

Abstract
RAX was originally discovered as the unique cellular activator for the dsRNA-dependent, interferon-inducible protein kinase PKR. Recent findings indicate that RAX is also a critical component of the RNA-induced silencing complex and a regulator of transcription. Here we report novel phenotypes for both fruit flies carrying a transposon insertion in the 5’ UTR of dRax (independently identified as loqs/R3D1) and mice with a deletion of the entire Rax gene. In Drosophila we observe a high level of dRax expression in the developing nerve cord. Mutant fly embryos homozygous for the insertion dRax[f00791] display highly abnormal commissural axon structure of the CNS and 70% of the flies homozygous for the mutant allele die prior to adulthood. Surviving male flies have reduced fertility and female flies are sterile. Furthermore, these flies appear to have a severe defect in nervous system coordination or neuromuscular function resulting in significantly reduced locomotion. Mice were also generated that are heterozygous for a deletion of the entire Rax gene (exons 1–8). While mice that are heterozygous for the mutant allele are viable and appear normal, we are unable to obtain mice homozygous for this mutant allele. Furthermore, we have not observed any embryo obtained by mating heterozygous mice at either E3.5, 7, or 14 that is nullizygous for the Rax gene. Since Rax is expressed in preimplantation blastocysts, these data indicate that deletion of the entire Rax gene is embryonic lethal in mice at a preimplantation stage of development. Collectively, these findings in two different species illustrate the importance of RAX for embryonic development.
Keywords: RAX, PKR, dsRNA, fly, mouse, embryogenesis, knockout
1. Introduction
RAX and its human ortholog PACT were independently discovered as the cellular activator for the interferon-inducible, dsRNA-dependent protein kinase PKR (Ito et al., 1999a; Ito et al., 1999b; Patel et al., 2000; Patel and Sen, 1998). In this role, cellular stresses such as IL-3 withdrawal from factor-dependent hematopoietic cells, treatment with inflammatory cytokines or viral infection induce RAX phosphorylation to promote activation of PKR and inhibition of eIF2α-dependent protein synthesis (Bennett et al., 2006; Bennett et al., 2004; Ito et al., 1994; Ito et al., 1999b). While PKR has been primarily studied for its anti-viral and anti-proliferation effects via its capacity to inhibit protein synthesis, it has also been reported to regulate several transcription factors, including p53, NF-κB and STAT1 (Bonnet et al., 2000; Cuddihy et al., 1999a; Cuddihy et al., 1999b; D'Acquisto and Ghosh, 2001; Fremont et al., 2006; Gil et al., 2000; Wong et al., 1997; Yeung and Lau, 1998; Zamanian-Daryoush et al., 2000). Thus, the RAX-PKR stress-signaling pathway is necessary for the cellular response to a broad range of apoptosis-inducing stress by functioning as a regulator of both transcription and translation.
RAX and PACT are 98% identical in amino acid structure and consist of three conserved dsRNA binding domains (Figure 1A). The N-terminal first and second domains bind dsRNA and are necessary for RAX/PACT association with PKR, whereas the third C-terminal domain is apparently not necessary for dsRNA binding or interaction with PKR but is required for activating PKR’s kinase activity (Huang et al., 2002; Peters et al., 2001). It has been demonstrated that RAX/PACT can efficiently bind to and activate PKR in vitro (Ito et al., 1999b; Patel and Sen, 1998). However, in vivo RAX/PACT mediated PKR activation is dependent on a stress application to cells (Ito et al., 1999b; Patel et al., 2000). Although both RAX/PACT and PKR are dsRNA-binding proteins, PKR activation may not require dsRNA binding since in vitro activation by RAX/PACT does not require dsRNA.
Figure 1. CG6866 is a Drosophila ortholog to either mammalian RAX/PACT or TRBP.
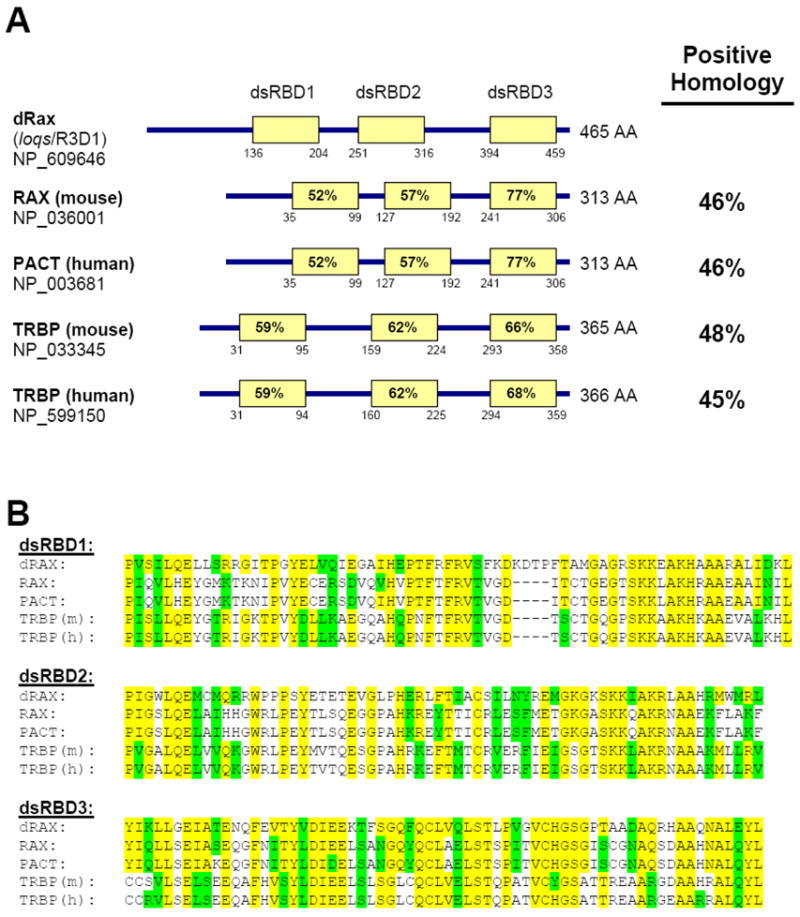
A. Comparison of the domain structures of dRax protein to RAX, PACT and TRBP proteins. The length of each protein is given in amino acids (AA) and the position of each dsRNA-binding domain (dsRBD) is indicated below the corresponding box. The dsRBDs were predicted using ScanProsite. Positive homology (identical and conserved residues) to dRax was determined using BLASTp. Homology between each mammalian dsRBD and its fly counterpart is indicated within each dsRBD box. B. Amino acid sequence alignment between dsRNA binding domains of the fly and mammalian orthologs. Residues identical to dRax are highlighted in yellow while conserved homology is highlighted in green.
In addition to its role as an activator of PKR, RAX has recently been reported to be an integral component of the RNA-induced silencing complex (RISC) (Kok et al., 2007; Lee et al., 2006). RAX associates with a ~500 kDa complex containing Tar binding protein (TRBP), Dicer, and Argonuate-2 that is capable of pre-miRNA processing and target cleavage (Lee et al., 2006). Furthermore, depletion of RAX from HeLa cells inhibits the siRNA-mediated silencing of a reporter gene and prevents the accumulation of mature miRNA (Lee et al., 2006). Interestingly, the third C-terminal dsRNA-binding domain of RAX interacts with the N-terminal helicase motif of Dicer and it has been proposed that RAX has a function in RISC assembly (Lee et al., 2006). The D. melanogaster ortholog of RAX (also named loqs or R3D1) is required for normal pre-miRNA processing and interacts with Dicer-1 (Forstemann et al., 2005; Jiang et al., 2005; Saito et al., 2005). It has been reported that female flies homozygous for the insertion of a transposon in the first exon of dRax are sterile due to a defect in ovary germline stem cell maintenance (Forstemann et al., 2005; Jiang et al., 2005). Furthermore, dRax is required for silencing of the endogenous Stellate locus by Suppressor of Stellate in the testes of male flies (Forstemann et al., 2005).
Recently, it has been reported that disruption of the mouse Rax gene causes developmental defects in the otic system and mice are smaller in size (Rowe et al., 2006). This Rax targeted mouse was generated by replacing the last exon of the Rax gene, exon 8, with a neomycin resistance cassette in a strategy designed to delete all of the coding sequence for the C-terminal, third dsRNA binding domain of RAX (Rowe et al., 2006). Thus, mice homozygous for this mutant allele produce an aberrant transcript containing the first seven exons of the Rax ORF but apparently no protein product is observed that corresponds to the C-terminal truncation mutant (Rowe et al., 2006).
Concurrent with the aforementioned studies using the Drosophila Rax mutant strain and a mouse with a deletion of Rax exon 8, our laboratory initiated studies of the dRax fly mutant and was in the process of generating a Rax knockout mouse to ascertain the physiologic function of RAX. Here we report that flies homozygous for dRax[f00791] not only have reduced fertility as observed by others but also display defects in central nervous system development. Furthermore, we find that deletion of exons 1 – 8, comprising the entire ORF of the murine Rax allele, is embryonic lethal at the preimplantation stage. These findings demonstrate the importance of RAX for embryogenesis.
2. Results
2.1. Drosophila ortholog of Rax (dRax) is required for central nervous system development
A Drosophila gene, CG6866, was identified as the ortholog of Rax/Pact based on sequence comparison. Like RAX, the dRax protein has three predicted double strand RNA binding domains (dsRBDs, Figure 1A). The same gene was identified independently by Forstemann et al (Forstemann et al., 2005) and Jiang et al (Jiang et al., 2005), whose work reveals that dRax (loqs) is indeed a dsRNA binding protein required for miRNA maturation and germ cell development. The Drosophila RAX ortholog has similar amino acid homology to either the mammalian proteins RAX/PACT or Tar binding protein, TRBP (Figure 1A). Significantly, the dsRNA binding domains of these proteins share considerable (52 – 77%) homology (Figure 1B).
Using a cRNA probe, we performed in situ hybridization to monitor the expression of dRax during embryogenesis. dRax mRNA is deposited in the eggs prior to the onset of zygotic expression. At stage 9–10, high level of dRax expression is observed in the developing nerve cord (Figure 2A). This stage corresponds to formation of the early nervous system, when the neuroblasts delaminate from the ectoderm to form the nerve cord. Thus, nerve cord specific expression of dRax suggests that dRax function may play a role in nervous system differentiation. The strong dRax mRNA signal in the central nervous system (CNS) persists through embryogenesis even after the early differentiation stage. The CNS and some neuroganglia are the only tissues with a strong dRax signal at the end of embryogenesis (Figure 2B).
Figure 2. Drosophila ortholog of Rax (dRax) is required for nervous system development.
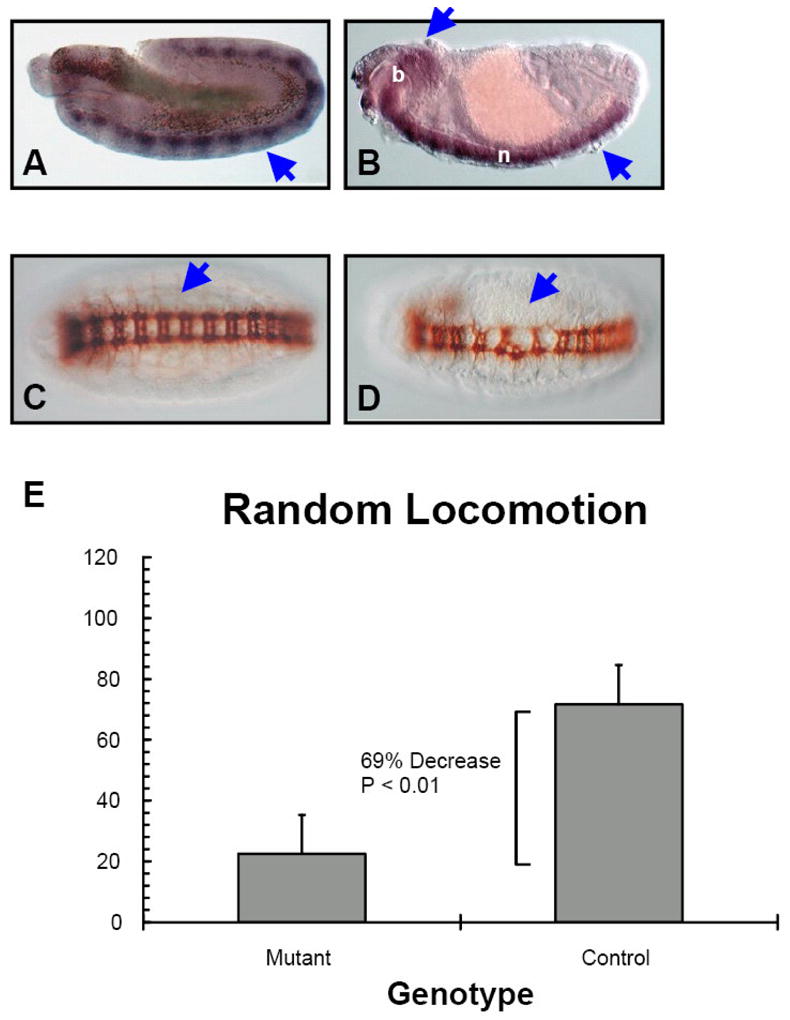
A. Stage 9–10 wild type embryo, sagittal view. dRax is specifically expressed in the early differentiating nerve cord (indicated by arrowhead). B. Stage 15 wild type embryo, sagittal view. In later stage embryos, dRax expression persists in the differentiating/differentiated central nervous system (arrowheads indicate elevated dRAX expression: b, brain; n, nerve cord). C. Wild type embryo stained with anti-102 to visualize the commissural axon structure (arrowhead). D. The commissural axon structure is disrupted in homozygous dRax mutant embryos (arrowhead). E. Compared to heterozygous (dRax[f00791]/cyo) control flies, homozygous dRax mutant flies have significantly reduced locomotor activity (data represented as Avg.+Stdev.; n=12).
Correspondingly, we found that dRax is required for proper nervous system development during embryogenesis. In mutant embryos homozygous for the insertion dRax[f00791], both the longitudinal and commissural axon structures of the CNS are highly abnormal (Figure 2C vs. D), reflecting the disorganization of the nervous system. The mutant is a PBac insertion in the first exon of dRax directly upstream of the translation start site that creates a hypomorphic mutant allele by attenuating its transcription (Forstemann et al., 2005). In situ hybridization analysis of the dRax[f00791] mutant embryos demonstrates a significant decrease of dRax expression in the developing nerve cord (data not shown). However, this is not a null mutant since there are detectable levels of dRax expression in the nerve cord of the mutant embryos (data not shown). Nonetheless, about 70% of homozygous animals die before adulthood while surviving males have reduced fertility and females are sterile. The CNS abnormality observed for mutant embryos manifested at the end of embryogenesis. A general, albeit subtler, abnormality in the CNS could also be detected using markers of specific neurons, such as Futsch (22C10) and Elav (data not shown). However, of the markers tested, BP102 staining that labels all axons in the CNS revealed the most striking phenotype (Figure 2C and D). Interestingly, we observe that the surviving homozygous flies are often stuck and unable to escape from food, which indicates a severe defect in nervous system coordination or neuromuscular function. Furthermore, using a Random Locomotion Test we observed that male homozygous dRax mutants collected 3 days after eclosion have a 69% decrease in locomotor activity compared to heterozygous control flies (P < 0.01, Figure 2E). Taken together these results point to the requirement of dRax for nervous system development.
2.2. Deletion of the Rax gene in mice results in preimplantational embryonic lethality
In order to investigate the function of RAX in mice, we generated a novel Rax null allele in which the entire RAX coding sequence is deleted. A 2.2 Kb genomic fragment corresponding to the Rax promoter and the first 130 bps of Rax exon 1 (just prior to the Rax AUG start codon), and a 7 Kb genomic region corresponding to the last 300 bps of Rax exon 8 (non-coding) and 6.7 Kb of intergenic sequence were used as 5′ and 3′ homology arms of the knockout vector, respectively (Figure 3A). This strategy avoids a “partial” or incomplete knockout of Rax that might produce a hypomorph and complicate data interpretation. In addition, there are no known/predicted genes overlapping the Rax locus that might be affected by removal of the targeted sequence.
Figure 3. Targeted disruption of the mouse Rax gene by homologous recombination.
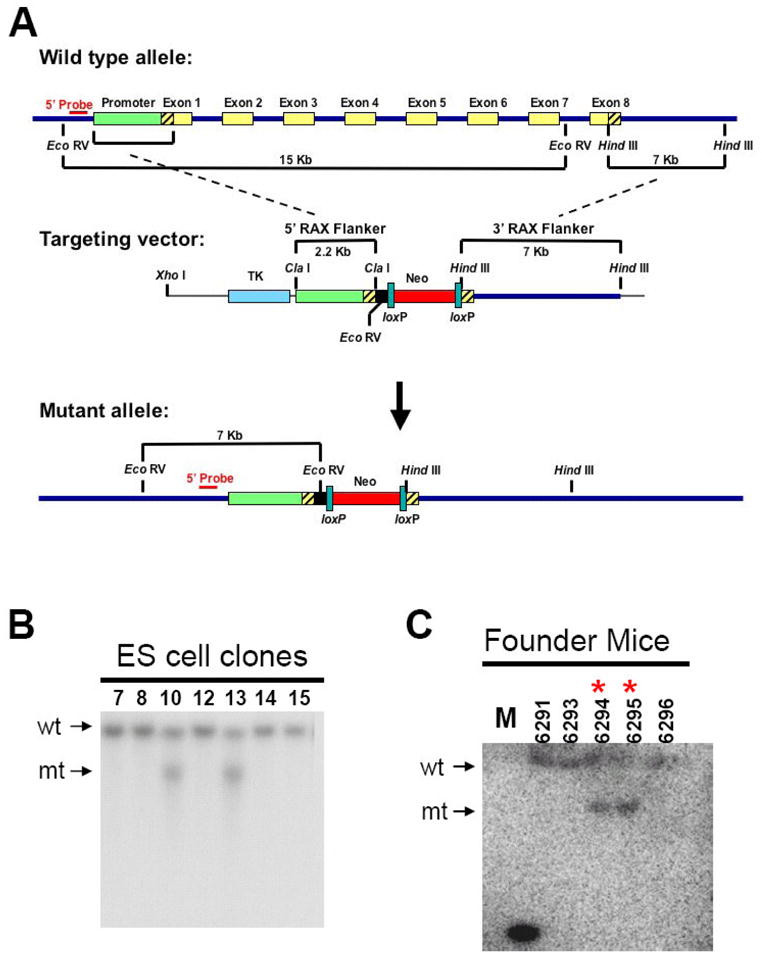
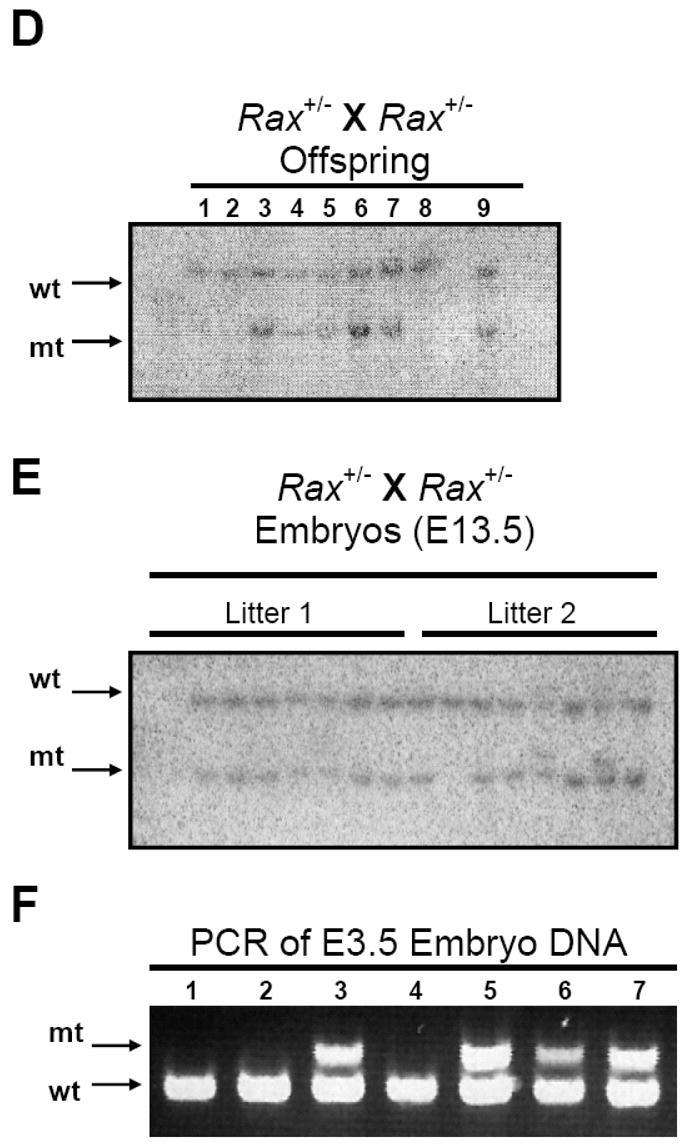
A. (top) The wild type Rax mouse allele contains a 15 Kb EcoR V fragment and a 7 Kb Hind III fragment. Exons are indicated by yellow boxes while noncoding regions of exon 1 and 8 are indicated by the diagonal lines. (middle) The Rax targeting vector was constructed using the 2.2 Kb Rax promoter region and the 7 Kb Hind III Rax fragment. (bottom) The mutant allele generated after homologous recombination and the diagnostic EcoR V restriction fragments are indicated. B. Southern blot analysis of EcoR V-cleaved genomic DNA isolated from targeted and selected ES cell clones using a 32P-labeled probe 5′ to the region of homologous insertion. wt, 15 Kb EcoR V fragment from wild type allele. mt, 7 Kb restriction fragment from mutant allele. C. Analysis of tail DNA from progeny of chimeric mice crossed with wild type mice demonstrates germline transmission of the disrupted Rax allele. M, marker. * indicates Rax+/− mice. D. Genotypes of offspring generated from mating of Rax+/− mice were determined by southern blotting. E. Southern blot using DNA isolated from E13.5 embryos generated from mating Rax+/- mice. F. Genotypes of E3.5 embryos generated from mating Rax+/− mice as determined by PCR. mt: mutant allele, wt: wild type allele
The Rax targeting vector was linearized and electroporated into mouse embryonic stem (ES) cells. DNA was isolated from ES cell clones and analyzed by Southern blotting using a 5′ probe to detect proper insertion by a shift in the size of the genomic DNA EcoR V fragment from 15 Kb to 7 Kb. Two ES cell clones positive for correct targeting into the Rax locus (Figure 3B) were injected into C57BL/6J blastocysts, and implanted into pseudo-pregnant females. Offspring coat color was examined to determine chimerism. Five chimeric mice with significant brown agouti coat color were generated (3 male, 2 female). These chimeric offspring were further mated with C57BL/6J wild type mice and maintained in a 129/B6 hybrid background. Germline transmission of the Rax knockout allele was confirmed by Southern blot analysis (Figure 3C). Mice heterozygous for the Rax mutant allele (Rax+/−) were intercrossed to produce Rax-null (Rax−/−) mice. Significantly, among a total of 76 mice from Rax+/− crosses (12 litters; average litter size of 6 – 7 mice) at weaning age, 17 and 59 mice were identified as Rax+/+ and Rax+/− mice, respectively, but no Rax−/− mice were recovered (P < 0.05, Figure 3D and Table I). We have not observed any postnatal lethality of the Rax+/− mice. Furthermore, mating heterozygous Rax+/− mice of either sex with wild type mice results in an approximately 1:1 ratio of wild type to heterozygous offspring, demonstrating that there is no defect in production and/or transmission of the zygote carrying the mutant allele (data not shown). Taken together, these results indicate that deletion of the entire Rax gene is embryonic lethal.
Table I.
Genotypes of progeny from Rax+/− X Rax+/− matings
| Total mice | No. of Litters | Avg. Litter size | Rax+/+ | Rax+/− | Rax−/− |
|---|---|---|---|---|---|
| 76 viable adultsa | 12 | 6.3 | 17 | 59 | 0 |
|
| |||||
| 23E13.5a | 3 | 7.6 | 2 | 21 | 0 |
| 36 E7.5a | 5 | 7.2 | 13 | 23 | 0 |
| 33 E3.5b | 6 | 5.5 | 9 | 24 | 0 |
Genotype determined by both PCR and southern blotting of genomic DNA
Genotype determined by PCR of genonnic DMA
In order to determine the embryonic stage at which lethality occurred for Rax−/− mice, embryos derived by Rax+/− crosses were isolated at various gestational stages, and their genotypes were determined by both Southern blotting and PCR analyses (Figure 3E and F). Both methods of genotyping yielded the same result. No embryos were identified as Rax−/− at either embryonic day (E)13.5 or E7.5 (Table I). Significantly, upon examination of 33 blastocysts (E3.5 embryos) isolated from 6 litters of Rax+/− crosses by PCR, we detected 9 Rax+/+, 24 Rax+/− and 0 Rax−/− embryos (P < 0.05, Table I and Figure 3F). Taken together, these results suggest that deletion of the Rax gene is embryonic lethal at a preimplantation stage of development.
In an effort to generate a Rax null embryonic stem (ES) cell line, we used the targeted ES cells heterozygous for the Rax mutant allele and high G418 concentration (2mg/ml) to select, by gene conversion, ES cells devoid of both Rax alleles. However, after three separate attempts, we were unable to select any ES cell clone with both alleles carrying the Rax deletion mutant (data not shown). Since Rax expression can be detected in wild type ES cells by RT-PCR (data not shown), these data suggest that RAX may be required for ES cell growth.
Rax is ubiquitously expressed throughout the mouse (UniProtKB/SwissProt entry Q9WTX2). To demonstrate that mice with the Rax-knockout allele produce less Rax mRNA and protein than Rax+/+ littermates, we compared lung and brain tissues, two tissues that express high levels of RAX, from Rax+/+ or Rax+/− mice by quantitative real-time PCR and by Western blotting. Significantly, an average of 15% fewer Rax transcripts are detected in the brains and lungs from Rax+/− mice than those from Rax+/+ mice (Figure 4A, P = 0.05). In addition, lysates generated from the brains of Rax+/− mice have an average of 33% less RAX protein compared to lysates from Rax+/+ brains as determined by densitometry of western blots (Figure 4B and C, P = 0.04). These data indicate that the Rax knockout allele does not produce RAX-coding transcripts.
Figure 4. Rax+/− mice have reduced Rax gene expression and protein level.
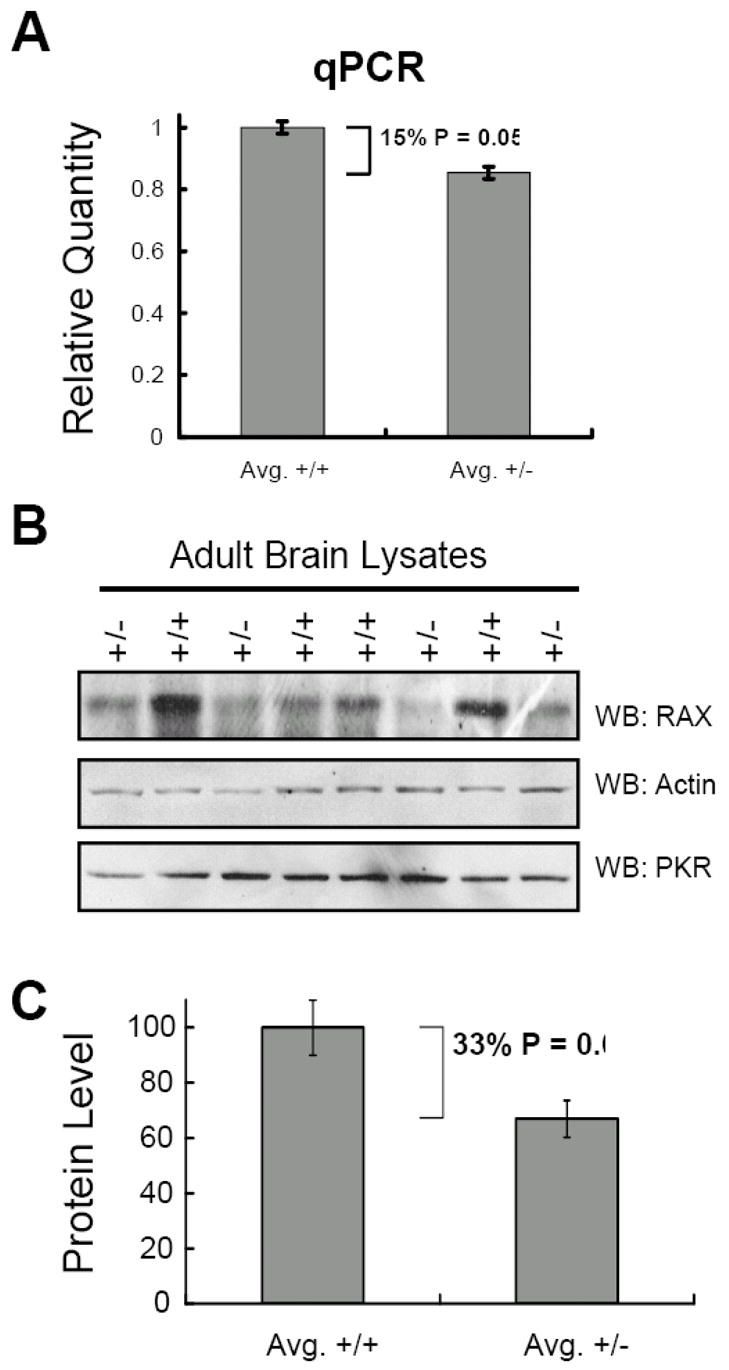
A. Quantitative real-time PCR was performed using RNA isolated from the lungs and brains of 5 mice heterozygous for the Rax knockout allele and of 5 Rax+/+ littermate mice. The average relative quantity of RAX mRNA is compared between normal (Avg. +/+) vs. heterozygous (Avg. +/−) mice using β-actin as an endogenous reference. B. Western blots of lysates generated from adult mouse brain tissue using antibody specific for RAX, actin, and PKR. Normal littermate (+/+) and heterozygous (+/−) lysates are indicated. C. The level of RAX and actin proteins were quantified by densitometry analysis of Western blots using lysates from the lungs and brains of 5 mice heterozygous for the Rax knockout allele and of 5 Rax+/+ littermate mice. The average level of RAX protein relative to actin is depicted for both normal (Avg. +/+) and heterozygous (Avg. +/-) tissue lysates. Statistical analysis was performed using ANOVA.
Finally, by RT-PCR with primers specific for Rax mRNA, we have detected Rax gene expression in wild type mouse blastocysts and ES cells (data not shown). Thus, Rax is expressed at a pre-implantation stage of embryogenesis.
3. Discussion
The results of these studies establish the requirement for the dsRNA-binding protein RAX during early development and embryogenesis of both the fly and mouse. Significantly, our analysis of the Drosophila ortholog of Rax, dRax (loqs, R3D1), indicates that dRax is expressed during nervous system development and required for nervous system organization. Interestingly, flies homozygous for the dRax[f00791] transposon insertion are unable to escape after having landed on food and have a severe reduction in locomotion activity, indicating either a neurological or neuromuscular defect. Similar to reports from other laboratories, we observed that the homozygous dRax[f00791] mutant is female sterile, a phenotype previously attributed to dRax’s role in processing miRNAs specific for germ cell maturation during normal oogenesis (Czech et al., 2008; Forstemann et al., 2005; Jiang et al., 2005; Park et al., 2007). Thus, our findings confirm those of others and extend them by indicating that dRax is also required for maturation of the drosophila nervous system. Future work may now include detailed analysis of the nervous system abnormalities in the dRax mutant flies to determine whether the mechanism for this defect is dependent on nervous system specific miRNA processing, cell proliferation and/or apoptosis.
The Rax gene is expressed throughout all tissues of the adult mouse and mRNA is detectable even at the preimplantation, blastocyst stage of embryogenesis. To investigate the function of murine Rax, we generated mice carrying a mutant Rax allele where exons 1 through 8, comprising the entire Rax ORF, have been deleted. Heterozygous Rax+/- mice appear normal in size, display no overt abnormal behavior and there is no statistically significant difference between the littersize of embryos and live born mice. Importantly, when Rax+/− mice are mated with wild type mice the expected 1:1 distribution of wild type to heterozygous offspring is observed, confirming that there is not a defect in transmission of the mutant allele. While the Rax+/− heterozygous mice are viable, we were unable to obtain any viable Rax−/− homozygous mice or embryos even at the blastocyst stage, indicating that deletion of the entire Rax gene is crucial for early embryonic development. Taken together, these results demonstrate that RAX is required for normal mouse embryonic development.
Significantly, tissues from Rax+/− mice produce more than half of the Rax transcripts and protein level present in the Rax+/+ tissues. In addition, we observed that the level of RAX protein varied from mouse to mouse relative to control proteins. These findings suggest that Rax+/− mice may have increased expression of the remaining wild type Rax allele by some uncharacterized autoregulatory feedback mechanism.
In contrast to the findings presented here, it was recently reported that mice homozygous for a deletion in the last exon of the Rax gene (exon 8) are viable but display a developmental defect of the ear, auditory deficiency and are smaller in size than normal littermates (Rowe et al., 2006). While these null mice produce a RAX transcript encoding a C-terminal deletion mutant of RAX that would be missing the third dsRNA binding domain, the authors did not detect any protein product by Western blotting (Rowe et al., 2006). However, it remains a formal possibility that a partially functional mutant RAX protein, which remains able to bind dsRNA and/or other proteins, could be produced to cause a hypomorphic phenotype. Such a RAX mutant product may not be detectable with the antibody used and could account for any discrepancy with results reported here.
Since Rax is ubiquitously expressed even during early mouse embryogenesis and there is no other homolog that may compensate any loss of expression, it is not surprising that complete deletion of Rax is early embryonic lethal in the mouse. In addition to functioning as an activator of PKR, recent evidence reveals that RAX is also a component of the RISC (Kok et al., 2007; Lee et al., 2006). Thus, while the Rowe et al mutant Rax allele may not produce a detectable protein product capable of activating PKR, any truncated RAX produced may remain able to perform other less well-characterized signal transduction and/or RNA interference pathway functions. In support of this notion, Pkr knockout mice are viable and do not display an overt phenotype, suggesting that RAX may function as more than just an activator of PKR (Yang et al., 1995). In addition, our laboratory and others have observed that RAX and TRBP can from a heterodimer that is dependent on their first and second dsRNA binding domains (data not shown; Kok et al., 2007; Laraki et al., 2008). It has been suggested that this RAX-TRBP heterodimer may not function to regulate PKR activity, but instead interacts with dicer to contribute to the RNA interference pathway (Kok et al., 2007; Laraki et al., 2008). Significantly, mice deficient for several other dsRNA binding proteins involved in RNA interference have also been reported to display catastrophic developmental phenotypes similar to what we observe for the dRax mutant fly and the Rax knockout mouse (Bernstein et al., 2003; Zhong et al., 1999). For example, deletion of the mouse ortholog of TRBP yields viable born mice that either die immediately after weaning or, in the case of any surviving males, have a defect in sperm maturation (Zhong et al., 1999). Moreover, Dicer−/− null mice are early embryonic lethal (Bernstein et al., 2003). Interestingly, Rax is more highly evolutionarily conserved than Pkr since a PKR ortholog has not been identified in Drosophila (Murphy et al., 2008). Collectively these results support the importance of RNA interference during embryogenesis and may indicate that RAX is a necessary regulator of RNA interference as well as PKR activation. In addition, mammalian RAX may be a functional link between any miRNA-mediated silencing and PKR-dependent inhibition of protein synthesis that occurs during cellular stress and/or development. In support of this notion, it has been recently reported that members of the RISC can associate with translation initiation proteins present in the ribosome to inhibit protein synthesis (Chendrimada et al., 2007; Thermann and Hentze, 2007). Future work will now be required to determine whether and how RAX-dependent PKR activation is regulated by miRNA produced during cellular stress to inhibit protein synthesis.
In summary, our findings illustrate the importance of the Rax gene to fly and mouse development. In addition, our data suggest that the previously reported Rax-knockout mouse allele may be hypomorphic. Unfortunately, we are unable to further assess the role of RAX as a regulator of RNA interference or an activator of PKR during early development due to the very early embryonic lethality of our Rax-null mice. Future work may now include generation of tissue-specific or inducible Rax-knockout systems to further elucidate the in vivo function of RAX.
4. Experimental Procedures
4.1. Drosophila work
The fly strain, PBac{WH}[f00791]/Cyo, was obtained from the Bloomington stock center. The insertion site was verified via In Verse PCR and sequencing. cDNA for dRax was isolated from an embryonic cDNA library by PCR, verified by sequencing, subcloned into pBluescript, and used as template for synthesizing the cRNA probe. In situ hybridization and histological analysis using anti-102 (visualizing the axon structure) were performed as previously described (Zhou et al., 1997). To perform the Random Locomotion Test male dRax homozygous mutants and their control littermates of genotype dRax[f00791]/Cyo were collected daily after eclosion. At day 3 they were subjected to the random locomotion test as described (Kane et al., 1997). F test, and subsequently Welch’s t-test were performed using R.
4.2. Generation of the Rax knockout mouse
The targeting vector for the Rax knockout was constructed using pTK-loxNeo as a vector backbone with the neomycin resistance (neor) cassette flanked by fragments of the Rax gene. To isolate a portion of the 3′ end of the Rax gene, a mouse 129/SV genomic DNA BAC library was screened by Genome Systems to obtain a BAC clone containing Rax genomic DNA. DNA sequencing of this BAC clone revealed that it contained about a 15Kb genomic fragment corresponding to exons 4 through 8 of the Rax genomic sequence. A 7 Kb Hind III fragment consisting of approximately the last 300 bps of Rax exon 8 (non-coding) and 6.7 Kb of non-transcribed and non-coding sequence downstream of the Rax gene was inserted into the Hind III site of pTK-loxNeo (downstream of the neor cassette) to create the vector pTK-3’RAX. To generate a DNA fragment for the 5′ end of the Rax gene, BAC clone RPCI-22 68D6 (Invitrogen) containing the entire Rax gene (Rowe and Sen, 2001), was used as a template to amplify a 2.2 Kb portion of the Rax gene corresponding to the Rax promoter and the first 120 bps of Rax exon 1 (just prior to the Rax AUG start codon) by PCR. This 5′ flanking fragment was inserted into the Cla I site of pTK-3′RAX between the human thymidine kinase gene (hTK; for positive selection) and the neor cassette to generate the final targeting vector, pKO-RAX.
60μg of the targeting vector pKO-RAX was linearized with Xho I and electroporated into J1 ES cells. Cells were grown in ES medium (DMEM, 20% FBS, 1% nonessential amino acids, 0.5% Penicillin-Streptomycin, 105 U/ml LIF) containing 400 mg/ml G418 and 250 nM FIAU. DNA was isolated from the resulting clones, digested with EcoR V, and homologous insertion was determined by Southern blotting using a 32P-labeled 520 bp probe with sequence corresponding to a region 5′ to the site of homologous insertion (chr2: 76490155 – 76490676). Two out of 360 ES cell clones were identified as positive for the RAX knockout allele, injected into C57BL/6J blastocysts, and implanted into psuedopregnant females. Chimeric offspring were then mated with C57BL/6J wild type mice (Charles River laboratories) and germline transmission of the Rax mutant was confirmed by Southern blot analysis using the 5′ probe (Figure 2).
In addition, PCR was performed on tail or embryo genomic DNA to confirm mouse genotype using primers specific to Rax wild type and mutant alleles, (Figure 2). Primer sequences used for genotyping are as follows (5′ – 3′): RAX1-FWD, TCTGAGAAGCTCTGGGAACCCAACTAG; RAX1-REV, GATGCCTGCTATGGGACATGGCGAGAAG; NeoF, ATGAACTGCAGGACGAGGCAGCG; NeoR, GGCGATAGAAGGCGATGCGCTG.
4.3. PCR and western blotting with mouse tissues
Lung and Brain tissues were isolated from mice, flash frozen in liquid nitrogen and stored at -80o C. Tissues were disrupted by mortar and pestle and homogenized using a QIAshredder (Qiagen, Valencia CA). DNA, RNA, and protein extracts were prepared from whole tissues using the Allprep DNA/RNA/Protein isolation kit (Qiagen, Valencia CA). RNA integrity and concentration were determined by nanodrop and bioanalyzer QC analysis. Quantitative real-time PCR was performed by the University of Florida, real-time PCR core facility using TaqMan gene expression assay Mm00478737_m1 for PRKRA (RAX) and mouse β-actin as endogenous control (Applied Biosystems, Foster City CA). Western blotting was performed using rabbit polyclonal antibody to RAX as described (Bennett et al., 2004). Individual bands in Western blots were quantified using Imagequant 5.0 (GE healthcare) and statistical significance determined using ANOVA data analysis in Microsoft Excel.
In addition, mRNA from 20 pooled C57BL/6J blastocysts was isolated using a Dynabeads mRNA DIRECT Micro kit (Invitrogen, Carlsbad, CA) and cDNA was prepared using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City CA). The primers used to detect RAX mRNA were: RAX2-3FWD: 5′-TGCACAGGTGAAGGTACGAG - 3′ and RAX6-7REV 5′ – CACGTTCGTTAGAGAAATGTGG – 3′.
Acknowledgments
This work was supported by NIH grant 5R01HL054083 and Bankhead-Coley award 06BB-14. We wish to thank Dr. Peter Ruvolo, Dr. Edward Scott and Gary Brown for their assistance in designing the RAX knockout mouse strategy, Zhigang Jiang for technical assistance during targeting of the mouse ES cells. Dr. Zhou is supported by CA95542.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bennett RL, Blalock WL, Abtahi DM, Pan Y, Moyer SA, May WS. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy and viral infection. Blood. 2006 doi: 10.1182/blood-2005-11-006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RL, Blalock WL, May WS. Serine 18 phosphorylation of RAX, the PKR activator, is required for PKR activation and consequent translation inhibition. J Biol Chem. 2004;279:42687–93. doi: 10.1074/jbc.M403321200. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol Cell Biol. 2000;20:4532–42. doi: 10.1128/mcb.20.13.4532-4542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chendrimada TP, Finn KJ, Ji X, Baillat D, Gregory RI, Liebhaber SA, Pasquinelli AE, Shiekhattar R. MicroRNA silencing through RISC recruitment of eIF6. Nature. 2007;447:823–8. doi: 10.1038/nature05841. [DOI] [PubMed] [Google Scholar]
- Cuddihy AR, Li S, Tam NW, Wong AH, Taya Y, Abraham N, Bell JC, Koromilas AE. Double-stranded-RNA-activated protein kinase PKR enhances transcriptional activation by tumor suppressor p53. Mol Cell Biol. 1999a;19:2475–84. doi: 10.1128/mcb.19.4.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddihy AR, Wong AH, Tam NW, Li S, Koromilas AE. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene. 1999b;18:2690–702. doi: 10.1038/sj.onc.1202620. [DOI] [PubMed] [Google Scholar]
- Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, Perrimon N, Kellis M, Wohlschlegel JA, Sachidanandam R, Hannon GJ, Brennecke J. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008 doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Acquisto F, Ghosh S. PACT and PKR: turning on NF-kappa B in the absence of virus. Sci STKE. 2001;2001:RE1. doi: 10.1126/stke.2001.89.re1. [DOI] [PubMed] [Google Scholar]
- Forstemann K, Tomari Y, Du T, Vagin VV, Denli AM, Bratu DP, Klattenhoff C, Theurkauf WE, Zamore PD. Normal microRNA maturation and germline stem cell maintenance requires Loquacious, a double-stranded RNA-binding domain protein. PLoS Biol. 2005;3:e236. doi: 10.1371/journal.pbio.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont M, Vaeyens F, Herst CV, De Meirleir KL, Englebienne P. Double-stranded RNA-dependent protein kinase (PKR) is a stress-responsive kinase that induces NFkappaB-mediated resistance against mercury cytotoxicity. Life Sci. 2006;78:1845–56. doi: 10.1016/j.lfs.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Gil J, Alcami J, Esteban M. Activation of NF-kappa B by the dsRNA-dependent protein kinase, PKR involves the I kappa B kinase complex. Oncogene. 2000;19:1369–78. doi: 10.1038/sj.onc.1203448. [DOI] [PubMed] [Google Scholar]
- Huang X, Hutchins B, Patel RC. The C-terminal, third conserved motif of the protein activator PACT plays an essential role in the activation of double-stranded-RNA-dependent protein kinase (PKR) Biochem J. 2002;366:175–86. doi: 10.1042/BJ20020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Jagus R, May WS. Interleukin 3 stimulates protein synthesis by regulating double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91:7455–9. doi: 10.1073/pnas.91.16.7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Warnken SP, May WS. Protein synthesis inhibition by flavonoids: roles of eukaryotic initiation factor 2alpha kinases. Biochem Biophys Res Commun. 1999a;265:589–94. doi: 10.1006/bbrc.1999.1727. [DOI] [PubMed] [Google Scholar]
- Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem. 1999b;274:15427–32. doi: 10.1074/jbc.274.22.15427. [DOI] [PubMed] [Google Scholar]
- Jiang F, Ye X, Liu X, Fincher L, McKearin D, Liu Q. Dicer-1 and R3D1-L catalyze microRNA maturation in Drosophila. Genes Dev. 2005;19:1674–9. doi: 10.1101/gad.1334005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane NS, Robichon A, Dickinson JA, Greenspan RJ. Learning without performance in PKC-deficient Drosophila. Neuron. 1997;18:307–14. doi: 10.1016/s0896-6273(00)80270-6. [DOI] [PubMed] [Google Scholar]
- Kok KH, Ng MH, Ching YP, Jin DY. Human TRBP and PACT directly interact with each other and associate with dicer to facilitate the production of small interfering RNA. J Biol Chem. 2007;282:17649–57. doi: 10.1074/jbc.M611768200. [DOI] [PubMed] [Google Scholar]
- Laraki G, Clerzius G, Daher A, Melendez-Pena C, Daniels S, Gatignol A. Interactions between the double-stranded RNA-binding proteins TRBP and PACT define the Medipal domain that mediates protein-protein interactions. RNA Biol. 2008:5. doi: 10.4161/rna.5.2.6069. [DOI] [PubMed] [Google Scholar]
- Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. Embo J. 2006;25:522–32. doi: 10.1038/sj.emboj.7600942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D, Dancis B, Brown JR. The evolution of core proteins involved in microRNA biogenesis. BMC Evol Biol. 2008;8:92. doi: 10.1186/1471-2148-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JK, Liu X, Strauss TJ, McKearin DM, Liu Q. The miRNA pathway intrinsically controls self-renewal of Drosophila germline stem cells. Curr Biol. 2007;17:533–8. doi: 10.1016/j.cub.2007.01.060. [DOI] [PubMed] [Google Scholar]
- Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem. 2000;275:37993–8. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. Embo J. 1998;17:4379–90. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Hartmann R, Qin J, Sen GC. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol. 2001;21:1908–20. doi: 10.1128/MCB.21.6.1908-1920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe TM, Rizzi M, Hirose K, Peters GA, Sen GC. A role of the double-stranded RNA-binding protein PACT in mouse ear development and hearing. Proc Natl Acad Sci U S A. 2006;103:5823–8. doi: 10.1073/pnas.0601287103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe TM, Sen GC. Organizations and promoter analyses of the human and the mouse genes for PACT, the protein-activator of the interferon-induced protein kinase, PKR. Gene. 2001;273:215–25. doi: 10.1016/s0378-1119(01)00588-1. [DOI] [PubMed] [Google Scholar]
- Saito K, Ishizuka A, Siomi H, Siomi MC. Processing of pre-microRNAs by the Dicer-1-Loquacious complex in Drosophila cells. PLoS Biol. 2005;3:e235. doi: 10.1371/journal.pbio.0030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R, Hentze MW. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature. 2007;447:875–8. doi: 10.1038/nature05878. [DOI] [PubMed] [Google Scholar]
- Wong AH, Tam NW, Yang YL, Cuddihy AR, Li S, Kirchhoff S, Hauser H, Decker T, Koromilas AE. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. Embo J. 1997;16:1291–304. doi: 10.1093/emboj/16.6.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M, Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. Embo J. 1995;14:6095–106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MC, Lau AS. Tumor suppressor p53 as a component of the tumor necrosis factor-induced, protein kinase PKR-mediated apoptotic pathway in human promonocytic U937 cells. J Biol Chem. 1998;273:25198–202. doi: 10.1074/jbc.273.39.25198. [DOI] [PubMed] [Google Scholar]
- Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20:1278–90. doi: 10.1128/mcb.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Peters AH, Lee K, Braun RE. A double-stranded RNA binding protein required for activation of repressed messages in mammalian germ cells. Nat Genet. 1999;22:171–4. doi: 10.1038/9684. [DOI] [PubMed] [Google Scholar]
- Zhou L, Schnitzler A, Agapite J, Schwartz LM, Steller H, Nambu JR. Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc Natl Acad Sci U S A. 1997;94:5131–6. doi: 10.1073/pnas.94.10.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]