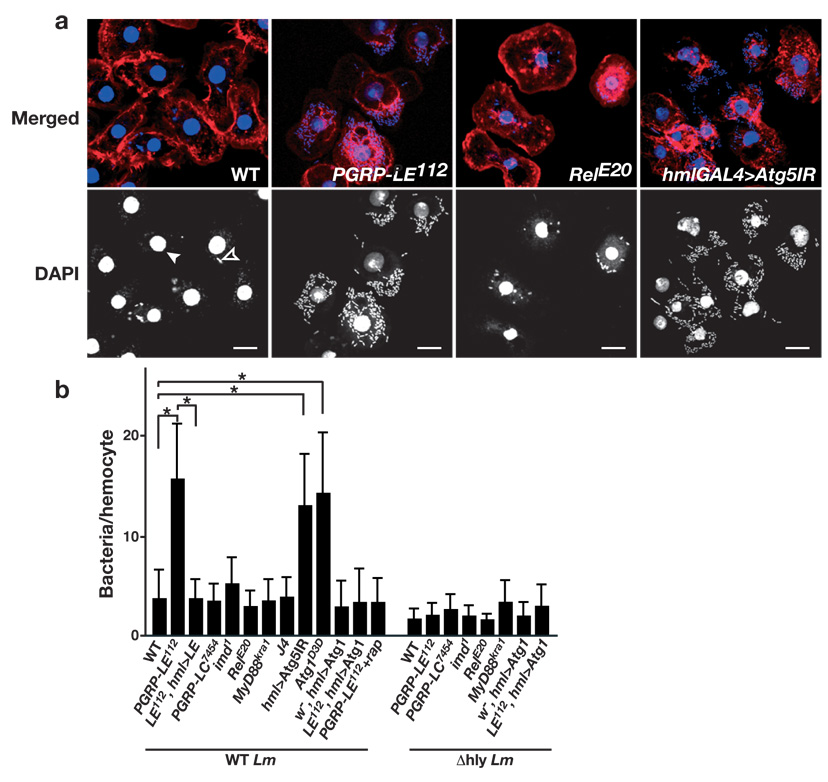
PGRP-LE and autophagy, but not the Toll and IMD pathways, are required to suppress the intracellular growth of L. monocytogenes in hemocytes. (a) Hemocytes from third instar larvae of the indicated genotype were cultured ex vivo and infected with wild-type L. monocytogenes. Hemocyte nuclei (filled arrowhead) and DNA of L. monocytogenes (open arrowhead) were visualized by DAPI staining (blue or white), and the actin cytoskeleton was stained with rhodamine-labeled phalloidin (red). For the induction of RNAi against Atg5, an inverted repeat sequence of Atg5 (Atg5IR) was expressed using hml-GAL4 (hmlGAL4>Atg5IR). Bars represent 10 µm. (b) The number of intracellular wild-type (WT Lm) or Δhly (Δhly Lm) L. monocytogenes per hemocyte of the indicated genotype was manually counted by DAPI staining of the infected hemocytes. Bars indicate standard deviation of triplicate measurements of at least three independent experiments. *P < 0.001 (t-test) each genotype versus wild-type. Genotype abbreviations used: PGRP-LE112,ywPGRP-LE112; LE112,hml>LE, ywPGRP-LE112;UAS-PGRP-LE/+;hml-GAL4/+; PGRP-LC7454, w;;PGRP-LC7454; RelE20, RelishE20; J4, Df(2L)J4; hml>Atg5IR, ywUAS-Atg5IR/+;;hml-GA4/+; w−,hml>Atg1, w−;;UAS-Atg16B/hml-GAL4, LE112,hml>Atg1, ywPGRP-LE112;;UAS-Atg16B/hml-GAL4
Several recent studies reported that autophagy functions in innate immune responses against intracellular pathogens in mammalian cells13–18. As AMPs do not appear necessary to control intracellular L. monocytogenes, we investigated whether autophagy is required for the inhibition of L. monocytogenes growth in hemocytes by expressing an RNAi transgene targeting drosophila Atg5 (CG1643), an essential factor for autophagy26, 27, with an hml-GAL4 driver, which efficiently reduced Atg5 expression in hemocytes (Supplementary Fig. 1, online). Ex vivo-cultured hemocytes expressing Atg5 RNAi contained higher numbers of bacteria (Fig. 2a,b). The numbers of bacteria were similarly increased in hemocytes from the Atg1Δ3D mutant (Fig. 2b), in which starvation-induced autophagy is completely abolished26. Consistent with a recent report demonstrating that autophagy limits the intracellular growth of L. monocytogenes in mouse embryonic fibroblast cells28, these results indicate the essential role of autophagy in defending against L. monocytogenes infection. Moreover, the induction of autophagy by rapamycin treatment29 and by the forced expression of Atg130 using the hml-GAL4 driver reduced the numbers of bacteria in PGRP-LE112 cells, suggesting that PGRP-LE acts upstream of autophagy in the resistance against L. monocytogenes infection (Fig. 2b).
The function of autophagy in hemocytes during innate immune responses against L. monocytogenes was confirmed in vivo by examining survival rates of flies in which the RNAi transgene against Atg5 was expressed in hemocytes. Atg5 RNAi flies were susceptible to wild-type L. monocytogenes infection, similar to PGRP-LE112 mutants, whereas they were totally resistant to the Δhly strain of L. monocytogenes (Fig. 3a,b). In contrast, the susceptibility of the Atg5 RNAi line and PGRP-LE112 to Erwinia carotovora, an extracellular Gram-negative bacteria, was not different from that of the control flies (Fig. 3c). The PGRP-LC7454 mutant was more susceptible to E. carotovora. This is the first demonstration that autophagy is crucial for host survival against intracellular pathogens.
Figure 3.
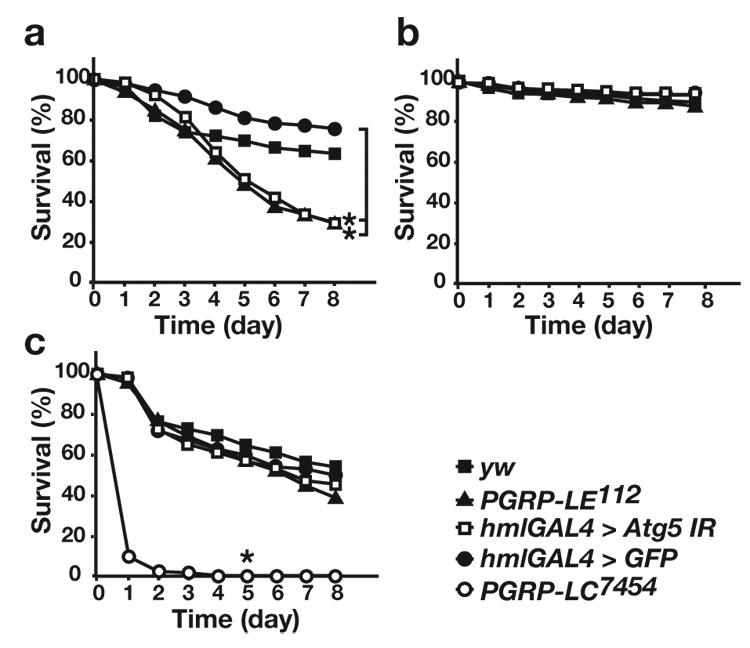
Autophagy is crucial for host survival against L. monocytogenes infection. (a–c) Survival rate of yw flies, PGRP-LE112, Atg5 RNAi (hml-GAL4>Atg5IR), and control hml-GAL4-carrying flies (hml-GAL4>GFP) after injection of (a) wild-type or (b) Δhly L. monocytogenes, or (c) E. carotovora (Ecc15) into adult flies. The average survival rate of four independent experiments is shown. * P < 0.01 (Wilcoxon-Mann-Whitney test versus hmlGAL4>GFP in (a), versus yw in (c)) (P=0.0024, 0.0007, hml-GAL4>GFP versus hml-GAL4>Atg5IR, PGRP-LE112, respectively in (a) P=0.0055, yw versus PGRP-LC7454 flies in (c))
Drosophila S2 cells are macrophage-like cells and are an excellent model for studying intracellular infections, especially L. monocytogenes infection31, 32. S2 cells had no detectable expression of PGRP-LE (Supplementary Fig. 4, online), thus we compared L. monocytogenes infection in S2 cells and S2 cells stably transfected with a metallothionein promoter-PGRP-LE expression plasmid5. Incubation for 1.5 h with wild-type L. monocytogenes, followed by additional incubation for 6 h in medium containing gentamicin and 100 µM CuSO4 to induce PGRP-LE reduced the number of bacteria within the cells to numbers similar to that observed following infection with the Δhly strain (Fig. 4a). Atg5 knockdown by RNAi in S2 cells expressing PGRP-LE (Supplementary Fig. 4b, online) restored the numbers of bacteria to those seen in S2 cells (Fig. 4a). Infection of S2 cells expressing PGRP-LE with wild-type L. monocytogenes induced AMP expression in a PGRP-LE dependent manner (Supplementary Fig. 5a, online). This induction was totally dependent on imd, a key factor in the Imd pathway (Supplementary Fig. 5b, online). In contrast to AMP induction, PGRP-LE mediated suppression of bacterial growth was not affected by the knockdown of Relish or imd (Fig. 4a and data not shown). PGRP-LE–mediated suppression of bacterial growth was also not affected by the knockdown of Dif and dl (Fig. 4a). On the other hand, AMP induction was not affected by Atg5 knockdown, suggesting that the autophagy pathway is not essential for AMP induction in L. monocytogenes infection (Supplementary Fig. 5b, online). Knockdown of Relish, Dif/dl, and Atg5 expression had no effect on the number of Δhly strain bacteria in either S2 cells or S2 cells expressing PGRP-LE (Fig. 4a), suggesting that these genes were not involved in phagocytic entry of the bacteria. These results suggest that the autophagy pathway but not NF-κB transcription factors (Relish, Dif, Dorsal) is required for PGRP-LE–mediated suppression of intracellular L. monocytogenes growth. Consistent with the results suggesting the importance of the autophagy pathway in the PGRP-LE–mediated suppression of intracellular Listeria growth, a time-course evaluation of the intracellular growth of the bacteria in S2 cells expressing PGRP-LE revealed a decrease in the number of intracellular wild-type, but not Δhly, L. monocytogenes, 2.5 hours after infection (0.5 h infection plus 2 h with gentamicin). At later time points, the bacteria remained at low numbers in the S2-PGRP-LE cell line, but multiplied robustly in S2 cells that did not express PGRP-LE (Supplementary Fig. 6, online).
Figure 4.
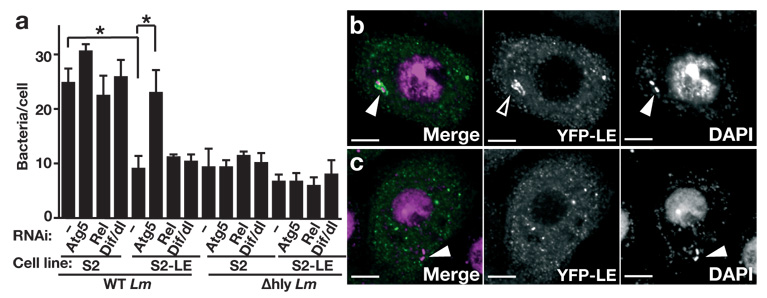
Autophagy, but not the IMD or Toll pathways, is crucial for PGRP-LE–mediated suppression of intracellular growth of L. monocytogenes in S2 cells. (a) S2 cells (S2) or S2 cells expressing PGRP-LE (S2-LE) were transfected with double-stranded RNA specific for Atg5, Rel or Dif/dl (RNAi, below graph) and infected with wild-type (WT Lm) or Δhly (Δhly Lm) L. monocytogenes for 1.5 h, followed by 6 h incubation in CuSO4- and gentamicin-containing medium, and L. monocytogenes growth was quantified by determining colony-forming units by plate assay. Bars indicate the variance of two independent experiments. *P < 0.001 (t-test) (b, c) Co-localization of PGRP-LE with wild-type L. monocytogenes in S2 cells. S2 cells engineered to express YFP-PGRP-LE were infected with (b) WT Lm or (c) Δhly Lm for 0.5 h, followed by 1 h incubation in gentamicin-containing medium, DAPI staining, and visualization by fluorescence confocal microscopy. YFP-PGRP-LE (green) and DAPI (magenta) are shown in the merged panel. Closed arrowheads show L. monocytogenes, and the open arrowhead indicates YFP-PGRP-LE accumulated around the bacteria. Scale bar, 5 µm.
PGRP-LE functions as an intracellular receptor by detecting peptidoglycan fragments5, and is essential for suppressing intracellular bacterial growth. To test the possibility that PGRP-LE detects bacteria that invade host cell cytoplasm, S2 cells expressing yellow fluorescent protein (YFP)-tagged PGRP-LE5 were infected with wild-type or Δhly strain L. monocytogenes, and assayed by confocal fluorescence microscopy (Fig. 4b,c). With no bacterial infection, YFP-tagged PGRP-LE was dispersed throughout the cytoplasm5. With wild-type bacterial infection, however, YFP-tagged PGRP-LE accumulated around the bacteria (Fig. 4b); in contrast, infection with the Δhly strain, which cannot enter the cytoplasm, did not result in the accumulation of YFP-tagged PGRP-LE around bacteria (Fig. 4c). These results suggest that PGRP-LE directly detects invading L. monocytogenes after it enters the cytoplasm.
Autophagy induction via the cytoplasmic sensor PGRP-LE
These data suggest that intracellular PGRP-LE is responsible for detecting invading L. monocytogenes and for inducing autophagy. To test this possibility, we examined whether L. monocytogenes invading the cell cytoplasm is surrounded by autophagosomes in a PGRP-LE–dependent manner. A fusion protein of LC3 with green fluorescent protein (GFP)33 was expressed under the control of an actin promoter in S2 cells or S2 cells expressing PGRP-LE, and the distribution of GFP-LC3 was examined using confocal fluorescence microscopy. GFP-LC3 is an autophagosome-specific membrane marker that is detected in ring- or dot- shaped structures when autophagosomes are formed in mammalian cell cytoplasm13, 14, 33 and in drosophila fat body cells27. In contrast to a previous report that TLR4-dependent induction of autophagy by lipopolysaccharide treatment in a murine macrophage cell line is maximal 12 to 16 h after LPS stimulation19, dot-shaped or ring-shaped GFP-LC3 signals were observed in S2 cells expressing PGRP-LE after only 1.5 h incubation (0.5 h of the initial incubation plus an additional 1 h in the presence of gentamicin) with wild-type L. monocytogenes (Fig. 5a,c). The number of GFP-LC3 dots observed in Δhly strain-infected cells was near the background quantity (Fig. 5a). In S2 cells, in which no detectable PGRP-LE is expressed, wild-type L. monocytogenes infection did not increase GFP-LC3 dot formation (Fig. 5a). These results suggest that PGRP-LE is essential for the GFP-LC3 dot formation caused by the invasion of wild-type L. monocytogenes into the cell cytoplasm. Treatment of Rapamycin, an inhibitor of autophagy inhibitory factor TOR29, of S2 cells induced a similar frequency of GFP-LC3 dots as in S2 cells expressing PGRP-LE (Fig. 5a), suggesting that dot or ring-shaped GFP-LC3 formation per se was not affected by the lack of PGRP-LE expression. In S2 cells expressing PGRP-LE and infected with wild-type L. monocytogenes, one or several bacteria were often surrounded by ring-shaped GFP-LC3 dots (Fig. 5b,c). Colocalization of the wild-type bacteria with GFP-LC3 dots in S2 cells expressing PGRP-LE was detected after 0.5 h incubation with bacteria, and the frequency of the colocalization after 1.5 h incubation (0.5 h incubation with bacteria plus 1 h in the presence of gentamicin) was 57.7±5.3%, which was significantly higher than that in wild-type bacteria-infected S2 cells (7.9± 6.9%) (Fig. 5b).
Figure 5.
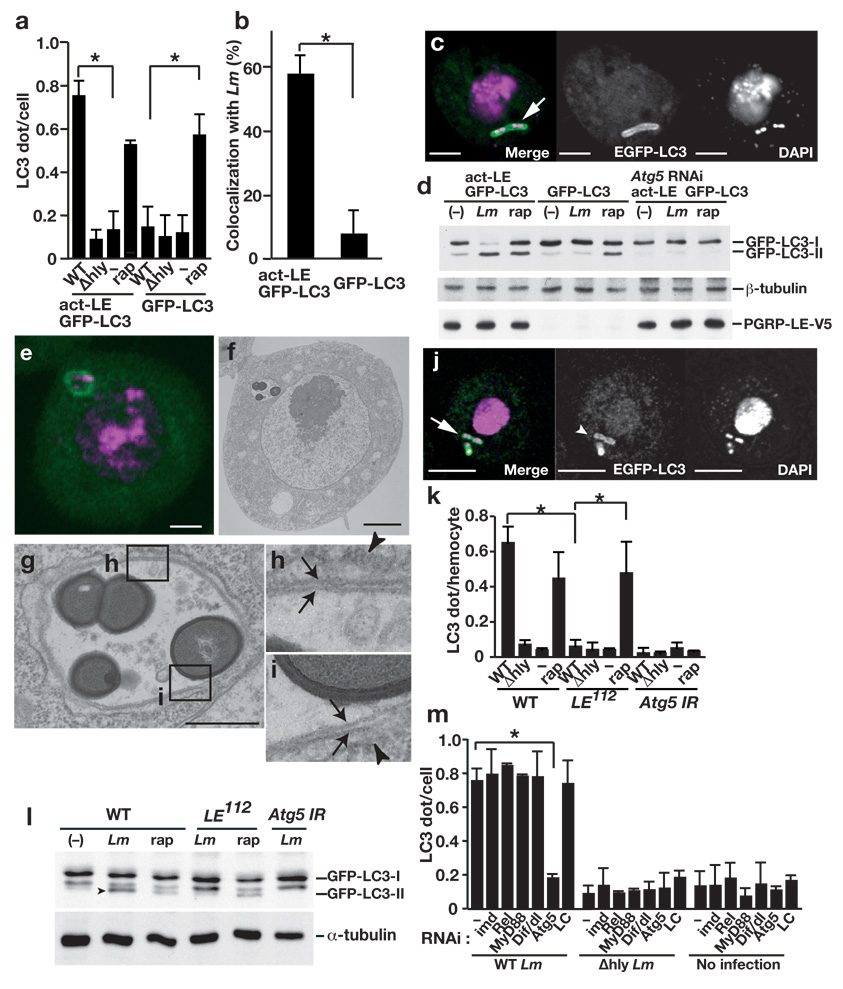
PGRP-LE mediates autophagosome formation in S2 cells and hemocytes. (a) The number of dot- or ring-shaped GFP-LC3 signals per cell was quantified after wild-type (WT) or Δhly L. monocytogenes infection or without infection (−) of S2 cells expressing both PGRP-LE and GFP-LC3 or only GFP-LC3, or after 1.5 h incubation with 5 µM rapamycin (rap). Bars indicate standard deviation of triplicate measurements. (b) The number of GFP-LC3 dots colocalized with wt L. monocytogenes in indicated S2 cells was quantified using confocal microscopy images. Bars indicate standard deviation of triplicate measurements. (c) Confocal microscopy images of WT L. monocytogenes infected S2 cells expressing PGRP-LE and GFP-LC3. GFP-LC3 (green), DAPI (magenta). Scale bar, 5 µM. Arrow indicates co-localization of GFP-LC3 and L. monocytogenes. (d) Indicated S2 cells were infected with WT L. monocytogenes (Lm), or treated with 5 µM rapamycin (rap) and indicated proteins in lysates were detected by immunoblotting. (e–i) Ultrastructural analysis of WT L. monocytogenes-infected S2 cells expressing PGRP-LE and GFP-LC3. (e,f) Fluorescence microscopy (e) and electon microscopy (f) images of cells expressing GFP-LC3 (green) stained with DAPI (magenta). Scale bars, 1 µm. (g) Magnified image of a bacteria-containing vacuole shown in (f). Scale bar, 500 nm. (h, i) Magnified images of the fields indicated in (g). Arrows indicate double-membrane structure that surrounds the bacteria. Arrowheads indicate endoplasmic reticulum–like membrane. (j) Confocal microscopy images of WT L. monocytogenes-infected hemocytes. GFP-LC3 (green), DAPI (magenta). (k) The number of dot- or ring-shaped GFP-LC3 signals per cell in ex vivo-cultured hemocytes expressing GFP-LC3 infected or treated as indicated. Bars indicate standard deviation of triplicate measurements. (l) Hemocytes from third instar larvae of the indicated genotype were cultured ex vivo and infected or treated as indicated and lysates were probed with antibodies specific for the indicated antibodies. Arrowhead indicates processed form of GFP-LC3. (m) S2 cells expressing PGRP-LE and GFP-LC3 under the control of an actin promoter were transfected with double-stranded RNA specific for indicated transcripts (RNAi, below graph) and infected with L. monocytogenes for 0.5 h. After 1 h incubation in gentamicin-containing medium, GFP-LC3 dot formation was quantified by confocal microscopy. Bars indicate standard deviation of at least triplicate measurements. * P < 0.001 (t-test).
During the autophagic processes, cytosolic LC3 (LC3-I) is conjugated on its C-terminus with phosphatidylethanolamine, and the lipidated LC3 (LC3-II) localizes to the autophagic membrane34. Therefore, the amount of LC3-II correlates with the number of autophagosomes. After infection, the amount of GFP-LC3-II increased markedly in S2 cells expressing PGRP-LE, whereas there was no change in the amount of GFPLC3-II in infected parental S2 cells that did not express PGRP-LE (Fig. 5d). Rapamycin treatment induced an increase in the amount of GFP-LC3-II in both cell types (Fig. 5d). These observations suggest that PGRP-LE is not essential for autophagosome formation per se, but that autophagosome formation is induced via PGRP-LE when L. monocytogenes invades the cytoplasm. As expected, Atg5 knockdown prevented the conversion of LC3-I to LC3-II upon bacterial infection or rapamycin treatment (Fig. 5d).
Next, we investigated whether the GFP-LC3-expressing structures that surround L. monocytogenes have the typical characteristics of autophagosomes. One hour after infection (0.5 h of the initial incubation plus an additional 0.5 h in the presence of gentamicin), we examined GFP-LC3 dots in S2 cells expressing PGRP-LE and GFP-LC3 by fluorescence microscopy (Fig. 5e); the same fields were also observed under electron microscopy (Fig. 5f–i). We noted double-membrane structures engulfing the bacteria (Fig. 5g–i), suggesting that PGRP-LE recognition of L. monocytogenes in the cytoplasm induces conventional autophagy around the bacteria.
We then determined whether endogenously expressed PGRP-LE is essential for inducing autophagy following L. monocytogenes infection of hemocytes. GFP-LC3 was expressed using a heat shock-GAL4 driver in the third instar larvae, and hemocytes from the larvae were cultured ex vivo with L. monocytogenes. After 1 h incubation with wild-type L. monocytogenes, followed by an additional 1 h incubation in gentamicin-containing medium, GFP-LC3 dots were observed in L. monocytogenes-infected wild-type hemocytes, and confocal microscopic analysis revealed that infected L. monocytogenes were surrounded by GFP-LC3-expressing structures (Fig. 5j, k). In contrast, GFP-LC3 dot formation frequency was similar in uninfected and infected PGRP-LE112 hemocytes (Fig. 5k). This result was not due to decreased phagocytic activity in PGRP-LE112 hemocytes, because similar numbers of Δhly strain L. monocytogenes infected PGRP-LE112 and wild-type hemocytes (Fig. 2b). Similarly, this result was not due to a generalized defect in autophagosome formation in PGRP-LE112 hemocytes as the frequency and morphology of the GFP-LC3 dots induced by rapamycin treatment was similar in PGRP-LE112 and wild-type hemocytes (Fig. 5k and Supplementary Fig. 7, online). GAL4-dependent RNAi transgene targeting of Atg5 greatly decreased the number of GFP-LC3 dots per infected cells to that in uninfected cells (Fig. 5k). GFP-LC3 dots were rarely detected in Δhly strain-infected hemocytes (Fig. 5k). Together, these data indicate that L. monocytogenes invasion of the cytoplasm induces autophagy in a manner dependent on endogenously expressed functional PGRP-LE.
PGRP-LE-dependent induction of autophagy in hemocytes was confirmed by immunoblotting studies that revealed the modification of GFP-LC3. Wild-type and PGRP-LE112 hemocytes expressing GFP-LC3, or hemocytes that express a RNAi transgene targeting Atg5 were incubated ex vivo with wild-type L. monocytogenes for 0.5 h, followed by an additional incubation for 2 h in gentamicin-containing medium; thereafter modification of GFP-LC3 was analyzed by immunoblotting using an anti-GFP antibody. L. monocytogenes infection or rapamycin treatment of wild-type hemocytes enhanced the intensity of the processed form of GFP-LC3, whereas infection had no effect on GFP-LC3 processing in the PGRP-LE112 mutant or Atg5 RNAi hemocytes (Fig. 5l). These results indicate that PGRP-LE is required for the induction of autophagy in response to L. monocytogenes infection.
PGRP-LE–mediated AMP induction in response to L. monocytogenes infection in S2 cells is dependent on the IMD pathway (Supplementary Fig. 5, online). PGRP-LE–mediated suppression of intracellular bacterial growth in S2 cells and hemocytes, however, was not dependent on the IMD and Toll pathways. We therefore examined whether PGRP-LE–mediated autophagosome formation was dependent on the IMD and/or Toll signaling pathways. The number of GFP-LC3 dots per cell was quantified in wild-type Listeria-infected S2 cells that expressed both PGRP-LE and GFP-LC3. Knockdown of imd, Relish, MyD88, or both Dif and dorsal by RNAi in wild-type Listeria-infected S2 cells expressing PGRP-LE and GFP-LC3 did not affect the formation of GFP-LC3 dots, but Atg5 knockdown decreased the number of LC3 dots per cell (Fig. 5m). Knockdown of PGRP-LC, the membrane-associated PGRP that senses bacteria on the surface of the cell membrane, also did not affect GFP-LC3 dot formation (Fig. 5m). These results suggest that the PGRP-LE–mediated autophagosome formation is independent of IMD and Toll pathways, consistent with other data indicating that PGRP-LE–mediated suppression of intracellular bacterial growth does not involve with these pathways (Fig. 4a).
Autophagy induced by DAP-type PGN
PGRP-LE recognizes TCT and DAP-containing PGN4, 5. We next examined whether TCT and PGN induce autophagy in the cell cytoplasm. Highly purified TCT35, highly purified DAP-type PGN, or highly purified lysine-type PGN was transfected into S2 cells expressing PGRP-LE and GFP-LC3 using a calcium phosphate transfection method, and GFP-LC3 dots were quantified after 2 h of incubation. TCT, DAP-, or lysine-type PGN increased the number of GFP-LC3 dots per cell to a frequency similar to that in L. monocytogenes-infected cells (Fig. 6a). In contrast, lysine-type PGN but not TCT or DAP-type PGN induced an increase in the number of GFP-LC3 dots in S2 cells lacking PGRP-LE (Fig. 6a). Similar results were observed using chemically synthesized TCT (data not shown). Transfection of synthetic desmuramylpeptide, a peptidoglycan fragment containing DAP, FK156 (d-lactyl-l-Ala-γ-d-Glu-meso-DAP-Gly)36, or its derivative, FK565 (haptanoyl-γ-d-Glu-meso-DAP-d-Ala), did not increase the number of GFP-LC3 dots in S2 cells expressing PGRP-LE (data not shown). Correlative FM-EM analysis revealed that the TCT-induced GFP-LC3–expressing structures have double-membrane structures typical of autophagosomes (Supplementary Fig. 8a,b, online). These results suggest that autophagy induction mediated by TCT and DAP-type PGN, but not lysine-type PGN, is dependent on PGRP-LE. We also examined GFP-LC3 dot formation in response to TCT, DAP-type, or lysine-type PGN in ex vivo-cultured hemocytes, although the precise mechanism of cytoplasmic delivery of PGN in hemocytes is not clear. The TCT- and DAP-type PGN-dependent formation of GFP-LC3 dots was suppressed in PGRP-LE112 mutant hemocytes compared to wild-type counterparts, but lysine-type PGN-dependent formation of GFP-LC3 dots was not affected in PGRP-LE112 mutant hemocytes (Fig. 6b, Supplementary Fig. 8c–h). These results suggest that PGRP-LE is crucial for the induction of autophagosome formation in response to TCT and DAP type-PGN, but not to lysine-type PGN. Together, these findings indicate that PGRP-LE likely recognizes DAP-type PGN of L. monocytogenes to induce autophagy.
Figure 6.
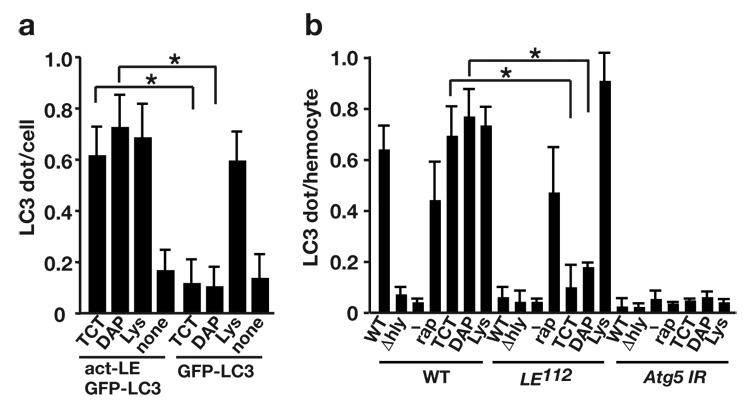
PGRP-LE is responsible for TCT and DAP-type PGN-induced autophagy. (a) S2 cells expressing PGRP-LE (act-LE) and GFP-LC3, or GFP-LC3 only, were treated with 100 nM TCT, 100 µg/ml highly purified DAP-type peptidoglycans from L. plantarum (DAP), or lysine-type peptidoglycans from S. epidermidis (Lys). After 2 h incubation, GFP-LC3 dot formation was quantified. Bars indicate the variance of two independent experiments. (b) The number of dot- or ring-shaped GFP-LC3 signals per hemocyte of indicated genotype was quantified after wild-type (WT) or Δhly L. monocytogenes infection, 5 µM rapamycin treatment (rap), or treatment with 100 nM TCT (TCT), 100 µg/ml highly purified DAP-type PGN from L. plantarum (DAP), or lysine-type PGN from S. epidermidis (Lys). Bars indicate variance of two independent experiments. Genotypes: w−; UAS-GFP-LC3/UAS-Gal4; heat-shock-GAL4/+ (WT), PGRP-LE112 ; UAS-GFP-LC3/UAS-Gal4; heat-shock-GAL4/+ (LE112), UAS-Atg5 IR/+, UAS-GFP-LC3/UAS-Gal4; heat-shock-GAL4/+ (Atg5 IR). * P < 0.001 (t-test).
DISCUSSION
Recent evidence indicates the importance of autophagy as a defense against intracellular pathogens, and the mechanism(s) underlying the induction of autophagy during pathogen infection is a central issue. The results of the present study demonstrate that PGRP-LE, through its ability to induce autophagy and prevent the intracellular bacterial growth, is essential in drosophila for resistance against L. monocytogenes. These findings indicate that the induction of autophagy, as an innate defense mechanism targeting intracellular pathogens, is activated by intracellular microbial sensors. In mammalian cells, the NOD-like receptors (or other intracellular microbial receptors) may sense bacterial invasion of the cytoplasm and induce autophagy in response to cytoplasm-invading bacterial infection. In fact, autophagy induction during L. monocytogenes infection of mouse embryonic fibroblast cells requires the expression of listeriolysin O28, suggesting that the sensing of L. monocytogenes in mammalian cells also occurs in the cytoplasm.
In primary hemocytes and S2 cells, both DAP-type and lysine-type PGN induced autophagy, although PGRP-LE was responsible only for the autophagy induction stimulated by the DAP-type PGN. These results strongly suggest that other cytoplasmic sensor(s) detect invading bacteria with cell walls containing lysine-type PGN. In mammals, autophagy has also been linked to the resistance against parasites, such as Toxoplasma gondii, and viruses12. Although the fundamental mechanisms linking these pathogens with the autophagy machinery might be similar to that used in bacterial infections, it is likely that the host cells utilize distinct sensors to induce autophagy in response to each type of pathogen.
Although the molecular mechanisms and signaling pathways leading to starvation-induced autophagy are well-studied, the signaling pathways that induce autophagy upon pathogen infection is just beginning to be clarified. In contrast to AMP induction, PGRP-LE-mediated induction of autophagy was independent of the Toll and IMD pathways. However, autophagy inducted by LPS through TRIF-dependent TLR4 signaling overcomes an M. tuberculosis-mediated phagosome block and promotes its colocalization with autophagosomes19. PGRP-LE has an RHIM-like motif, which is similar to the receptor-interacting protein homotypic interaction motif present in TRIF and receptor-interacting protein 1 (RIP1)5. This motif is required for the interaction between these two proteins, and for TRIF- and TLR3-induced NF-κB activation37, 38. It is possible that an unidentified factor with an RHIM-like motif interacts with PGRP-LE to activate the signaling pathway to induce autophagy.
Autophagy has been shown to be essential for the defense against several kinds of pathogens in cultured cells13–17. Thus, this report opens the door to investigating the connection between intracellular pattern recognition receptors and autophagy as an immune response, and the signaling pathways involved in both insects and mammals.
Methods
Fly strains
Stocks were raised on a standard cornmeal-yeast agar medium at 25°C. The following strains used in this study are described elsewhere: ywPGRP-LE112 (PGRP-LE112)3, w;;PGRP-LC7454 (PGRP-LC7454)39, RelishE20 (RelishE20)3, w;;Atg1Δ3D (Atg1Δ3D)26, w;;hml-GAL4 (hml-GAL4)25, w;cg-GAL4 (cg-GAL4)30, w;UAS-PGRP-LE (UAS-PGRP-LE) 4, ywUAS-Atg5 IR (UAS-Atg5 IR)26, yw;;UAS-Atg16B (UAS-Atg1)30, and w;UAS-GFP-LC3 (UAS-GFP-LC3)27.
Survival experiments
Bacterial infections were performed by injecting approximately 70 nl per fly of a dilution of cultured bacterial strains (1/100,000 dilution of an L. monocytogenes suspension or E. carotovora suspension; A600 = 1.0), which resulted in the injection of approximately 10 bacteria per fly. All survival experiments were performed with 30 flies for each genotype tested at 28°C for the L. monocytogenes injections, or 25°C for the E. carotovora injections. Surviving flies were transferred daily into fresh vials.
L. monocytogenes infection of ex vivo-cultured hemocytes
Third instar larvae were dissected in 10% fetal bovine serum-containing Schneider’s Drosophila medium mounted on concanavalin A-treated glass slides. After a few minutes incubation to allow the hemocytes to attach to the glass slide, the medium was exchanged with new media, and a suspension of L. monocytogenes grown to mid-log phase was added to the culture (approximately 20 bacteria per cell), followed by 1 h incubation at 28°C. Cells were washed and further incubated at 28°C for the indicated time in Schneider’s Drosophila medium containing 10% fetal bovine serum and 10 µg/ml gentamicin. Cells were then fixed with 2% paraformaldehyde for immunohistochemical analyses. For the induction of GFP-LC3, third instar larvae were heat-shocked (at 37°C for 20 min) 12 h before hemocyte isolation.
Bacterial strains
The following L. monocytogenes strains were used in this study: wild-type strain, 10403S; Δhly strain, DP-L216131; GFP-expressing wild-type strain, DH-L1039; and GFP-expressing Δhly strain, DH-L113732. E. carotovora carotovora 15 was used in the survival experiments.
Peptidoglycans
The DAP-type and the Lys-type peptidoglycans were highly purified from Lactobacillus plantarum ATCC 8014 and Staphylococcus epidermidis ATCC 155, respectively, according to the previously reported method40. Purity was confirmed by amino acid analysis. Highly purified TCT was described previously35. Chemically synthesized TCT was prepared using an orthogonally protected meso-diaminopimelic acid (meso-DAP) containing tetrapeptide (l-Ala-γ-d-Glu(OBn)-meso-DAP(N-Z, OBn)-d-Ala(OBn)) and a disaccharide (4,6-O-benzylidene-3-O-benzyl-GlcNAc-(β1-4)-MurNAc(anh)). After condensation of the disaccharide and the tetrapeptide with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (water-soluble carbodiimide hydrochloride; WSCD•HCl), 1-hydroxybenzotriazole (HOBt), triethylamine, in N,N-dimethyl formamide (DMF), deprotection of all the protecting groups by hydrogenation (H2 (20 kg/cm2), Pd(OH)2, in tetrahydrofuran) gave TCT. The molecular weight of TCT was confirmed with ESI-TOF-MS (negative) m/z 920.4 [M−H]−; HRMS-ESI TOF-MS (positive) m/z calcd. for C29H48N6O16, [M+H]+ 922.3893, found 922.3898. The synthetic desmuramylpeptide, a peptidoglycan fragment containing DAP, FK156 36 (d-lactyl-l-Ala-γ-d-Glu-meso-DAP-Gly) and its derivative, FK565 (haptanoyl-γ-d-Glu-meso-DAP-d-Ala) was supplied by Astellas Pharmaceutical (formerly Fujisawa and Yamanouchi).
RNA interference in S2 cells
The double-stranded RNA used in the RNAi experiments was synthesized as previously described5. Templates for double-stranded Atg5 RNA were amplified with the primers 5’-TAATACGACTCACTATAGGGCAAATAAGGAACATGGCC-3’ and 5’-TAATACGACTCACTATAGGGTCAGGCAGGACATGTAG-3’. Double-stranded RNA was transfected into S2 cells as previously described5.
L. monocytogenes infection of S2 cells and measurement of the number of intracellular bacteria
Cultures (1 × 106 cells/ml) were incubated for 18 h, and 50 nM water-soluble cholesterol was added to the culture as previously described30. After 30 min incubation, a suspension of L. monocytogenes (approximately 20 bacteria/cell unless otherwise described) was added and the cells were incubated at 28°C for 1.5 h in the case of infection with 20 bacteria/cell. Cells were then washed with phosphate buffered saline (PBS), and incubated in Schneider’s drosophila medium containing 100 µM CuSO4 and 10 µg/ml gentamicin for 6 h. After washing with PBS, cells were dispersed in water to release bacteria from the cytoplasm of the S2 cells. Samples were then plated on Brain-Heart-Infusion plates to measure colony-forming units.
Immunohistochemistry
Fixed hemocytes or S2 cells were washed with PBS and stained with anti-GFP (Medical & Biological Laboratories) in PBS containing 1% bovine serum albumin and 0.1% Triton X-100 for 1 h, and then with fluorescein isothiocyanate-labeled anti-rabbit IgG (Molecular Probes), rhodamine-labeled phalloidin (Molecular Probes) for 1 h when required, followed by 10 min DAPI staining (Sigma).
Immunoblotting
S2 cells were infected with wild-type L. monocytogenes (approximately 500 bacteria/cell) for 0.5 h at 28°C, and further incubated in gentamicin-containing medium for 1 h at 28°C. Ex vivo-cultured hemocytes were infected with wild-type L. monocytogenes (approximately 200 bacteria/cell) for 1 h at 28°C, washed with PBS, and further incubated in gentamicin-containing medium for 2 h at 28°C. For immunoblotting, cells were washed with PBS, and lysed with buffer containing 50 mM Tris-HCl, 2% sodium dodecyl sulfate, 10% glycerol, and 100 mM β-mercaptoethanol. Lysate from the same number of cells was loaded on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene membranes, and subjected to immunoblot analysis using antibodies against either GFP (Medical & Biological Laboratories), α-tubulin (Santa Cruz Biotechnology), β-tubulin (Hybridoma Bank), or V5-tag (Invitrogen). Blots were visualized using ECL-Western Blotting Analysis System (GE Healthcare).
Correlative FM-EM
Correlative fluorescence microscopy-electron microscopy (FM-EM) allows individual cells to be examined both in an overview with FM and in a detailed subcellular structure view with EM. For observation by FM-EM, S2 cells cultured on glass-bottom dishes (MatTek) were fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4) for 2 h, and then examined using a confocal laser scanning microscope. The same specimens were further incubated with 2.5% glutaraldehyde and 2% formaldehyde in 0.1 M sodium cacodylate (pH 7.4) at 4°C overnight. After three washings with 0.1 M sodium cacodylate (pH 7.4) containing 7% sucrose, the samples were postfixed with 1% osmium tetroxide and 0.5% potassium ferrocyanide in the same buffer for 1 h, washed with distilled water three times, dehydrated in ethanol, and embedded in Epon812 (TAAB Laboratories Equipment, Ltd.). Ultrathin sections of the cell (70 nm thick) were stained with saturated uranyl acetate and Reynolds lead citrate solution. Electron micrographs were taken with a JEOL JEM-1011 transmission electron microscope (JEOL, Ltd.).
Supplementary Material
Acknowledgements
We thank D. A. Portnoy (University of California, Berkeley) and D. E. Higgins (Harvard Medical School) for L. monocytogenes strains; L. W. Cheng (University of California, Berkeley) for the S2 cell L. monocytogenes infection protocol; T. P. Neufeld (University of Minnesota) for Atg1Δ3D, UAS-Atg5 IR, UAS-Atg1, and UAS-GFP-LC3; D. Hultmark (Umeå University) for RelishE20; K. Anderson (Cornell University) for PGRP-LC7454; A. Goto for hml-GAL4; the Bloomington Stock Center, Drosophila Genetic Resource Center at the Kyoto Institute of Technology and the Genetic Strain Research Center of National Institute of Genetics for fly stocks; and S. Iwanaga (a professor emeritus, Kyushu University), M. Mitsuyama (Kyoto University), A. Yamamoto (Nagahama Institute of Bio-Science and Technology), and S. Natori (a professor emeritus, The University of Tokyo) for helpful discussions. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Japan Society for the Promotion of Science; the Program for the Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN); the NIH grant (AI074958); and the Naito Foundation and from the NIH (AI60025 and AI074958) to N.S.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- 2.Ferrandon D, Imler J-L, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signaling during bacterial and fungal infections. Nat. Rev. Immunol. 2007;7:862–874. doi: 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- 3.Takehana A, et al. Peptidoglycan recognition protein (PGRP)-LE and PGRP-LC act synergistically in Drosophila immunity. EMBO J. 2004;23:4690–4700. doi: 10.1038/sj.emboj.7600466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takehana A, et al. Overexpression of a pattern-recognition receptor, peptidoglycan-recognition protein-LE, activates imd/relish-mediated antibacterial defense and the prophenoloxidase cascade in Drosophila larvae. Proc. Natl. Acad. Sci. USA. 2002;99:13705–13710. doi: 10.1073/pnas.212301199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaneko T, et al. PGRP-LC and PGRP-LE have essential yet distinct functions in the drosophila immune response to monomeric DAP-type peptidoglycan. Nat. Immunol. 2006;7:715–723. doi: 10.1038/ni1356. [DOI] [PubMed] [Google Scholar]
- 6.Lemaitre B, Reichhart J-M, Hoffmann JA. Drosophila host defense: Differential induction of antimicrobial peptide genes after infection by various classes of microorganisms. Proc. Natl. Acad. Sci. USA. 1997;94:14614–14619. doi: 10.1073/pnas.94.26.14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanneganti T-D, Lamkanfi M, Núñez G. Intracellular NOD-like receptor in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Chaput C, Boneca IG. Peptidoglycan detection by mammals and flies. Microbes Infect. 2007;9:637–647. doi: 10.1016/j.micinf.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Delbridge LM, O’Riordan MXD. Innate recognition of intracellular bacteria. Curr. Opin. Immunol. 2007;19:10–16. doi: 10.1016/j.coi.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Hsu YM, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- 11.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 12.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakagawa I, et al. Autophagy defends cells against invading Group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 14.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 15.Ogawa M, et al. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 16.Ling YM, et al. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrade RM, Weaaendarp M, Gubbels M-J, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Invest. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008 March 13; doi: 10.1038/emboj.2008.31. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 22.Mansfield BE, Dionne MS, Schneider DS, Freitag NE. Exploration of host-pathogen interactions using Listeria monocytogenes and Drosophila melanogaster. Cell Microbiol. 2003;5:901–911. doi: 10.1046/j.1462-5822.2003.00329.x. [DOI] [PubMed] [Google Scholar]
- 23.Hamon M, Bierne H, Cossart P. Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 2006;4:423–434. doi: 10.1038/nrmicro1413. [DOI] [PubMed] [Google Scholar]
- 24.Jones S, Portnoy DA. Characterization of Listeria monocytogenes pathogenesis in a strain expressing Perfringolysin O in place of Listeriolysin O. Infect. Immun. 1994;62:5608–5613. doi: 10.1128/iai.62.12.5608-5613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goto A, Kadowaki T, Kitagawa Y. Drosophila hemolectin gene is expressed in embryonic and larval hemocytes and its knock down causes bleeding defects. Dev. Biol. 2003;264:582–591. doi: 10.1016/j.ydbio.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Rusten TE, et al. Programmed autophagy in the Drosophila fat body is induced by ecdysome through regulation of the PI3K pathway. Dev. Cell. 2004;7:179–192. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3:117–125. doi: 10.4161/auto.3618. [DOI] [PubMed] [Google Scholar]
- 29.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998;273:3963–396. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 30.Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng LW, Portnoy DA. Drosophila S2 cells: an alternative infection model for Listeria monocytogenes. Cell. Microbiol. 2003;5:875–885. doi: 10.1046/j.1462-5822.2003.00327.x. [DOI] [PubMed] [Google Scholar]
- 32.Agaisse H, et al. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science. 2005;309:1248–1251. doi: 10.1126/science.1116008. [DOI] [PubMed] [Google Scholar]
- 33.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaneko T, et al. Monomeric and polymeric gram-negative peptidoglycan but not purified LPS stimulate the Drosophila IMD pathway. Immunity. 2004;20:637–649. doi: 10.1016/s1074-7613(04)00104-9. [DOI] [PubMed] [Google Scholar]
- 36.Kitaura Y, et al. N2-(-D-glutamyl)-meso-2L,2'D-diaminopimelic acid as the minimal prerequisite structure of FK-156: its acyl derivatives with potent immunostimulating activity. J. Med. Chem. 1982;25:335–337. doi: 10.1021/jm00346a001. [DOI] [PubMed] [Google Scholar]
- 37.Sun X, Yin J, Starovansnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 2002;277:9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 38.Meylan E, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κB activation. Nat. Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 39.Gottar M, et al. The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature. 2002;416:640–644. doi: 10.1038/nature734. [DOI] [PubMed] [Google Scholar]
- 40.Kotani S, Watanabe Y, Shimono T, Kinoshita F, Narita T. Immunoadjuvant activities of peptidoglycan subunits from the cell walls of Staphylococcus aureus and Lactobacillus plantarum. Biken J. 1975;18:93–103. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.