

Abstract
Transgenic overexpression of Fli-1 in normal mice leads to SLE-like disease and increased expression was reported in SLE-affected human and murine lymphocytes. Reducing Fli-1 expression in MRL/lpr mice decreased antibody production, proteinuria, renal pathology, and mortality. Compared to those with wild-type expression of Fli-1, we report here that proliferative responses of Fli-1-deficient naïve B cells to several mitogens were reduced in lupus-prone and control mice. Expression of mitogen receptors, including BCR, TLR4, and TLR9, was not significantly impacted in Fli-1-deficient naïve B cells. IL12a transcripts were upregulated and NFAT transcripts were downregulated in Fli-1-deficient MRL/lpr B cells. These results demonstrate that Fli-1 deficiency affects B cell proliferative responses to mitogens, independent of BCR and TLR expression. IL12a and NFAT, known to influence proliferation, were identified as potential mediators of this effect. This may be a mechanism by which overexpression of Fli-1 contributes to B cell hyperactivity and subsequent SLE pathogenesis.
Keywords: B cell, BCR, Fli-1, IL12, NFAT, proliferation, SLE, TNF beta, TLR
Introduction
Systemic lupus erythematosus (SLE) is a complex chronic autoimmune disease characterized by numerous immunological abnormalities [1–3]. The multifaceted etiology of this disease is incompletely understood, but is mainly attributed to genetic susceptibility, while epigenetics, stochastic events, infections, exposure to toxins, and medications are thought to also be influential factors [4–16]. The immunological abnormalities observed in SLE can be categorized into three types: production of pathogenic autoantibodies, impaired ability to process and eliminate immune complexes, and lack of regulation regarding T and B lymphocyte function. B cell hyperactivity, both in vivo and in vitro, is a classic characteristic of SLE, and is likely a consequence of abnormal signaling events [17–19].
Friend leukemia insertion site 1 (Fli-1) is a DNA-binding protein capable of transcriptional activation or repression [20–23]. Based on the high homology of its DNA binding domain with that of Ets-1, and the central GGAA motif of the sequence to which it binds, Fli-1 is classified as a member of the ETS family of transcription factors [24]. C57BL/6 mice possessing a single targeted disruption of Fli-1 (Fli-1+/−) express 50% less Fli-1, but display a normal phenotype, while Fli-1−/− embryos suffer neural tube hemorrhage and die by 12.5 days of gestation [25]. Evidence suggests dysregulation of genes encoding extracellular matrix proteins that contribute to vascular integrity, and those that encode genes related to megakaryocyte development and function is responsible for in utero death. Interestingly, humans bearing a chromosomal deletion, which includes Fli-1, have thrombocytopenia [26–29]. Mice with a disrupted Fli-1 functional domain (Fli-1ΔCTA/ΔCTA) are viable and produce a truncated protein, lacking the carboxy-terminus transactivation domain, which exhibits 50% less transactivation activity than native Fli-1 [21]. This model is useful because the similarity of many ETS binding sites may allow one ETS factor to substitute for another when it is absent or deficient. For example, Erg-3 has a DNA-binding sequence that is 98% homologous to that of Fli-1. In the case of IgH regulation, Erg-3 and Fli-1 bind the enhancer with seemingly equal affinity and activate it with equal efficiency [30]. In the Fli-1ΔCTA/ΔCTA model, Fli-1 retains the ability to bind its target genes, minimizing the possibility of other ETS factors influencing expression of Fli-1 target genes.
Transgenic outbred CD1 mice that overexpress Fli-1 developed spontaneous glomerulonephritis, and died prematurely due to renal failure similar to standard models of spontaneous SLE [31]. B cells of mice possessing the Fli-1 transgene exhibited in vivo hyperplasia and hyperproliferation in response to in vitro stimuli. Baseline expression of Fli-1 was elevated in lymphocytes of murine models of SLE and human patients [32, 33]. In patients, Fli-1 expression correlated with disease severity. Autoantibody production, proteinuria, and renal pathology were significantly reduced, and survival was markedly extended in Fli-1+/−MRL/lpr mice [33]. These results confirm a key role for Fli-1 in murine SLE.
To determine potential mechanisms of the role of Fli-1 in disease, we evaluated the effect of Fli-1 deficiency on B cell proliferation, mitogen receptor expression, and the expression of their related signaling and effector proteins in three models – lupus-prone Fli-1+/− MRL/lpr mice, and non-diseased Fli-1+/− and Fli-1ΔCTA/ΔCTA C57BL/6 mice.
Understanding the role of Fli-1 in B cell function may lead to novel methods of treatment, targeting Fli-1 itself or, more likely, its pertinent target genes.
Materials and methods
Manipulation of the murine Fli-1 gene
Derivation of Fli-1+/− MRL/lpr, Fli-1+/− C57BL/6, and Fli-1ΔCTA/ΔCTA C57BL/6 mice was previously described [25, 33]. Briefly, a floxed neo cassette was used to disrupt exon IX of Fli-1. Resulting C57BL/6 Fli-1+/− mice were backcrossed to the MRL/lpr (The Jackson Laboratory, Bar Harbor, Maine) background, using speed congenic techniques to monitor inheritance of disease susceptibility loci for seven generations, to develop MRL/lpr Fli-1+/−mice. LoxP-/Cre-mediated excision of the disrupted exon was used to generate C57BL/6 Fli-1ΔCTA/ΔCTA mice, expressing the truncated protein. Table 1 outlines the strains, genotypes, and disease status of mice used in the studies described here. As no gender-specific effects were noted in any of the three Fli-1-deficient models, both male and female mice were used in all experiments.
Table 1.
Mouse strains, genotypes, and disease status.
| strain | MRL/lpr | C57BL/6 | |||
|---|---|---|---|---|---|
| Fli-1 genotype | +/+ | +/− | +/+ | +/− | ΔCTA/ΔCTA |
| SLE | YES | NO | |||
DNeasy DNA purification kit (Qiagen) was used to extract DNA from tail snips according to the manufacturer’s protocol [34]. Extracted DNA was subject to PCR as previously described [25].
B cell isolation
Under sterile conditions, lymphocyte suspensions were prepared by crushing fresh spleens between frosted glass slides in RPMI 1640 cell culture medium (Mediatech). Following a 5 min centrifugation at 1200 rpm, medium was removed, and red blood cells were lysed by resuspension in a 9:1 mixture of 0.16M ammonium chloride (Sigma) and 0.17M Tris, pH 7.6. Cells were washed twice, resuspended in RPMI 1640, and counted.
For proliferation and FACS assays, naïve B cells were isolated by negative selection with CD43 Microbeads (Miltenyi Biotec) according to the manufacturer’s directions [35]. For PCR arrays and real-time RT-PCR assays, splenic B cells were isolated by negative selection using CD90 Microbeads (Miltenyi Biotec) according to manufacturer’s directions and cultured with or without 10 g/ml LPS for ~2.5 days [36].
Purity of isolated cells was confirmed by staining with PerCP- or APC-labeled anti-B220 or anti-CD19 (BD Pharmingen) and PE-labeled anti-CD43 (Miltenyi Biotec or BD Pharmingen) and detection by a FACSCalibur™ instrument (BD Biosciences). Each sample was determined to consist of ≥90% naïve B cells.
Proliferation assays
Naïve B cell cultures were prepared, as published elsewhere, in duplicate at 2×105 cell/ml with 0.5, 1, 5, or 10 μg/ml LPS (Sigma), 1, 5, or 10 μg/ml anti-IgM (Jackson ImmunoResearch), 10 ng/ml phorbol 12-myristate 13-acetate (PMA) (Sigma) and 0.25, 0.5, or 1 μg/ml ionomycin (Sigma), or 1, 5, or 10 μg/ml CpG oligonucleotides (Cell Sciences) [37, 38]. Cultures were incubated at 37°C in 5% CO2 for 48 hours prior to addition of 5 μCi/ml tritiated thymidine and incubation at 37°C in 5% CO2 for 14 hours [39]. Cells were harvested and proliferation determined by a Packard TopCount scintillation counter (PerkinElmer).
FACS analysis of receptor expression
2×107 naïve B cells/ml were resuspended in sterile FACS buffer (PBS with 1% sodium azide (Sigma) and 0.1% FBS). 1 μl anti-CD16/CD32 (BD Pharmingen) per 106 cells was added and incubated on ice for 20 minutes. 0.5 μg/ml PE-labeled anti-CD43 (Miltenyi Biotec, BD Pharmingen), APC-labeled anti-CD19 (BD Pharmingen), PerCP-labeled anti-B220 (BD Pharmingen), APC-labeled anti-IgM (eBioscience, BD Pharmingen), unlabeled or APC-labeled anti-CD79a (Anaspec, BD Pharmingen), biotin- or FITC-labeled anti-TLR4 (eBioscience, Imgenex), and/or unlabeled or FITC-labeled anti-TLR9 (eBioscience, Imgenex) were added to 0.5–1×106 cells and incubated on ice for 20 minutes, protected from light. Following two washes in FACS buffer, 0.5 μg/ml FITC-labeled anti-rabbit IgG (Abcam), and/or APC-labeled streptavidin (BD Biosciences) were added where appropriate and incubated on ice for 20 minutes, protected from light. Following two washes in FACS buffer, fluorescence was detected by a FACSCalibur™ flow cytometer.
For intracellular staining, cells were resuspended and blocked as described above, then stained with 0.5 μg/ml PE-labeled anti-CD43 and APC-labeled anti-CD19 or PerCP-labeled anti-B220 (BD Pharmingen). Following two washes in FACS buffer, cells were fixed, permeabilized, and stained with unlabeled or FITC-labeled anti-TLR9 according to manufacturer’s protocol [40, 41]. Staining with FITC-labeled anti-rabbit IgG followed, when appropriate.
Real-time RT-PCR
RNA was extracted from B cells with Trizol® reagent (Invitrogen) according to the manufacturer’s protocol [42]. RNA was diluted in TE:8 buffer (10mM Tris-hydrochloride, 1mM EDTA, pH 8.0) and scanned by a SmartSpec™ Plus (Bio-Rad) spectrophotometer to determine RNA concentration and purity according to the manufacturer’s instructions [43].
mRNA was amplified according to Invitrogen’s SuperScript Indirect RNA Amplification kit protocol, doubling length of incubations throughout [44]. Resulting aRNA yield was determined as described above. RNA or aRNA was converted to cDNA according to manufacturer’s instructions for the RT2 PCR Array First Strand Kit (SuperArray) [45]. cDNA was subjected to a Mouse RT2 RNA QC PCR Array (SuperArray) on a MyiQ real-time PCR detection system (Bio-Rad) according to manufacturer’s protocol, to determine reverse transcription efficiency, amplification efficiency, genomic DNA content, and DNA contamination [46]. cDNA was then applied to a Mouse Toll-Like Receptor Signaling Pathway RT2 Profiler™ PCR Array (SuperArray) according to manufacturer’s protocol [45]. cDNA was also subject to single RT2 qPCR primer assays in triplicate, using primers for murine csf3, elf1, il12a, lta, nfat1c, nfkbia, nfkb1, or tlr9 (SuperArray) according to manufacturer’s protocol [47].
Identification of overrepresented transcription factor binding motifs
MDFrame software was used to identify overrepresented transcription factor binding motifs located in conserved promoter regions of differentially expressed genes [48, 49].
Statistical analysis
Analysis of proliferation data by Mann-Whitney test was performed using Prism software (GraphPad). A p value of ≤0.05 was considered significant. PCR array data was scaled and quantile normalized prior to analysis by T test. A p value of 0.05 and a fold-change greater than 2 was considered significant.
Results
Fli-1 deficiency restricts B cell proliferation
To investigate the effects of reduced Fli-1 expression on B cell proliferation in the context of murine SLE, naïve B cells from Fli-1+/− and Fli-1+/+ MRL/lpr mice were stimulated with LPS, anti-IgM, CpG DNA, or PMA and ionomycin (PMA/ion), and proliferation was assessed. Presumably, more activated B cells are present in wild-type mice, as disease is more severe and/or progresses faster [33]. Use of naïve B cells for these studies ensures that proliferative activity observed is a result of controlled in vitro stimulation, as opposed to in vivo activation by unknown stimuli, allowing fair comparisons. Mice were sacrificed at three ages (6, 12, and 20 weeks) representing different stages of disease (pre-, mild, and severe disease, respectively) [50, 51]. At 6 weeks of age, naïve B cells from Fli-1+/− MRL/lpr mice proliferated significantly less in response to LPS, and PMA/ion than those from Fli-1+/+ MRL/lpr mice (Figure 1A). Significantly reduced proliferation of Fli-1+/− MRL/lpr B cells in response to anti-IgM and LPS at 12 weeks of age, and anti-IgM at 20 weeks of age was also observed (Figure 1B and 1C). Proliferation of naive Fli-1+/− MRL/lpr B cells in response to anti-IgM and CpG DNA at 6 weeks, CpG DNA at 12 weeks, and CpG DNA and LPS at 20 weeks of age was also reduced, though these differences did not reach statistical significance (Figure 1).
Figure 1.
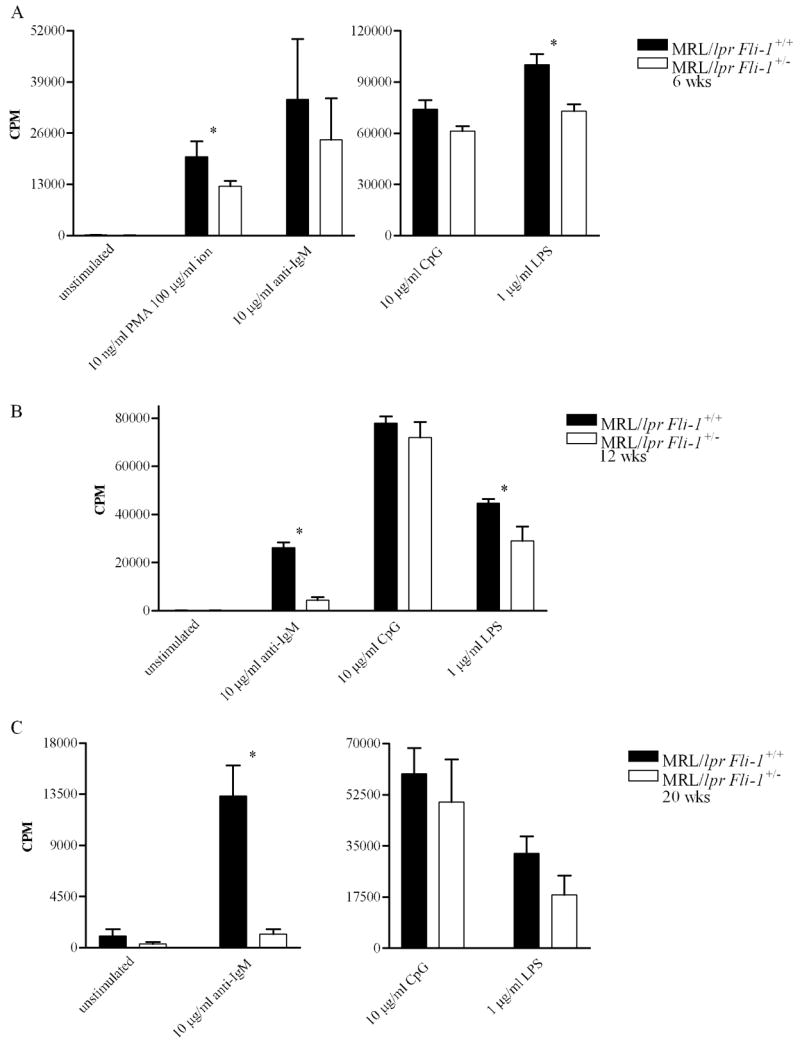
Proliferative response of MRL/lpr Fli-1+/+ (n = 2 or 3) versus Fli-1+/− (n = 2 or 3) B cells to mitogens at (A) 6, (B) 12, or (C) 20 weeks of age. Data from 2 or 3 independent experiments is shown as mean CPM (counts per minute) ± SEM (standard error of the mean). * = p ≤ 0.05.
C57BL/6 mice were sacrificed at ages corresponding to those at which MRL/lpr mice were sacrificed, though age was not expected to affect the activity of C57BL/6 B cells. At 6 weeks of age, naïve B cells from Fli-1+/− C57BL/6 mice proliferated significantly less in response to LPS and anti-IgM than naïve B cells from Fli-1+/+ C57BL/6 mice (Figure 2A). Heterozygous and wild-type responses to CpG DNA, however, were similar. Compared to wild-type, heterozygous cells also proliferated significantly less in response to LPS, CpG DNA, and anti-IgM at 12 weeks of age (Figure 2B). Twenty-week-old Fli-1 heterozygous B cells proliferated significantly less than wild-type cells in response to PMA/ion and anti-IgM, but not in response to LPS and CpG DNA (Figure 2C).
Figure 2.
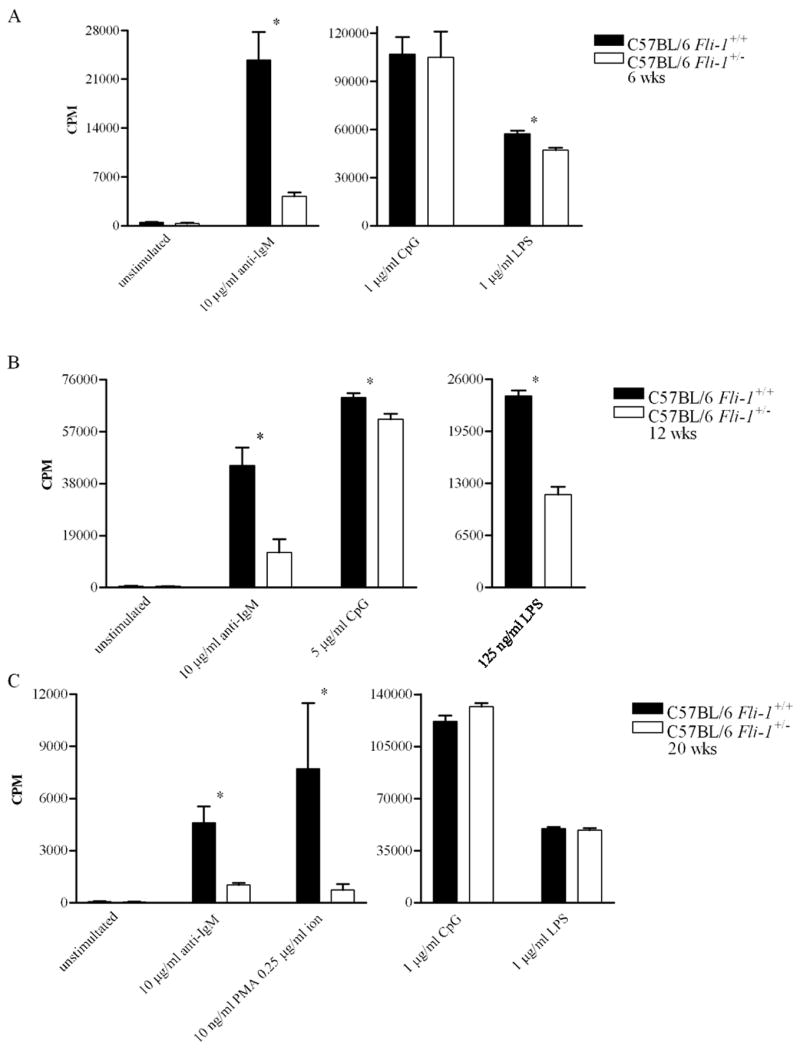
Proliferative response of C57BL/6 Fli-1+/+ (n = 2 or 3) versus Fli-1+/− (n = 2 or 3) B cells to mitogens at (A) 6, (B) 12, or (C) 20 weeks of age. Data from 2 or 3 independent experiments is shown as mean CPM ± SEM. * = p ≤ 0.05.
Naïve B cells from 6-week-old Fli-1ΔCTA/ΔCTA C57BL/6 mice proliferated significantly less, compared to Fli-1-wild-type C57BL/6 B cells, in response to anti-IgM and CpG DNA, but not LPS (Figure 3A). At 12 weeks of age, naïve B cells from Fli-1ΔCTA/ΔCTA C57BL/6 mice proliferated less than those from Fli-1+/+ C57BL/6 mice in response to LPS, while differences in response to PMA/ion and CpG DNA did not reach statistical significance (Figure 3B). Proliferation of 20-week-old Fli-1ΔCTA/ΔCTA C57BL/6 B cells was significantly reduced in response to anti-IgM, CpG DNA, and LPS (Figure 3C).
Figure 3.
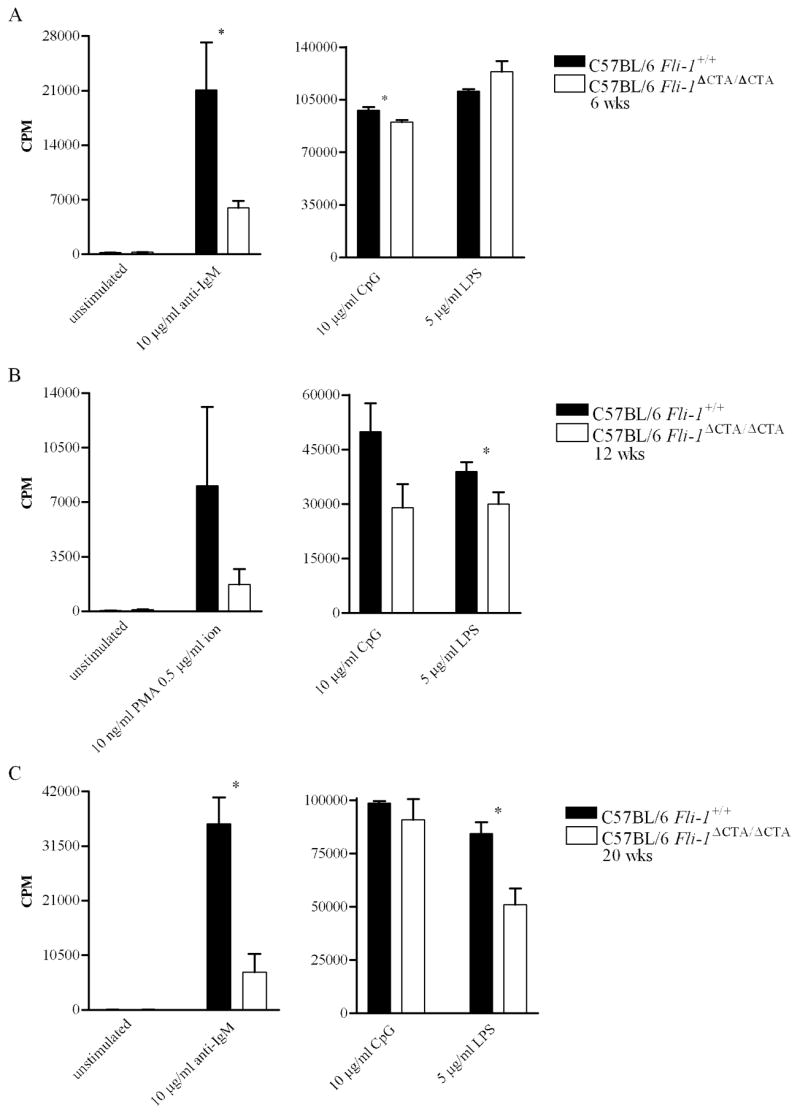
Proliferative response of C57BL/6 Fli-1+/+ (n = 2 or 3) versus Fli-1ΔCTA/ΔCTA (n = 3) B cells to mitogens at (A) 6, (B) 12, or (C) 20 weeks of age. Data from 2 or 3 independent experiments is shown as mean CPM ± SEM. * = p ≤ 0.05.
BCR expression is independent of Fli-1 expression or activity
Given the decreased B cell proliferative response in Fli-1-deficient mice, we next examined whether Fli-1 deficiency affected mitogen receptor expression. To examine the effect of Fli-1 deficiency on BCR expression, IgM and Igα expression on naïve B cells from 6-, 12-, and 20-week-old Fli-1+/− versus Fli-1+/+ MRL/lpr mice was examined. IgM expression was slightly reduced, and Igα was unchanged, on Fli-1+/− cells from 20-week-old mice, while Igα was slightly reduced, and IgM was unchanged, compared to wild-type, on Fli-1+/− B cells from 6-week-old mice (Figure 4 and data not shown). Neither IgM nor Igα expression appeared to be affected by Fli-1 deficiency at 12 weeks of age (data not shown). IgM and Igα are expressed together as the BCR, but Igα is also known to associate with IgD on naïve B cells, and dissimilar IgD expression may account for the observed differences in Igα expression while IgM expression remains unaffected [52]. IgD expression was not altered on Fli-1 deficient cells from MRL/lpr mice of any age (Figure 4 and data not shown).
Figure 4.
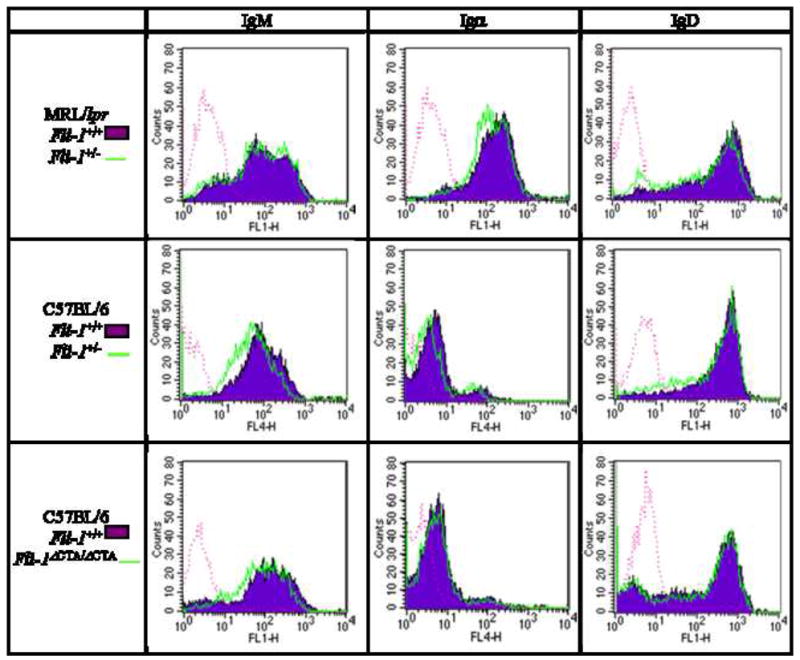
Expression of IgM, Igα, and IgD on naïve B cells from 6-week-old MRL/lpr Fli-1+/+ (area filled, n = 12, 7, and 8 respectively) versus Fli-1+/− (area open, n = 11, 6, and 7 respectively), C57BL/6 Fli-1+/+ (area filled, n = 15, 12, and 13 respectively) versus Fli-1+/− (area open, n = 14, 11, and 14 respectively), and C57BL/6 Fli-1+/+ (area filled, 11, 11, and 3 respectively) versus Fli-1ΔCTA/ΔCTA (area open, n = 11, 11, and 3 respectively) mice. Data is representative of 2-5 independent experiments. Isotype controls are represented by dotted lines.
At 20 weeks of age, IgM expression was slightly reduced on C57BL/6 Fli-1+/− B cells compared to wild-type, but appeared to be similar at 6 and 12 weeks of age (Figure 4 and data not shown). IgD expression was also slightly reduced on B cells from 12-week-old C57BL/6 Fli-1+/− mice compared to those from wild-type C57BL/6 mice, but was similar at 6 and 20 weeks of age (Figure 4 and data not shown). Igα expression was slightly reduced on Fli-1+/− B cells from 6- and 20- week-old C57BL/6 mice, compared to wild-type, but was similar on B cells from 12-week-old mice (Figure 4 and data not shown).
Upon comparison of B cells from C57BL/6 Fli-1+/+ and Fli-1ΔCTA/ΔCTA mice, minimal differential expression of IgM, Igα, and IgD was observed at 6 weeks of age (Figure 4). IgM, Igα, and IgD expression on Fli-1+/+ and Fli-1ΔCTA/ΔCTA cells was similar at 12 weeks of age (data not shown). At 20 weeks, Igα and IgD expression was similar, while IgM expression was slightly increased on Fli-1ΔCTA/ΔCTA cells compared to wild-type (data not shown). Similar to BCR expression, Ca2+ influx in response to anti-IgM in cells from 12-week-old Fli-1ΔCTA/ΔCTA mice was not affected (data not shown).
TLR4 and TLR9 expression is independent of Fli-1 expression or activity
To examine the effect of Fli-1 deficiency on expression of TLR4 and TLR9, expression of these receptors on naïve B cells from Fli-1+/+ versus Fli-1+/− MRL/lpr mice at 6, 12, and 20 weeks of age was determined. Slightly reduced expression of TLR4 was noted on Fli-1+/− B cells, compared to wild-type B cells, at the ages of 6 and 20 weeks, while it appeared to be slightly increased at 12 weeks (Figure 5 and data not shown). Much of the current literature asserts that TLR9 is expressed primarily intracellularly, and CpG DNA must be taken into the endosome to initiate signaling [53–55]. While experimental evidence of this has been established in macrophages, dendritic cells, and B cell lines, TLR9 has also been detected on the surface of primary B cells [56, 57]. Therefore, both surface and intracellular expression of TLR9 were assessed. Compared to Fli-1-wild-type B cells, surface expression of TLR9 on B cells from Fli-1+/− MRL/lpr mice was slightly reduced at all three ages and intracellular expression of TLR9 was reduced in Fli-1-deficient cells from mice 12 and 20 weeks of age, and unchanged in Fli-1 deficient cells from mice 6 weeks of age (Figure 5 and data not shown).
Figure 5.
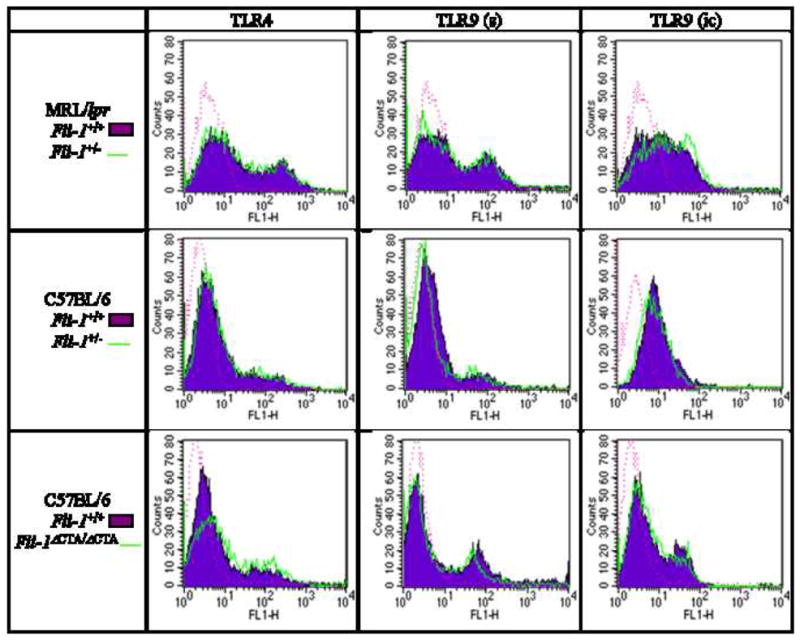
Expression of TLR4 and TLR9 (s - surface, ic - intracellular) on naïve B cells from 6-week-old MRL/lpr Fli-1+/+ (area filled, n = 7, 4, and 1 respectively) versus Fli-1+/− (area open, n = 6, 4, and 1 respectively), C57BL/6 Fli-1+/+ (area filled, n = 12, 8, and 5 respectively) versus Fli-1+/− (area open, n = 11, 9, and 6 respectively), C57BL/6 Fli-1+/+ (area filled, n = 11, 10, and 10 respectively) versus Fli-1ΔCTA/ΔCTA (area open, n = 11, 10, and 10 respectively) mice. Data is representative of 2-5 independent experiments. Isotype controls are represented by dotted lines.
Comparison of TLR4 expression on Fli-1+/+ versus Fli-1+/− C57BL/6 naïve B cells demonstrated a slight reduction on Fli-1-deficient B cells at 6 and 20 weeks of age, comparable to the observations made evaluating TLR4 expression on Fli-1+/+ versus Fli-1+/− MRL/lpr B cells (Figure 5 and data not shown). TLR4 expression appeared unaffected on Fli-1-deficient cells at 12 weeks of age (data not shown). Surface expression of TLR9 was also slightly reduced on Fli-1-deficient B cells at the age of 6 weeks, but similar to wild-type at 12 and 20 weeks, while intracellular expression of TLR9 was slightly reduced at all ages (Figure 5 and data not shown).
Only TLR9 surface expression was slightly reduced on Fli-1ΔCTA/ΔCTA C57BL/6, compared to wild-type B cells, at 6 and 20 weeks of age (Figure 5 and data not shown). Unlike Fli-1+/− MRL/lpr and C57BL/6 B cells, Fli-1ΔCTA/ΔCTA cells demonstrated no discernible reduction in TLR4 or intracellular TLR9 expression at any age (Figure 5 and data not shown).
Il12a, lta, nfatc1, and tlr9 transcript expression is affected by Fli-1 deficiency
Because Fli-1 genotype had little effect on BCR or TLR4/9 expression, we assessed downstream signaling pathways from BCR and TLR for differences that may account for the observed proliferation differences in B cells with variant Fli-1 expression. The expression of 84 genes related to TLR signaling was determined in LPS-stimulated or unstimulated splenic B cells isolated from 6-week-old Fli-1+/+ and Fli-1+/− MRL/lpr mice. Several of these genes are also known to participate in BCR signaling. Genes identified as more than 2-fold differentially expressed and statistically significant by these arrays, and genes coding for transcription factors which bind the promoters of these differentially expressed genes (csf3, il12a, lta, tlr9, nfkb1, nfkbia, elf1, and nfatc1) were subject to individual RT PCR analysis, using total RNA from stimulated or unstimulated B cells of 6-week-old Fli-1+/+ versus Fli-1+/− MRL/lpr mice. Fold change differences in expression from confirmation assays are summarized in Table 2. Expression of il12a and lta in unstimulated cells and tlr9 in stimulated cells demonstrated greater than twofold increases in confirmation assays. Furthermore, expression of nfat1c demonstrated a greater than two-fold decrease in stimulated B cells.
Table 2.
Expression of selected genes in Fli-1+/+ (n = 5) versus Fli-1+/− (n = 5) MRL/lpr B cells. One triplicate experiment per gene.
| gene | Fold change in MRL/lpr Fli-1+/− B cells | |
|---|---|---|
| Unstimulated | LPS stimulated | |
| csf3 | 1.5 | −0.97 |
| elf1 | 1.96 | 1.13 |
| il12a | 2.2 | 1.2 |
| lta | 2.58 | 1.56 |
| mphosph1 | 2.6 | 2.1 |
| nfatc1 | −1.01 | −2.03 |
| nfkb1 | 1.78 | −0.9 |
| nfkbia | −0.5 | 1.0 |
| tlr9 | 1.58 | 2.62 |
Discussion
Fli-1 is overexpressed in lymphocytes of SLE patients and murine models of SLE. Transgenic overexpression in normal mice leads to SLE-like disease [31, 32]. Reduction of Fli-1 expression in the MRL/lpr mouse model of SLE results in decreased immunoglobulin production, proteinuria, and renal pathology; and markedly increased survival [33]. These findings suggest that Fli-1 activity may be a central contributor to SLE pathogenesis. Investigation of this role and discovery of Fli-1 target genes may provide novel therapeutic targets.
Transgenic mice that overexpress Fli-1 exhibit innate B cell hyperplasia and exaggerated in vitro proliferation of B cells in response to LPS and anti-IgM [31]. Also, spleens from MRL/lpr Fli-1+/− mice demonstrated lower percentages of B cells and serum exhibited reduced immunoglobulin concentration [33]. It was therefore hypothesized that Fli-1 deficiency may impair B cell proliferation. Additionally, it was predicted that Fli-1 activity is integral to the process in normal immunity, outside the context of SLE. Because overexpression of Fli-1 preceded the manifestation of disease in Fli-1 transgenic mice, significant effects of Fli-1 deficiency early in the progression of MRL/lpr disease was anticipated [31].
Effects of Fli-1 deficiency on B cell proliferation were evaluated in three models – Fli-1+/−MRL/lpr, Fli-1+/− C57BL/6, and Fli-1ΔCTA/ΔCTA C57BL/6 mice. Proliferative responses of naïve B cells from Fli-1-deficient mice to several mitogens were significantly reduced compared to those of Fli-1-wild-type littermates (Figures 1–3). Proliferation initiated by all these mitogens was affected, suggesting that Fli-1 regulates B cell genes which directly or indirectly influence proliferation.
It was noted that statistical significance of differences in proliferation were observed more frequently in younger MRL/lpr groups than the 20-week-old group (Figure 1). Differences in proliferation of MRL/lpr B cells of varying Fli-1 genotype may be less prominent at 20 weeks because disease in the heterozygotes has progressed to a stage more equivalent to that of wild-type mice. Alternatively, and more likely, significant differences may not be observed because, as wild-type MRL/lpr mice age, the ability of their B cells to respond to proliferative stimuli decreases, potentially equalizing the proliferative responses of Fli-1+/− and wild-type cells [58–60].
Proliferation of B cells from Fli-1+/− and Fli-1ΔCTA/ΔCTA C57BL/6 mice demonstrated impairment similar to that of B cells from Fli-1+/− MRL/lpr mice, indicating Fli-1 participates in B cell proliferation under non-disease conditions (Figures 1 and 2). In the Fli-1-heterozygous model, limited availability of Fli-1 protein may allow binding sites to be occupied and activated by other ETS factors, such as Erg-3. As the mutant Fli-1 is assumed to bind the majority of available Fli-1 binding sites, compensation for Fli-1 deficiency may explain why fewer statistically significant differences in B cell proliferation were observed in Fli-1+/− C57BL/6 mice than in Fli-1ΔCTA/ΔCTA C57BL/6 mice compared to Fli-1+/+ C57BL/6 mice (Figures 2 and 3). The effect of Fli-1 deficiency on B cell proliferation appeared to be accentuated in MRL/lpr mice, perhaps suggesting that aspects of SLE contribute to this effect. Differing genetic background or the lack of functional Fas receptor on MRL/lpr B cells may also influence the extent of the effects of Fli-1 deficiency.
B cell proliferation was evaluated in each model of Fli-1-deficiency when they were 6, 12, and 20 weeks old – ages that correspond to progressive stages of MRL/lpr disease [50, 51]. Proliferative responses of Fli-1-deficient MRL/lpr B cells were significantly reduced compared to wild-type B cell responses beginning at the pre-disease stage at 6 weeks of age (Figure 1). Given the development of SLE-like disease in Fli-1-overexpressing transgenic mice, the observed impact of Fli-1 deficiency prior to disease onset suggests that abnormal expression of Fli-1 is likely a contributing factor to causal events, rather than a consequence of disease.
As a transcription factor, Fli-1 is able to regulate expression of multiple genes. Altered expression of mitogen receptors corresponding to the mitogens used to evaluate proliferation was also investigated as a mechanism behind the decreased B cell proliferation associated with Fli-1 deficiency.
A higher proportion of B cells expressing the CpG DNA-binding TLR9 is found in SLE patients with active, versus remitted, disease [61]. Expression of TLR9 also correlates with levels of anti-dsDNA. It was therefore hypothesized that Fli-1 deficiency results in decreased or incomplete expression of these receptors. Surface expression of BCR components IgM and Igα, TLR4, and TLR9, and intracellular expression of TLR9 was only slightly and inconsistently reduced in naïve B cells of Fli-1-deficient mice compared to Fli-1-wild-type mice of similar strain (Figure 5 and data not shown). Slight reduction of Igα, without concomitant reduction of IgM or IgD, or vice versa, was also occasionally observed (Figure 4 and data not shown). Were the reduced expression of Igα due to faulty association with IgD or IgM exclusively, normal expression of either IgM or IgD and reduced expression of the other would be expected. As this was not observed, it is unlikely that Fli-1 deficiency affects Igα expression in this manner.
Because Igα is known to be regulated by Fli-1, a more impressive reduction of Igα expression was expected [62]. As BCR expression is essential to B cell development and survival, expression of Igα may not be sensitive to the level to which Fli-1 activity is decreased in our models. This may be true not only for Igα, but also for the other receptors, either because there is enough residual Fli-1 activity for near normal expression, or because Fli-1 plays a minor or nonessential role in regulating expression. Alternatively, our method of analysis may be insufficient to detect subtle, but significant, differences in expression, though we believe this is unlikely.
Additionally, differential expression of a particular receptor did not always coincide with differential B cell proliferation response to the corresponding mitogen. It is thus unlikely that reduced expression of mitogen receptors is the mechanism driving the large differences in B cell proliferation observed. If these results accurately reflect in vivo BCR expression, the idea that this minimally reduced expression may contribute to the observed proliferation assay results cannot be completely discounted, though to what extent can not be speculated.
Significantly reduced proliferative responses to all mitogens tested, including PMA/ion, which bypass interaction with receptors, suggests that multiple pathways are affected by Fli-1 deficiency. Therefore the mechanism is likely multifactorial and may or may not be similar for all affected pathways. Minimal decreases in mitogen receptor expression on Fli-1-deficient cells suggest that more profound effects of Fli-1 deficiency may be downstream of receptor expression. Transcript expression of several TLR and BCR signaling-related genes was evaluated in MRL/lpr Fli-1+/+ versus Fli-1+/− B cells. IL12a and TNFβ transcripts were upregulated in unstimulated Fli-1-deficient cells (Table 3). TLR9 transcripts were upregulated and NFAT transcripts were downregulated in stimulated Fli-1-deficient B cells (Table 3).
As Fli-1 is generally recognized as a transcription activator, it was unexpected to find upregulation of genes in Fli-1 deficient cells. There are, however, a number of published examples of Fli-1 acting as a repressor of genes including those that code for collagen type 1 and the retinoic acid receptor [63, 64]. Fli-1 itself may not interact with upregulated genes identified here, but may activate expression of a repressor that inhibits their transcription.
IL12a upregulation may have a protective effect as transcript levels are below normal in SLE PMBC [65]. Also, treatment of SLE PMBC and MRL/lpr mice with IL12 reduced anti-dsDNA production [66, 67]. In treated mice, lymphadenopathy, splenomegaly, proteinuria, and glomerulonephritis were also alleviated [68]. These effects of IL12 on SLE parameters may contribute to the overall phenotype of Fli-1+/− MRL/lpr mice. Upregulation of IL12 may also contribute to the effect of Fli-1 deficiency on B cell proliferation, as IL12 treatment reduces proliferation and tumorgenicity of malignant B cells in which IL12 receptor expression is induced [69]. We were unable to detect serum IL12 in MRL/lpr mice of either Fli-1 genotype.
TNFβ upregulation may offset the overproduction of immunoglobulin in MRL/lpr disease, as reduction of TNFβ results in increased immunoglobulin production [70]. Decreased immunoglobulin levels were present in Fli-1+/− MRL/lpr sera, but TNFβ levels remain unknown. Low production of TNFβ was associated with SLE and lupus nephritis in a Korean population [71]. The relationship between elevated TNFβ transcripts and reduced B cell proliferation in Fli-1-deficient B cells requires further investigation, as literature regarding this cytokine and its effect on proliferation are conflicting.
Though the mechanism is unknown, absence of TLR9 in lupus-prone mice exacerbates disease, suggesting that upregulation of TLR9 is protective [72]. While transcripts were upregulated in Fli-1-deficient MRL/lpr B cells, assessment of receptor expression did not reflect this upregulation. Perhaps an alternative method of measuring TLR9 protein expression is needed to accurately address the question of the relationship of Fli-1 with TLR9, and whether this relationship influences B cell proliferation.
As NFAT expression is known to be induced in stimulated B cells and has an established role in lymphocyte proliferation, the finding of decreased NFAT transcripts in Fli-1-deficient B cells is of interest (Table 3) [73–75]. As a known regulator of c-myc and BAFF, decreased NFAT expression could directly result in decreased proliferation [76, 77]. In addition, NFAT is known to activate expression of CD23, the IgE Fc receptor [78]. The soluble form of this receptor is elevated in SLE and B-chronic lymphocytic leukemia (B-CLL) patient serum and enhances immunoglobulin production [79–81]. Interaction of soluble CD23 with its receptors stimulates a growth response [82]. Selective increase of surface CD23 prompts resting B-CLL cells to proceed through G1 and S phase of the cell cycle [83]. Blockade of CD23 ligation by anti-CD23 monoclonal antibodies inhibits proliferation of Epstein-Barr virus (EBV)-transformed B cells [84]. Interestingly, B220+ cells from Fli-1ΔCTA/ΔCTA C57BL/6 mice demonstrated reduced surface CD23 expression (unpublished data). Confirmation that NFAT expression is reduced in these mice may clarify the mechanism by which Fli-1 deficiency diminishes B cell proliferation.
The results of the studies presented here clearly demonstrate that Fli-1 deficiency affects B cell proliferative responses to mitogens, independent of BCR and TLR expression. It should be considered that the genes evaluated here are not an exhaustive representation of proteins involved in these pathways. However, the upregulation of IL12a and downregulation of NFAT transcripts are prospective points from which to continue exploration of the relationship between Fli-1, B cell proliferation, and SLE.
Acknowledgments
We would like to acknowledge individuals without whom this work would not be possible. Dr. Dennis Watson and members of his laboratory developed the Fli-1+/− and Fli-1ΔCTA/ΔCTA C57BL/6 mice. We are grateful to Jackie EuDaly, Michelle Lee, Meagan Mollenhauer, and Dr. Omar Moussa for technical assistance. We also thank Dr. Margie Peden-Adams, Dr. Makio Ogawa, and Dr. Haiqun Zeng for use of instruments in their laboratories. We also acknowledge personnel of the Ralph H. Johnson Veterans Affairs Medical Center animal facility for care of study mice.
Abbreviations
- CPM
counts per minute
- Fli-1
Friend leukemia insertion site 1
- NFAT
nuclear factor of activated T cells
- PMA
phorbol 12-myristate 13-acetate
- SEM
standard error of the mean
- SLE
systemic lupus erythematosus
References
- 1.Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and Estimated Population Burden of Selected Autoimmune Diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–43. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 2.Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, Domenech I, Aydintug AO, Jedryka-Goral A, de Ramon E, et al. Systemic Lupus Erythematosus: Clinical and Immunologic Patterns of Disease Expression in a Cohort of 1,000 Patients. The European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore) 1993;72:113–24. [PubMed] [Google Scholar]
- 3.McCarty DJ, Manzi S, Medsger TA, Jr, Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of Systemic Lupus Erythematosus. Race and Gender Differences. Arthritis Rheum. 1995;38:1260–70. doi: 10.1002/art.1780380914. [DOI] [PubMed] [Google Scholar]
- 4.Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for Impaired T Cell DNA Methylation in Systemic Lupus Erythematosus and Rheumatoid Arthritis. Arthritis Rheum. 1990;33:1665–73. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- 5.Mishra N, Brown DR, Olorenshaw IM, Kammer GM. Trichostatin a Reverses Skewed Expression of Cd154, Interleukin-10, and Interferon-Gamma Gene and Protein Expression in Lupus T Cells. Proc Natl Acad Sci U S A. 2001;98:2628–33. doi: 10.1073/pnas.051507098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reilly CM, Mishra N, Miller JM, Joshi D, Ruiz P, Richon VM, Marks PA, Gilkeson GS. Modulation of Renal Disease in Mrl/Lpr Mice by Suberoylanilide Hydroxamic Acid. J Immunol. 2004;173:4171–8. doi: 10.4049/jimmunol.173.6.4171. [DOI] [PubMed] [Google Scholar]
- 7.Eisenberg RA, Craven SY, Warren RW, Cohen PL. Stochastic Control of Anti-Sm Autoantibodies in Mrl/Mp-Lpr/Lpr Mice. J Clin Invest. 1987;80:691–7. doi: 10.1172/JCI113123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An Increased Prevalence of Epstein-Barr Virus Infection in Young Patients Suggests a Possible Etiology for Systemic Lupus Erythematosus. J Clin Invest. 1997;100:3019–26. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fawaz-Estrup F. Human Parvovirus Infection: Rheumatic Manifestations, Angioedema, C1 Esterase Inhibitor Deficiency, Ana Positivity, and Possible Onset of Systemic Lupus Erythematosus. J Rheumatol. 1996;23:1180–5. [PubMed] [Google Scholar]
- 10.Geier DA, Geier MR. A Case-Control Study of Serious Autoimmune Adverse Events Following Hepatitis B Immunization. Autoimmunity. 2005;38:295–301. doi: 10.1080/08916930500144484. [DOI] [PubMed] [Google Scholar]
- 11.Kowal C, Weinstein A, Diamond B. Molecular Mimicry between Bacterial and Self Antigen in a Patient with Systemic Lupus Erythematosus. Eur J Immunol. 1999;29:1901–11. doi: 10.1002/(SICI)1521-4141(199906)29:06<1901::AID-IMMU1901>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 12.Conrad K, Mehlhorn J, Luthke K, Dorner T, Frank KH. Systemic Lupus Erythematosus after Heavy Exposure to Quartz Dust in Uranium Mines: Clinical and Serological Characteristics. Lupus. 1996;5:62–9. doi: 10.1177/096120339600500112. [DOI] [PubMed] [Google Scholar]
- 13.Kilburn KH, Warshaw RH. Prevalence of Symptoms of Systemic Lupus Erythematosus (Sle) and of Fluorescent Antinuclear Antibodies Associated with Chronic Exposure to Trichloroethylene and Other Chemicals in Well Water. Environ Res. 1992;57:1–9. doi: 10.1016/s0013-9351(05)80014-3. [DOI] [PubMed] [Google Scholar]
- 14.Satoh M, Hamilton KJ, Ajmani AK, Dong X, Wang J, Kanwar YS, Reeves WH. Autoantibodies to Ribosomal P Antigens with Immune Complex Glomerulonephritis in Sjl Mice Treated with Pristane. J Immunol. 1996;157:3200–6. [PubMed] [Google Scholar]
- 15.Sarzi-Puttini P, Atzeni F, Capsoni F, Lubrano E, Doria A. Drug-Induced Lupus Erythematosus. Autoimmunity. 2005;38:507–18. doi: 10.1080/08916930500285857. [DOI] [PubMed] [Google Scholar]
- 16.Timbrell JA, Facchini V, Harland SJ, Mansilla-Tinoco R. Hydralazine-Induced Lupus: Is There a Toxic Metabolic Pathway? Eur J Clin Pharmacol. 1984;27:555–9. doi: 10.1007/BF00556891. [DOI] [PubMed] [Google Scholar]
- 17.Fauci AS, Moutsopoulos HM. Polyclonally Triggered B Cells in the Peripheral Blood and Bone Marrow of Normal Individuals and in Patients with Systemic Lupus Erythematosus and Primary Sjogren’s Syndrome. Arthritis Rheum. 1981;24:577–83. doi: 10.1002/art.1780240402. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki N, Sakane T. Induction of Excessive B Cell Proliferation and Differentiation by an in Vitro Stimulus in Culture in Human Systemic Lupus Erythematosus. J Clin Invest. 1989;83:937–44. doi: 10.1172/JCI113979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liossis SN, Kovacs B, Dennis G, Kammer GM, Tsokos GC. B Cells from Patients with Systemic Lupus Erythematosus Display Abnormal Antigen Receptor-Mediated Early Signal Transduction Events. J Clin Invest. 1996;98:2549–57. doi: 10.1172/JCI119073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klemsz MJ, Maki RA, Papayannopoulou T, Moore J, Hromas R. Characterization of the Ets Oncogene Family Member, Fli-1. J Biol Chem. 1993;268:5769–73. [PubMed] [Google Scholar]
- 21.Rao VN, Ohno T, Prasad DD, Bhattacharya G, Reddy ES. Analysis of the DNA-Binding and Transcriptional Activation Functions of Human Fli-1 Protein. Oncogene. 1993;8:2167–73. [PubMed] [Google Scholar]
- 22.Zhang L, Lemarchandel V, Romeo PH, Ben-David Y, Greer P, Bernstein A. The Fli-1 Proto-Oncogene, Involved in Erythroleukemia and Ewing’s Sarcoma, Encodes a Transcriptional Activator with DNA-Binding Specificities Distinct from Other Ets Family Members. Oncogene. 1993;8:1621–30. [PubMed] [Google Scholar]
- 23.Athanasiou M, Mavrothalassitis G, Sun-Hoffman L, Blair DG. Fli-1 Is a Suppressor of Erythroid Differentiation in Human Hematopoietic Cells. Leukemia. 2000;14:439–45. doi: 10.1038/sj.leu.2401689. [DOI] [PubMed] [Google Scholar]
- 24.Fisher RJ, Mavrothalassitis G, Kondoh A, Papas TS. High-Affinity DNA-Protein Interactions of the Cellular Ets1 Protein: The Determination of the Ets Binding Motif. Oncogene. 1991;6:2249–54. [PubMed] [Google Scholar]
- 25.Spyropoulos D, Pharr P, Lavenburg K, Jackers P, Papas T, Ogawa M, Watson DK. Hemorrhage, Impaired Hematopoiesis, and Lethality in Mouse Embryos Carrying a Targeted Disruption of the Fli1 Transcription Factor. Mol Cell Biol. 2000;20:5643–5652. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baud V, Lipinski M, Rassart E, Poliquin L, Bergeron D. The Human Homolog of the Mouse Common Viral Integration Region, Fli1, Maps to 11q23-Q24. Genomics. 1991;11:223–4. doi: 10.1016/0888-7543(91)90124-w. [DOI] [PubMed] [Google Scholar]
- 27.Hart A, Melet F, Grossfeld P, Chien K, Jones C, Tunnacliffe A, Favier R, Bernstein A. Fli-1 Is Required for Murine Vascular and Megakaryocytic Development and Is Hemizygously Deleted in Patients with Thrombocytopenia. Immunity. 2000;13:167–77. doi: 10.1016/s1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- 28.Breton-Gorius J, Favier R, Guichard J, Cherif D, Berger R, Debili N, Vainchenker W, Douay L. A New Congenital Dysmegakaryopoietic Thrombocytopenia (Paris-Trousseau) Associated with Giant Platelet Alpha-Granules and Chromosome 11 Deletion at 11q23. Blood. 1995;85:1805–14. [PubMed] [Google Scholar]
- 29.Raslova H, Komura E, Le Couedic JP, Larbret F, Debili N, Feunteun J, Danos O, Albagli O, Vainchenker W, Favier R. Fli1 Monoallelic Expression Combined with Its Hemizygous Loss Underlies Paris-Trousseau/Jacobsen Thrombopenia. J Clin Invest. 2004;114:77–84. doi: 10.1172/JCI21197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivera RR, Stuiver MH, Steenbergen R, Murre C. Ets Proteins: New Factors That Regulate Immunoglobulin Heavy-Chain Gene Expression. Mol Cell Biol. 1993;13:7163–9. doi: 10.1128/mcb.13.11.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Eddy A, Teng YT, Fritzler M, Kluppel M, Melet F, Bernstein A. An Immunological Renal Disease in Transgenic Mice That Overexpress Fli-1, a Member of the Ets Family of Transcription Factor Genes. Mol Cell Biol. 1995;15:6961–70. doi: 10.1128/mcb.15.12.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georgiou P, Maroulakou I, Green J, Dantis P, Romanospica V, Kottaridis S, Lautenberger J, Watson DK, Papas T, Fischinger P, Bhat N. Expression of Ets Family of Genes in Systemic Lupus Erythematosus and Sjogren’s Syndrome. Int J of Oncology. 1996;9:9–18. [PubMed] [Google Scholar]
- 33.Zhang XK, Gallant S, Molano I, Moussa OM, Ruiz P, Spyropoulos DD, Watson DK, Gilkeson G. Decreased Expression of the Ets Family Transcription Factor Fli-1 Markedly Prolongs Survival and Significantly Reduces Renal Disease in Mrl/Lpr Mice. J Immunol. 2004;173:6481–9. doi: 10.4049/jimmunol.173.10.6481. [DOI] [PubMed] [Google Scholar]
- 34. [(accessed 2007 Dec 28)];Qiagen, Dneasy Blood & Tissue Handbook. (updated 2006 July) http://www1.qiagen.com/literature/handbooks.
- 35.Miltenyi Biotec. [(accessed 2007 Dec 28)];Cd43 (Ly-48) Microbeads, Mouse Datasheet. (updated 2006) http://www.miltenyibiotec.com/download/datasheets/53/DS130-049-801.
- 36.Miltenyi Biotec. [(accessed 2008 Jan 21)];Cd90 (Thy1.2) Microbeads Datasheet. http://www.miltenyibiotec.com/download/datasheets/46/DS130-049-101.pdf.
- 37.Melchers F, Braun V, Galanos C. The Lipoprotein of the Outer Membrane of Escherichia Coli: A B-Lymphocyte Mitogen. J Exp Med. 1975;142:473–82. doi: 10.1084/jem.142.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peden-Adams M, Alonso K, Godard C, Skipper S, Mashburn W, Hoover J, Charbonneau C, Henshel D, Dickerson R. Effects of Environmentally Relevant Concentrations of 2,3,7,8-Tcdd on Domestic Chicken Immune Function and Cyp450 Activity: F1 Generation and Egg Injection Studies. Chemosphere. 1998;37:1923–39. doi: 10.1016/s0045-6535(98)00259-8. [DOI] [PubMed] [Google Scholar]
- 39.Dziarski R. Studies on the Mechanism of Peptidoglycan- and Lipopolysaccharide-Induced Polyclonal Activation. Infect Immun. 1982;35:507–14. doi: 10.1128/iai.35.2.507-514.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. [(accessed 2008 Jan 4)];eBioscience, Intracellullar Immunofluorescent Staining for Flow Cytometry. http://www.ebioscience.com/ebioscience/appls/FCI.htm.
- 41. [(accessed 2008 Jan 4)];eBioscience, Elisa, Cytokine Antibody Pairs. http://www.ebioscience.com/ebioscience/appls/ELISApr.htm.
- 42. [(accessed 2007 Dec 31)];Invitrogen, Guides: Linnea Protocols: Nucleic Acid Purification and Analysis: Isolation of Rna Using Trizol Reagent. http://www.invitrogen.com.
- 43.Bio-Rad Laboratories. [(accessed 2007 Dec 31)];Life Science Research: Literature/Software: Smartspec Plus Spectrophotometer Instruction Manual. http://www.bio-rad.com.
- 44.Invitrogen. [(accessed 2007 Dec 31)];Superscript Indirect Rna Amplification System User Manual. (updated 2006 sAug 9) http://www.invitrogen.com/content/sfs/manuals/superscript_indirecte_rnaamp_man.pdf.
- 45.SuperArray Bioscience. [(accessed 2007 Dec 31)];Rt2 Profiler Pcr Array System User Manual. (updated 2007 Nov 30) http://www.superarray.com/manual/pcrarray.pdf.
- 46.SuperArray Bioscience. [(accessed 2007 Dec 31)];Rt2 Rna Qc Pcr Arrays User Manual. (updated 2007 Aug 31) http://www.superarray.com/manual/rnaqcplate.pdf.
- 47.SuperArray Bioscience. [(accessed 2007 Dec 31)];Rt2 Qpcr Primer Assays User Manual. (updated 2007 Jun 22) http://www.superarray.com/manual/realtimePCR.pdf.
- 48.Mao L, Zheng WJ. Combining Comparative Genomics with De Novo Motif Discovery to Identify Human Transcription Factor DNA-Binding Motifs. BMC Bioinformatics. 2006;7(Suppl 4):S21. doi: 10.1186/1471-2105-7-S4-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. [(accessed 2007 Dec 31)];Mdframe: A Computational Framework for the Automatic Identification of Human Regulatory Motifs. http://genomebioinfo.musc.edu/MDframe.html.
- 50.Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous Murine Lupus-Like Syndromes. Clinical and Immunopathological Manifestations in Several Strains. J Exp Med. 1978;148:1198–215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dixon FJ, Andrews BS, Eisenberg RA, McConahey PJ, Theofilopoulos AN, Wilson CB. Etiology and Pathogenesis of a Spontaneous Lupus-Like Syndrome in Mice. Arthritis Rheum. 1978;21:S64–7. doi: 10.1002/art.1780210909. [DOI] [PubMed] [Google Scholar]
- 52.Parkhouse RM, Preece G, Sutton R, Cordell JL, Mason DY. Relative Expression of Surface Igm, Igd and the Ig-Associating Alpha(Mb-1) and Beta(B-29) Polypeptide Chains. Immunology. 1992;76:535–40. [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmad-Nejad P, Hacker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial Cpg-DNA and Lipopolysaccharides Activate Toll-Like Receptors at Distinct Cellular Compartments. Eur J Immunol. 2002;32:1958–68. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 54.Means TK, Luster AD. Toll-Like Receptor Activation in the Pathogenesis of Systemic Lupus Erythematosus. Ann N Y Acad Sci. 2005;1062:242–51. doi: 10.1196/annals.1358.027. [DOI] [PubMed] [Google Scholar]
- 55.Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. Tlr9 Is Localized in the Endoplasmic Reticulum Prior to Stimulation. J Immunol. 2004;173:1179–83. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dasari P, Nicholson IC, Hodge G, Dandie GW, Zola H. Expression of Toll-Like Receptors on B Lymphocytes. Cell Immunol. 2005;236:140–5. doi: 10.1016/j.cellimm.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 57.Eaton-Bassiri A, Dillon SB, Cunningham M, Rycyzyn MA, Mills J, Sarisky RT, Mbow ML. Toll-Like Receptor 9 Can Be Expressed at the Cell Surface of Distinct Populations of Tonsils and Human Peripheral Blood Mononuclear Cells. Infect Immun. 2004;72:7202–11. doi: 10.1128/IAI.72.12.7202-7211.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raveche ES, Steinberg AD, DeFranco AL, Tjio JH. Cell Cycle Analysis of Lymphocyte Activation in Normal and Autoimmune Strains of Mice. J Immunol. 1982;129:1219–26. [PubMed] [Google Scholar]
- 59.Kawanishi H, Joseph K. Effects of Phorbol Myristate and Ionomycin on in Vitro Growth of Aged Peyer’s Patch T and B Cells. Mech Ageing Dev. 1992;65:289–300. doi: 10.1016/0047-6374(92)90042-c. [DOI] [PubMed] [Google Scholar]
- 60.Frasca D, Nguyen D, Riley RL, Blomberg BB. Effects of Aging on Proliferation and E47 Transcription Factor Activity Induced by Different Stimuli in Murine Splenic B Cells. Mech Ageing Dev. 2003;124:361–9. doi: 10.1016/s0047-6374(03)00009-5. [DOI] [PubMed] [Google Scholar]
- 61.Papadimitraki ED, Choulaki C, Koutala E, Bertsias G, Tsatsanis C, Gergianaki I, Raptopoulou A, Kritikos HD, Mamalaki C, Sidiropoulos P, Boumpas DT. Expansion of Toll-Like Receptor 9-Expressing B Cells in Active Systemic Lupus Erythematosus: Implications for the Induction and Maintenance of the Autoimmune Process. Arthritis Rheum. 2006;54:3601–11. doi: 10.1002/art.22197. [DOI] [PubMed] [Google Scholar]
- 62.Maier H, Ostraat R, Parenti S, Fitzsimmons D, Abraham LJ, Garvie CW, Hagman J. Requirements for Selective Recruitment of Ets Proteins and Activation of Mb-1/Ig-Alpha Gene Transcription by Pax-5 (Bsap) Nucleic Acids Res. 2003;31:5483–9. doi: 10.1093/nar/gkg785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK, Trojanowska M. Fli-1 Inhibits Collagen Type I Production in Dermal Fibroblasts Via an Sp1-Dependent Pathway. J Biol Chem. 2001;276:20839–48. doi: 10.1074/jbc.M010133200. [DOI] [PubMed] [Google Scholar]
- 64.Darby TG, Meissner JD, Ruhlmann A, Mueller WH, Scheibe RJ. Functional Interference between Retinoic Acid or Steroid Hormone Receptors and the Oncoprotein Fli-1. Oncogene. 1997;15:3067–82. doi: 10.1038/sj.onc.1201503. [DOI] [PubMed] [Google Scholar]
- 65.Huang X, Hua J, Shen N, Chen S. Dysregulated Expression of Interleukin-23 and Interleukin-12 Subunits in Systemic Lupus Erythematosus Patients. Mod Rheumatol. 2007;17:220–3. doi: 10.1007/s10165-007-0568-9. [DOI] [PubMed] [Google Scholar]
- 66.Houssiau FA, Mascart-Lemone F, Stevens M, Libin M, Devogelaer JP, Goldman M, Renauld JC. Il-12 Inhibits in Vitro Immunoglobulin Production by Human Lupus Peripheral Blood Mononuclear Cells (Pbmc) Clin Exp Immunol. 1997;108:375–80. doi: 10.1046/j.1365-2249.1997.d01-1009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tyrrell-Price J, Lydyard PM, Isenberg DA. The Effect of Interleukin-10 and of Interleukin-12 on the in Vitro Production of Anti-Double-Stranded DNA Antibodies from Patients with Systemic Lupus Erythematosus. Clin Exp Immunol. 2001;124:118–25. doi: 10.1046/j.1365-2249.2001.01466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hagiwara E, Okubo T, Aoki I, Ohno S, Tsuji T, Ihata A, Ueda A, Shirai A, Okuda K, Miyazaki J, Ishigatsubo Y. Il-12-Encoding Plasmid Has a Beneficial Effect on Spontaneous Autoimmune Disease in Mrl/Mp-Lpr/Lpr Mice. Cytokine. 2000;12:1035–41. doi: 10.1006/cyto.1999.0662. [DOI] [PubMed] [Google Scholar]
- 69.Airoldi I, Di Carlo E, Banelli B, Moserle L, Cocco C, Pezzolo A, Sorrentino C, Rossi E, Romani M, Amadori A, Pistoia V. The Il-12rbeta2 Gene Functions as a Tumor Suppressor in Human B Cell Malignancies. J Clin Invest. 2004;113:1651–9. doi: 10.1172/JCI20303. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Kowalczyk D, Mytar B, Zembala M. Cytokine Production in Transient Hypogammaglobulinemia and Isolated Iga Deficiency. J Allergy Clin Immunol. 1997;100:556–62. doi: 10.1016/s0091-6749(97)70150-7. [DOI] [PubMed] [Google Scholar]
- 71.Lee SH, Park SH, Min JK, Kim SI, Yoo WH, Hong YS, Park JH, Cho CS, Kim TG, Han H, Kim HY. Decreased Tumour Necrosis Factor-Beta Production in Tnfb*2 Homozygote: An Important Predisposing Factor of Lupus Nephritis in Koreans. Lupus. 1997;6:603–9. doi: 10.1177/096120339700600708. [DOI] [PubMed] [Google Scholar]
- 72.Wu X, Peng SL. Toll-Like Receptor 9 Signaling Protects against Murine Lupus. Arthritis Rheum. 2006;54:336–42. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- 73.Venkataraman L, Francis DA, Wang Z, Liu J, Rothstein TL, Sen R. Cyclosporin-a Sensitive Induction of Nf-at in Murine B Cells. Immunity. 1994;1:189–96. doi: 10.1016/1074-7613(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 74.Healy JI, Dolmetsch RE, Lewis RS, Goodnow CC. Quantitative and Qualitative Control of Antigen Receptor Signalling in Tolerant B Lymphocytes. Novartis Found Symp. 1998;215:137–44. doi: 10.1002/9780470515525.ch10. discussion 144-5, 186–90. [DOI] [PubMed] [Google Scholar]
- 75.Yoshida H, Nishina H, Takimoto H, Marengere LE, Wakeham AC, Bouchard D, Kong YY, Ohteki T, Shahinian A, Bachmann M, Ohashi PS, Penninger JM, Crabtree GR, Mak TW. The Transcription Factor Nf-Atc1 Regulates Lymphocyte Proliferation and Th2 Cytokine Production. Immunity. 1998;8:115–24. doi: 10.1016/s1074-7613(00)80464-1. [DOI] [PubMed] [Google Scholar]
- 76.Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, Gress TM, Ellenrieder V. Overexpression of C-Myc in Pancreatic Cancer Caused by Ectopic Activation of Nfatc1 and the Ca2+/Calcineurin Signaling Pathway. Embo J. 2006;25:3714–24. doi: 10.1038/sj.emboj.7601246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fu L, Lin-Lee YC, Pham LV, Tamayo A, Yoshimura L, Ford RJ. Constitutive Nf-Kappab and Nfat Activation Leads to Stimulation of the Blys Survival Pathway in Aggressive B-Cell Lymphomas. Blood. 2006;107:4540–8. doi: 10.1182/blood-2005-10-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kneitz C, Goller M, Tony H, Simon A, Stibbe C, Konig T, Serfling E, Avots A. The Cd23b Promoter Is a Target for Nf-at Transcription Factors in B-Cll Cells. Biochim Biophys Acta. 2002;1588:41–7. doi: 10.1016/s0925-4439(02)00114-x. [DOI] [PubMed] [Google Scholar]
- 79.Bansal A, Roberts T, Hay EM, Kay R, Pumphrey RS, Wilson PB. Soluble Cd23 Levels Are Elevated in the Serum of Patients with Primary Sjogren’s Syndrome and Systemic Lupus Erythematosus. Clin Exp Immunol. 1992;89:452–5. doi: 10.1111/j.1365-2249.1992.tb06979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brizard A, Morel F, Lecron JC, Dreyfus B, Brizard F, Barra A, Preud’homme JL. Proliferative Response of B Chronic Lymphocytic Leukemia Lymphocytes Stimulated with Il2 and Soluble Cd23. Leuk Lymphoma. 1994;14:311–8. doi: 10.3109/10428199409049683. [DOI] [PubMed] [Google Scholar]
- 81.Klashman DJ, Martin RA, Martinez-Maza O, Stevens RH. In Vitro Regulation of B Cell Differentiation by Interleukin-6 and Soluble Cd23 in Systemic Lupus Erythematosus B Cell Subpopulations and Antigen-Induced Normal B Cells. Arthritis Rheum. 1991;34:276–86. doi: 10.1002/art.1780340305. [DOI] [PubMed] [Google Scholar]
- 82.Swendeman SL, Thorley-Lawson D. Soluble Cd23/Blast-2 (S-Cd23/Blast-2) and Its Role in B Cell Proliferation. Curr Top Microbiol Immunol. 1988;141:157–64. doi: 10.1007/978-3-642-74006-0_21. [DOI] [PubMed] [Google Scholar]
- 83.Fournier S, Rubio M, Delespesse G, Sarfati M. Role for Low-Affinity Receptor for Ige (Cd23) in Normal and Leukemic B-Cell Proliferation. Blood. 1994;84:1881–6. [PubMed] [Google Scholar]
- 84.Militi S, Chiapparino C, Testa U, Carminati P, De Santis R, Serlupi-Crescenzi O. Role of Il-6 and Cd23 in the Resistance to Growth Arrest and Apoptosis in Lcl41 B Lymphoma Cells. Cytokine. 2005;31:314–23. doi: 10.1016/j.cyto.2005.05.007. [DOI] [PubMed] [Google Scholar]