

Abstract
Certain kindreds with low-penetrant (lp) retinoblastoma carry mutant alleles which retain partial tumor suppressor activity and we previously showed that these alleles exhibit defective, temperature-sensitive binding in yeast. To investigate the molecular basis for incomplete penetrance, we studied three recurrent lp alleles and observed approximately 50% of wildtype activity measured by i) phosphorylation at key regulatory sites, S780, S795, S807/S811, ii) transcriptional co-activation, and iii) ‘flat-cell’ differentiation in mammalian cells in vivo. In addition, we studied a small-cell carcinoma that is homozygous for the R661W allele providing the first analysis of the effect of a naturally occurring lp allele in a human tumor. While we detected abundant expression of the R661W protein, we noted marked instability of both endogenous and recombinant R661W following treatment in vivo with the Hsp90 inhibitor, geldanamycin, and stabilization of R661W following heat shock. In addition, we observed a discordant phenotype in the tumor cells with induction of p16 and loss of cyclin D1 consistent with a null RB status combined with homozygous expression of mutant ras which had not been reported previously for RB (-) small-cell cancer. These findings show that a recurrent missense lp allele retains greater functional activity in vivo than predicted from earlier in vitro assays, proposing a role for stabilizing chaperone-like activity in vivo. In addition, these data suggest that reversible protein instability and the requirement for a cooperating mutation may provide a stochastic explanation for the molecular basis of incomplete penetrance in kindreds carrying these alleles.
Keywords: low penetrance, retinoblastoma, Hsp90 inhibition, geldanamycin, small-cell cancer
Introduction
While inheritance of an RB null allele is invariably associated with the early onset of multifocal retinal tumors, certain retinoblastoma families demonstrate a phenotype of low-penetrance (lp) with reduced or absent tumor formation in obligate carriers. We and others previously studied the biochemical properties of the lp R661W allele and observed that the recombinant mutant protein had unique functional features. For example, while RB mutants isolated from classic retinoblastoma or adult tumors were null for all parameters of RB function 1, the R661W mutant was defective for E2F1 binding and for inducing G1 cell cycle arrest, but retained the wildtype ability i) to partially suppress colony growth of RB(-) cells, ii) to induce parameters of cell differentiation, and iii) to weakly bind to SV40 large T. 2-4 These observations were the first demonstration that the cell cycle arrest and cell differentiation functions of the RB product were separable and might be mediated by differential protein binding patterns. Remarkably, we observed that two other RB mutants from different families with lp phenotypes, C712R and deletion of codon 480, exhibited identical biochemical properties in vitro which suggested two broad classifications for lp RB alleles. 2, 3 Class 1 alleles referred to genetic or epigenetic alterations which reduced the steady-state levels of wild type RB protein such as RB gene promoter mutations 5, 6 or defects in exon splicing which may be predicted to reduce, but not eliminate, full length mature mRNA. 7 In contrast, class 2 alleles corresponded to defective RB products, such as R661W, which retained partial wildtype activity. Subsequent analyses of additional splice mutations involving N-terminal exons has predicted lp alleles with an interplay of class 1 and class 2 features to i) regulate gene transcription, ii) modulate normal or near-normal in-frame splice site selection, or iii) allow for an alternate downstream translational initiation with retention of functional activity. 7-9 Although class 1 gene promoter mutations represent the simplest model system to test the hypothesis that a minimum level of RB protein binding activity is essential for tumor suppression 5, these mutant alleles are uncommon events in lp retinoblastoma families 9, 10 and these analyses do not provide insight into functional domains of the RB product. Conversely, missense class 2 RB alleles have been commonly reported in lp families (summarized in Table 1) and provide the opportunity to define more precisely tumor suppressor activity within the RB product. To further study the basis for the phenotype of incomplete penetrance, we tested if alterations in site-specific cyclin-mediated phosphorylation could be linked to the discordant functional properties observed with these missense RB alleles. In addition, since we had previously reported that these mutants were temperature-sensitive in yeast cells for SV40 T binding, we examined the effect of temperature on protein binding and transcriptional co-activation in mammalian cells and on the induction of flat cell differentiation in RB (-) cells in vivo. Finally, small-cell carcinoma has a distinctive genetic signature characterized by mutational inactivation of RB combined with the absence of Ras mutations. 11 We have now studied a small-cell carcinoma that carries an endogenous R661W mutant allele providing the first analysis of an lp allele in human tumor development. We detected abundant steady-state levels of the R661W product in vivo with loss of the wildtype allele and 2n copy of the mutant allele. However, as compared to wt RB, we observed loss of immunoblot signal for the mutant protein following serial freeze-thaw cycles in vitro and subsequently demonstrated that the R661W protein was highly unstable following treatment in vivo with the Hsp90 inhibitor, geldanamycin. We also investigated cooperating genetic events within the epistatic RB/p16/cyclin D pathway and within the ras pathway. 1, 12 We observed induction of CDKN2a/p16 protein levels and loss of cyclin D1 protein that is characteristic for RB (-) tumors 13, providing evidence that the signaling pathways downstream of RB can ‘register’ the abundantly expressed R661W protein as null. In contrast, the detection of a co-expressed mutant K-ras allele is a rare event in small-cell cancer 1, 11, which includes approximately 300 cases of small-cell cancers tabulated from the literature in the Catalogue of Somatic Mutation in Cancer database 14 without H-, N-, or K-ras mutations. In summary, these data provide the first evidence that certain lp RB alleles may be stabilized and retain greater functional activity in vivo than predicted from earlier in vitro assays. Therefore, the requirement for additional cooperating events to block this residual activity may provide a stochastic explanation for the molecular basis of incomplete penetrance in kindreds carrying these alleles. Finally, while GA has been proposed as an important tool to destabilize gain-of-function oncogene signaling pathways, these data unexpectedly suggests that GA may also serve to unmask important chaperone stabilizing effects of an otherwise silent mutant tumor suppressor gene.
Table 1. Kindreds with common class 2 lp RB alleles.
| Mutation | # fam | ob ca/i-fam | no t / uni / bi | references |
|---|---|---|---|---|
| 661W | 24 | 81 / 9 | 50/ 25 / 6 | 9, 35, 45-49 |
| 712R | 4 | 14 / 2 | 7/ 6 / 1 | 49-51 |
| Δ480 | 1 | 5 / 1 | 1/ 3 / 1 | 47 |
ob ca: # obligate carriers; i-fam: # informative families; no t/uni/bi: no, unilateral, bilateral retinal tumors in obligate carriers
Results
Low-penetrant RB alleles retain CDK4:cyclin D site-specific phosphorylation in vivo
We had previously shown that three lp RB alleles, which have now been detected in 29 different families (Table 1), exhibit loss of E2F1 binding and loss of G1 arrest activity, but retain the ability to induce certain parameters of cellular differentiation in RB (-) cells. 4 A series of tandem carboxyl-terminal phosphorylation sites at S780, S795, S807, and S811 have been shown to serve as specific substrates for the CDK4:cyclin D family in vivo and were shown to regulate the ability of RB to mediate induction of cell differentiation. 15-18 We were interested, therefore, to examine if retention of partial tumor suppression could be linked with a change in site-specific phosphorylation of individual regulatory serine residues. We tested cyclin D1, 2, and 3 and cyclin E mediated phosphorylation at these sites in wildtype (wt) RB and the lp RB mutants by transiently co-expressing the indicated plasmids into H2009 RB (-) cells and subjecting the lysates to sequential immunoprecipitation with a pan-RB monoclonal antibody followed by imunoblotting with phospho-specific antibodies. As expected, we observed that the null C706F mutant isolated from a small-cell cancer sample 19 showed abundant levels of steady-state protein with negligible phosphorylation at all serine sites tested (Figure 1A). In contrast, we observed that the Δ480 and 661W RB mutants showed phosphorylation at the CDK4:cyclin D sites, S-780, S-795, and S807/811 with a pattern that was qualitatively and quantitatively similar to wt RB. While we observed that the phosphorylation of the 712R mutant was reduced compared to the Δ480 and 661W plasmids, this was consistent with our earlier published binding data, reproduced in Fig 1B, that showed that the 712R allele had the weakest level of pocket binding activity to SV40 Tag when measured in a quantitative yeast 2-hybrid solution-based assay. 3 Since transient transfection can result in cells that are growth arrested at 48-72 hours with partial inhibition of endogenous CDK:cyclin activation, ectopic co-expression of cyclins D or E are required to efficiently score the ability to undergo cyclin-mediated phosphorylation. Therefore, to confirm site-specific phosphorylation of lp alleles in the absence of ectopic cyclins, we re-examined these mutants by testing them in a mass culture of stable G418 resistant lp allele transfectants. Using similar immunoblot analyses, we detected steady-state phosphorylation at the CDK sites S780, S795 and S807/S811 which were qualitatively similar for both wt RB and for each of the stable Δ480, 661W and 712R transfectants and absent phosphorylation for the C706F mutant (Fig. 2). These data suggest that defective RB pocket binding activity that can fluctuate around 10-20% of wildtype levels, as measured in a yeast-two hybrid binding system, is still sufficient to serve as substrate for cdk:cyclin-mediated phosphorylation at these key regulatory sites.
Figure 1.

In vivo RB phosphorylation at S780, S795 and S807/S811 following transient co-transfections with cyclins D and E. Panel A) H2009 RB(-) cells were transiently transfected with parental RB vector alone (vector), wildtype RB (wt), or indicated lp mutants along with parental cyclin vector alone (-) or indicated cyclin plasmids. At 72 hours, lysates were subjected to sequential immunoprecipitation with a pan-RB antibody (G3-245) followed by immunoblotting using G3-245 for total RB protein levels or the indicated RB phospho-specific antibodies. Panel B) Yeast two-hybrid assay of mutant and wt RB cDNA to SV40 large T antigen using previously reported beta-galactosidase binding assay. 3
Figure 2.
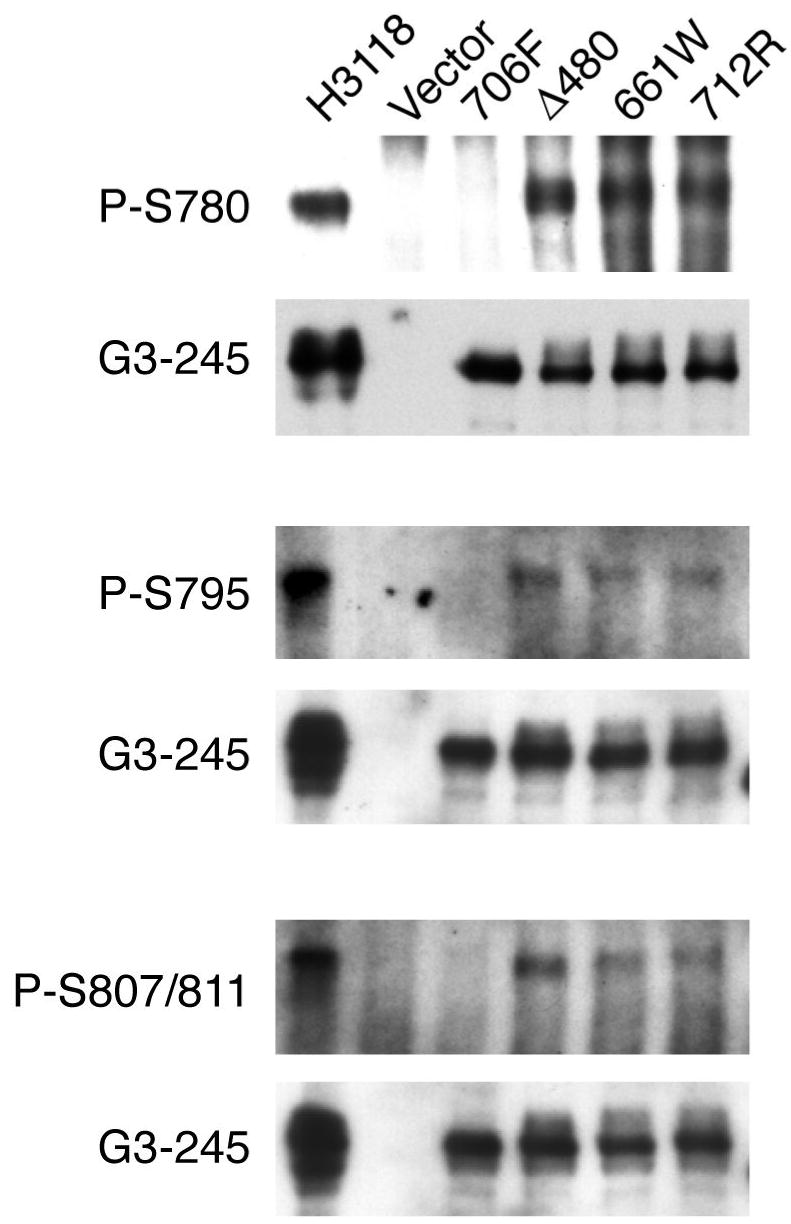
In vivo RB phosphorylation at S780, S795, and S807/S811 using stable low-penetrant transfectants in absence of ectopic cyclins. G418 resistant clones were propagated from stable transfectants using the indicated RB mutant plasmids and lysates were subjected to sequential immunoprecipitation with G3-245 followed by imunoblotting with the indicated pan-RB or phospho-specific antibodies.
lp RB alleles exhibit substantial functional activity in mammalian cells
To extend the observation that lp RB alleles were temperature-sensitive for SV40 T binding in yeast cells 3 and to test if these mutant alleles can show detectable binding activity in mammalian cells grown under physiological temperature conditions, we employed a mammalian two-hybrid assay and compared activity with wt RB and the null C706F mutant for binding with the myogenic transcription factor, MyoD. We selected MyoD since there is evidence linking the ability of RB, with an intact pocket binding domain, to serve as a co-activator of MyoD during cell differentiation 4,20-22, and we wished to examine if stress from minor alterations in incubation temperature could destabilize this residual functional activity. We observed that wt RB fused to the Gal4 binding domain (BD) co-transfected with an empty parental activation domain (AD) control (Figure 3) resulted in a 13-fold activation of the luciferase reporter when compared to the negligible levels obtained with the C706F-BD plasmid. Since the C706F plasmid differs from wt RB by only a single amino acid substitution that renders the RB ‘pocket’ binding null 19, this observation suggests that luciferase induction by wt RB is mediated by ‘pocket-dependent’ activating nuclear factor(s) present in the H2009 cell extract. In contrast to the 706F mutant, each of the three different lp RB plasmids, Δ480, 661W and 712R-BD, showed luciferase activation that was comparable to the levels observed for wt RB (70-80% of wt levels for each of the lp alleles). When wt RB-BD and MyoD-AD were co expressed, luciferase activity was increased approximately 2 fold, while expression of MyoD-AD with the null 706F-BD again showed negligible, background levels. This 2-fold increase in reporter activity is comparable to previous studies that have examined the co-activation of RB and myoD 23. Expression of MyoD-AD with each of the lp mutants again showed a weaker level of enhanced activity (Fig. 3, lanes 6, 8, 10). We repeated the assay with cells growing at different incubation temperatures, however, we were unable to detect variations in pocket binding levels. These data demonstrate that there is substantially greater binding activity for these lp alleles when measured in vivo in mammalian cells as compared to in vitro GST-based binding assays (5% binding compared to wt 2, 24) suggesting the presence of stabilizing chaperone-like elements. We also tested the ability of the lp mutants to induce morphological differentiation at different incubation temperatures by scoring for the phenotype of ‘flat cells’ 4 after 2 weeks of selection in G418. The lp mutants induced ‘flat cell’ morphology 25 at intermediate levels (40-60% of wt RB) as predicted, however, without significant alterations within the narrow temperature range which could be tested due to loss of cell viability in control cells at higher and lower temperatures.
Figure 3.

lp alleles retain significant functional activity. A) Luciferase activity of H2009 RB(-) cells co-transfected with the pG5-luciferase reporter, wildtype RB (wt) or the indicated low-penetrant RB mutants fused in-frame to a Gal4 binding domain, and either an empty VP16 activation domain (AD) or the AD fused in-frame with MyoD (AD-MyoD) as described in Material and Methods. Cells were incubated at the indicated temperatures for 72 hrs before luciferase double reporter assay measurements. Error bars indicate standard errors from two independent experiments performed in triplicate. B) SaOS2 RB(-) cells were transfected with the indicated RB plasmids and the number of ‘flat cells’ per 10 high power fields (100×) following 2 weeks of G418 selection was determined by manual counting and normalized to the number of ‘flat cells’ 25 induced by wild-type (wt) RB. Error bars indicate standard errors from three independent experiments performed in triplicate.
Effect of Hsp90 inhibition on a naturally occurring R661W allele in small-cell cancer
Samples from tumors in lp families from retinoblastoma registries have not been available to us for laboratory analyses partly due to the observation that children from these kindreds are aggressively screened and clinically managed with orbit-sparing treatments. An alternate approach to study the effect of a naturally occurring lp RB allele in human tumorigenesis is to identify sporadic tumors with an acquired somatic mutation. Small-cell lung cancer and extrapulmonary small-cell cancer are the only other tumor types, besides retinoblastoma, where the RB gene is frequently targeted for mutational inactivation 26, however, we were unable to detect a de novo lp mutation in our tumor collection samples. In contrast, we noted in the Sanger Cancer Mutation (COSMIC) database 14 that the R661W mutation was detected in an extrapulmonary small-cell cancer and we obtained the original tumor cell line (ECC4) from this patient. 27 ECC4 cells exhibit features of a neuroendocrine small-cell tumor with expression of elevated levels of creatine kinase-B and neuron-specific decarboxylase, show pure small-cell histology when grown as xenograft transplants 27, and grow in culture as tight spheroids consisting of approximately 100 cells characteristic of the growth patterns of retinoblastoma and small-cell cancer (data not shown). The presence of an lp allele in this tumor cell line, therefore, allowed us to test for the first time i) if the gene product is expressed during tumorigenesis or if the steady-state levels are reduced, ii) if the wt allele is retained or deleted in the tumor and if the locus shows gain or loss of the mutant allele, iii) if the epistatic RB/p16/cyclin D pathway ‘senses” the 661W product as null or wildtype, and iv) if there are cooperating mutational events that are normally not required in the setting of classic null RB mutations of small-cell carcinomas. We performed immunoblot analysis of two stocks of ECC4 cells and showed that the R661W product was abundantly expressed in these cells at steady-state levels that were comparable to levels detected in other lung tumor cell lines known to express wt RB (Fig 4A). However, we observed loss of R661W immunoblot signal following serial freeze-thaw cycles of the whole cell protein extracts when stored in vitro despite the addition of standard protease and phosphatase inhibitors, while no effect was observed with stored extracts of wildtype RB under identical conditions (data not shown). This result was in agreement with others who noted reduced levels of R661W protein 28 which suggested to us that protein instability in stored cellular extracts might partly explain the enhanced functional activity of lp alleles detected in vivo as compared to in vitro binding assays. In addition, this finding suggested the hypothesis that the stability of R661W in vivo may depend on cellular chaperones in vivo. To investigate this possibility, we harvested ECC4 cells and wt RB control cells (A549) after overnight incubation with the Hsp90 inhibitor geldanamycin (GA). We observed a marked decrease in steady-state R661W protein expression while no effect was detected with wt RB (Fig 4B). To test if this effect on protein instability could be detected after shorter periods of drug exposure, we incubated the cells to GA for 4, 8, and 16 h GA and observed a detectable effect on RB stability within 4 h of incubation (Fig 4C). In addition, we observed that incubation with the proteasome inhibitor, bortezomib (velcade, Vcd), had little effect on RB protein stability, nor did it reverse the effect of GA. Since GA specifically binds to Hsp90 to inhibit function of the Hsp90 super-chaperone complex 29, these observations suggest that Hsp90 participates in stabilizing mutated RB protein. We therefore tested the effect of up-regulating this chaperone complex by heat shocking cells for 30 min followed by a 4h recovery time and we observed a modest increase in the R661W immunoblot signal with no change or a slight decrease signal with wt RB.
Figure 4.

Effect of Hsp90 inhibition on R661W stability. A) RB protein immunoblot using lysates from two ECC4 aliquots (lanes 1,2) compared with whole cell extracts from three different lung cancer cell lines with known wildtype (lanes 3,4) or absent RB (H2009, lane 5) expression. B) Immunoblot of ECC4 or control wt RB cells (A549) treated for 16 h with DMSO vehicle or geldanamycin (GA). C) Immunoblot of A549 and ECC4 cells treated with indicated drugs or subjected to heat shock (HS) at 42°C for 30 min followed by 4 h recovery. D) RB (-) cells H2009 cells were transiently transfected with indicated plasmids and subjected to GA and/or velcade (Vcd) exposure for 16 h followed by harvesting and immunoblot analysis. E) Immunoblot of A549 and ECC4 cells incubated in the presence or absence of 100ug/ml CHX and/or 1uM GA for the indicated time points.
To independently confirm the effect of GA exposure on the stability of the mutant lp RB protein, we transfected an RB(-) cell line with either the CMV promoter driven expression vector alone, wt RB, C706F, or R661W plasmids and incubated the cells overnight with the indicated drugs prior to harvesting for immunoblot analysis. We again observed no effect of Hsp90 inhibition on the stability of wt RB, but found a detectable loss of signal for both missense point mutant RB products, confirming that both endogenous and recombinant mutant proteins are GA sensitive (Fig 4D). The proteasome inhibitor Vcd was again unable to rescue either RB mutant protein from the effects of GA. Finally, we estimated RB protein half-life by cycloheximide (CHX) incubation of A549 (wt RB) or ECC4 (R661W RB) cells in the presence or absence of GA followed by timed immunoblot analysis. We observed that the estimated half-life of the endogenous R661W RB protein in the absence of GA was > 8 hours (Figure 4E) which was comparable to the previously reported half-life of wt RB using 35S methionine labeling. 30 In contrast, incubation of ECC4 cells with GA resulted in a marked reduction in RB protein half-life to approximately 2 hours confirming the destabilizing effect of GA on the mutant RB product.
Co-expression of mutant K-ras with R661W RB in ECC4 cells
Since mutational inactivation of RB and CDKN2a/p16 are mutually exclusive events in human cancer 1, 12, we addressed the effect of the R661W mutation on the Cdk/Cyclin/p16 tumor suppressor pathway in ECC4 cells. Despite the abundant steady-state levels of R661W protein in these cells, we detected induction of CDKN2a/p16 protein and loss of cyclin D1 which is characteristic of the RB (-) phenotype of either small-cell carcinomas or E1A/HPV infected tumor cells 13, 31 and which suggest that the tumor cells ‘register’ the R661W allele as null 13 (Fig 5A, B). We performed DNA resequencing of the RB locus in ECC4 cells and confirmed the exon 20 C-to-T substitution (R661W) and absence of a wt signal showing loss of the wt allele (Fig 5C). We also performed oligonucleotide array-based CGH using the Agilent DNA microarray platform and showed 2n DNA content at the 13q14 locus suggesting that the mutant allele had undergone duplication to homozygosity (data not shown). In addition, no evidence for myc gene amplification or loss of chromosome 6 genes as previously reported for some retinoblastoma or small-cell tumors were detected by CGH (data not shown). Finally, to explain why co-expression of mutant K-ras has not been detected in RB (-) neuroendocrine small-cell tumors, prior studies have proposed that residual wt RB activity is required to exhibit the full tumorigenic effect of mutant ras. 32 However, in contrast to other neuroendocrine RB (-) small-cell tumors that have been tested to date, we confirmed the presence of a K-ras codon 61 mutation in the ECC4 cells (Fig 5D). This finding further supports the hypothesis that R661W is distinct from classic null RB mutations and retains residual wt activity.
Figure 5.
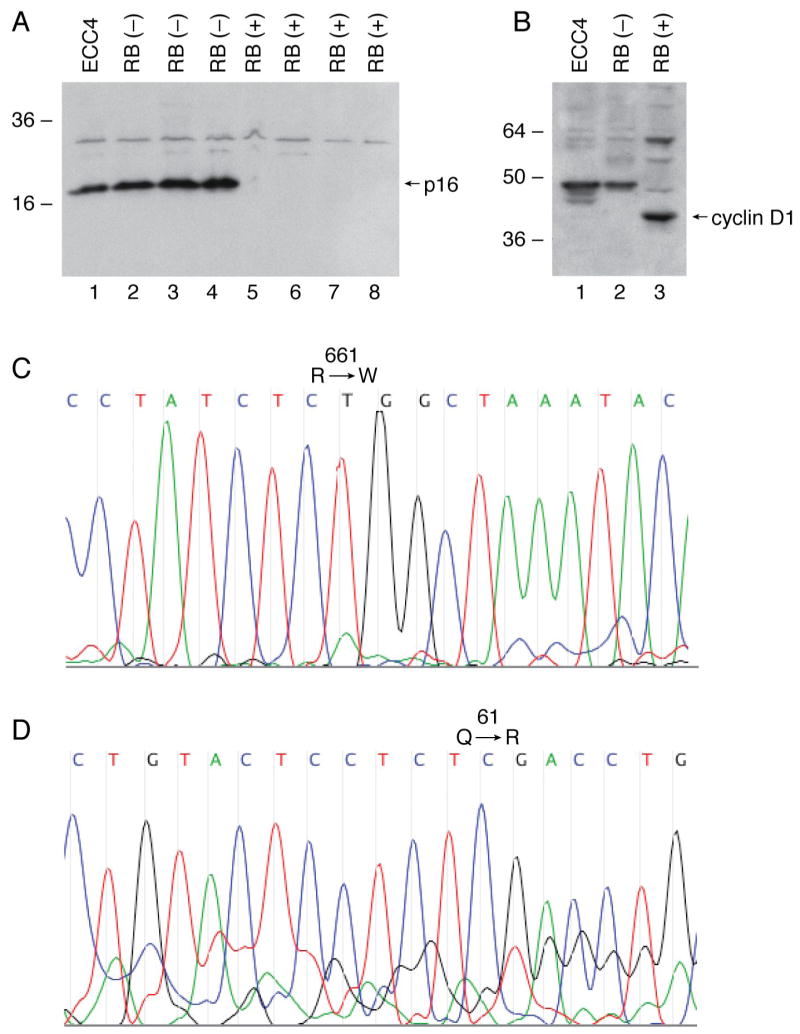
Discordant p16:Ki-ras cooperating events in ECC4 cells. Anti-CDKN2a/p16ink4a (Panel A) and anti-cyclin D1 (Panel D) immunoblots of ECC4 and a panel of lung cancer cells with known RB status as indicated. Re-sequencing chromatograph of exon 20 RB codon 661 (Panel C) and K-ras codon 61 (Panel D) in ECC4 small-cell tumor cells.
Discussion
Defining the molecular basis for the phenotype of incomplete penetrance of familial retinoblastoma is an important goal for both clinicians and geneticists. Although it has been argued that the prevalence of lp alleles might be overrepresented due to publication bias 33, the presence of cryptic lp germline alterations in ‘sporadic’ unilateral retinoblastoma may also be underestimated which can impact the optimal management strategy for these patients and families. 34 35 To study the phenotypic consequences of carrying lp germline genes, we have noted that these alleles separate into distinct biological categories which may need to be analyzed separately. For example, in-frame N- 36 or C-terminal 37 exon deletions located outside the central A-B pocket domain exhibit distinct biochemical properties from the del480, R661W, and C712R alleles which suggests heterogeneity within class 2 lp alleles. We have now shown that the most common lp alleles retain site-specific cyclin phosphorylation and functional activity in vivo that was substantially greater than predicted from earlier in vitro GST-precipitation methods. 24 Since the tumors that arise within lp retinoblastoma families are histologically and clinically as aggressive as tumors seen in patients with classic null RB 38 and since they often arise at a later age resembling the age of sporadic (two-hit) retinoblastoma 9, it is reasonable to predict that the reduced number of tumor foci in lp obligate carriers is due to the time required for an additional ‘event’, that is not otherwise required in patients with null alleles, to overcome this residual tumor suppressor activity. There are distinct types of genetic or epigenetic events that can be predicted including i) an alteration to further reduce RB gene transcription 5, ii) modulation of protein activity by disrupting a chaperone stabilizing function 3, or iii) the need for a cooperating mutation in another candidate gene to initiate an irreversible tumor phenotype 39. The study of the ECC4 tumor cell line provides an opportunity to begin to address some of these issues. For example, although we have shown that R661W is expressed in vivo at steady-state levels comparable to tumor cells with wt RB expression, the gene product is unstable following exposure to an Hsp90 inhibitor suggesting that reversible fluctuations in steady-state RB activity may participate in tumor development. In addition, we have observed a discordant cellular response in these cells with i) the loss of cyclin D1 protein and the induction of CDKN2a/p16 protein, characteristic of RB null cells 13, 40, and conversely ii) co-expression of mutant K-ras which is characteristic of RB wt cells and not observed in RB null small-cell tumors 14; Mitsudomi, 1991 #807}. Of note, prior studies suggested that the absence of Ras mutations in RB null tumors can be explained by data showing that escape from Ras-mediated senescence requires wt RB with loss of CDKN2a/p16 41, 42 and, alternatively, a recent study proposed that the full tumorigenic potential for mutant K-ras protein required the presence of wt RB activity. 32 While we have been unable to obtain lp tumor samples from retinoblastoma registries to test directly for additional candidate mutations, we did note a single study that detected Ras mutations in 4 retinoblastoma tumors (1 unilateral, 1 bilateral, and 2 samples with no further available clinical information). 43 In addition, a mouse knock-in model for the murine R661W equivalent has been generated and may provide in the future insight into lp tumorigenesis. 44 Ultimately, retinoblastoma registries should identify and collect tumor samples from all families with lp phenotypes to discover common patterns of cooperating genetic events and to define the spectrum of genotype:phenotype relationships within this important tumor suppressor pathway. In addition, while GA has been proposed as an important tool to destabilize gain-of-function oncogene signaling pathways, these data unexpectedly suggests that GA may also serve to unmask important chaperone stabilizing effects of an otherwise silent mutant tumor suppressor gene.
Materials and Methods
Plasmids
RB cDNA constructs representing full-length wildtype (wt), 706F, Δ480, 661W, 712R were generated by PCR-based site-directed mutagenesis and cloned into the pCI-neo vectors (Promega, Madison, WI) as previously described. 2 All recombinant constructs were confirmed by nucleotide sequencing. pBind-RB plasmids were constructed by subcloning the indicated mutant RB cDNA in-frame with the Gal4 DNA binding domain of pBind vector (Promega, Madison WI). A plasmid encoding MyoD was fused to the VP16 activation domain (pAct-MyoD) and the pG5luc vector containing the Gal4 binding site and the firefly luciferase gene were used as recommended by the manufacturer (Mammalian Checkmate, Promega). The Gal4 DNA-binding domain plasmid, pGBT9 (Clontech, Palo Alto, CA), containing the RB pocket domain was constructed as previously described. 3
In vivo RB1 phosphorylation analysis
The pCI-neo-RB plasmids were co-transfected with the indicated pRC-cyclin constructs (Invitrogen, Carlsbad, CA) into the RB (-) H2009 cell line. Protein extracts were harvested 72 hours post transfection and subjected to sequential immunoprecipitation using α-RB1 (G3-245, Pharmingen BD Biosciences, CA) followed by immunoblotting of the washed precipitates using the indicated phospho-site specific antibodies. Stable transfectants of SaOS2 cells in mass culture, in the absence of ectopic cyclins, were selected and expanded in the presence of 0.5mg/ml G418 and protein lysates were subjected to sequential immunoprecipitation and immunoblotting as described above. After immunostaining with phospho-specific antibodies, nitrocellulose membranes were stripped and re-blotted with α–RB1 (G3-245) to control for transfection efficiency. Phospho-S780, phospho-S795 and phospho-S807/S811 were purchased from New England Biolabs (Beverly, MA).
Mammalian in vivo assays
Cell lines were generated and propagated as previously described. 26 The ECC4 cell line was obtained from the Riken BioResource Center, Japan. RB (-) human H2009 cells or human SaOS2 cells were co-transfected using lipofectin reagent (Invitrogen) with the pBind-RB plasmids, pAct -MyoD and pG5luc. Cells were lysed at 72 hours post transfection and the luciferase activities were assayed as recommended by the manufacturer (Checkmate mammalian assay, Clontech). Three independent experiments were performed for each plasmid. The flat cell assay was performed as previously reported. 4 Briefly, SaOS2 cells were transfected with the indicated pCI-neo-RB plasmids and stable transfectants were selected in DMEM containing 10% FBS and G418 for 14 days at the indicated incubation temperatures. The minimal G418 concentration that would kill >99% of untransfected SAOS2 cells varied depending on the incubation temperature and was determined by titration at each temperature. The number of flat cells/ten high-power (100×) microscopic fields was determined manually. Each assay was performed in triplicate. To test protein stability in the presence of an Hsp90 inhibitor, wt RB control cells (A549) and mutant R661W cells were either treated with DMSO alone or 1 μM GA for 4, 8, or 16h (the GA was freshly diluted from a 10mM stock in DMSO). To test effects of a proteasome inhibitor, A549 and ECC4 cells were treated with DMSO alone or either 1 μM GA for 4 h, 50 nM velcade for 1 h, or both in combination. To test RB protein stability after heat shock, A549 and ECC4 cells were incubated at 42°C for 30 min (in temperature pre-equilibrated medium) and then allowed to recover at 37°C for 4 h. Following these treatments, cells were lysed and soluble protein was subjected to immunoblot analysis. To test protein half-lives, A549 and ECC4 cells were incubated in the presence or absence of 100ug/ml CHX and/or 1uM GA for the indicated time points. Cells were lysed and subjected to immunoblot analysis for Rb expression (Cell Signaling, cat #9309) and alpha-tubulin expression (Calbiochem, cat # CP06).
References
- 1.Kaye FJ. RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer. Oncogene. 2002;21:6908–14. doi: 10.1038/sj.onc.1205834. [DOI] [PubMed] [Google Scholar]
- 2.Otterson GA, Chen W, Coxon AB, Khleif SN, Kaye FJ. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc Natl Acad Sci U S A. 1997;94:12036–40. doi: 10.1073/pnas.94.22.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otterson GA, Modi S, Nguyen K, Coxon AB, Kaye FJ. Temperature-sensitive RB mutations linked to incomplete penetrance of familial retinoblastoma in 12 families. Am J Hum Genet. 1999;65:1040–6. doi: 10.1086/302581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG., Jr Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 1998;12:95–106. doi: 10.1101/gad.12.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakai T, Ohtani N, McGee TL, Robbins PD, Dryja TP. Oncogenic germ-line mutations in Sp1 and ATF sites in the human retinoblastoma gene. Nature. 1991;353:83–6. doi: 10.1038/353083a0. [DOI] [PubMed] [Google Scholar]
- 6.Ohtani-Fujita N, Dryja TP, Rapaport JM, Fujita T, Matsumura S, Ozasa K, Watanabe Y, Hayashi K, Maeda K, Kinoshita S, Matsumura T, Ohnishi Y, Hotta Y, Takahashi R, Kato MV, Ishizaki K, Sasaki MS, Horsthemke B, Minoda K, Sakai T. Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet Cytogenet. 1997;98:43–9. doi: 10.1016/s0165-4608(96)00395-0. [DOI] [PubMed] [Google Scholar]
- 7.Lohmann DR, Gallie BL. Retinoblastoma: revisiting the model prototype of inherited cancer. Am J Med Genet C Semin Med Genet. 2004;129:23–8. doi: 10.1002/ajmg.c.30024. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez-Sanchez F, Ramirez-Castillejo C, Weekes DB, Beneyto M, Prieto F, Najera C, Mittnacht S. Attenuation of disease phenotype through alternative translation initiation in low-penetrance retinoblastoma. Hum Mutat. 2007;28:159–67. doi: 10.1002/humu.20394. [DOI] [PubMed] [Google Scholar]
- 9.Taylor M, Dehainault C, Desjardins L, Doz F, Levy C, Sastre X, Couturier J, Stoppa-Lyonnet D, Houdayer C, Gauthier-Villars M. Genotype-phenotype correlations in hereditary familial retinoblastoma. Hum Mutat. 2007;28:284–93. doi: 10.1002/humu.20443. [DOI] [PubMed] [Google Scholar]
- 10.Fujita T, Ohtani-Fujita N, Sakai T, Rapaport JM, Dryja TP, Kato MV, Ishizaki K, Sasaki MS, Hotta Y, Maeda K, Kinoshita S, Ohnishi Y, Minoda K. Low frequency of oncogenic mutations in the core promoter region of the RB1 gene. Hum Mutat. 1999;13:410–1. doi: 10.1002/(SICI)1098-1004(1999)13:5<410::AID-HUMU10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 11.Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6:1353–62. [PubMed] [Google Scholar]
- 12.Otterson GA, Kratzke RA, Coxon A, Kim YW, Kaye FJ. Absence of p16INK4 protein is restricted to the subset of lung cancer lines that retains wildtype RB. Oncogene. 1994;9:3375–8. [PubMed] [Google Scholar]
- 13.Schauer IE, Siriwardana S, Langan TA, Sclafani RA. Cyclin D1 overexpression vs. retinoblastoma inactivation: implications for growth control evasion in non-small cell and small cell lung cancer. Proc Natl Acad Sci U S A. 1994;91:7827–31. doi: 10.1073/pnas.91.16.7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, Flanagan A, Teague J, Wooster R, Futreal PA, Stratton MR. Cosmic 2005. Br J Cancer. 2006;94:318–22. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331–42. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- 16.Pan W, Cox S, Hoess RH, Grafstrom RH. A cyclin D1/cyclin-dependent kinase 4 binding site within the C domain of the retinoblastoma protein. Cancer Res. 2001;61:2885–91. [PubMed] [Google Scholar]
- 17.Lees JA, Buchkovich KJ, Marshak DR, Anderson CW, Harlow E. The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. Embo J. 1991;10:4279–90. doi: 10.1002/j.1460-2075.1991.tb05006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. Embo J. 1996;15:7060–9. [PMC free article] [PubMed] [Google Scholar]
- 19.Kaye FJ, Kratzke RA, Gerster JL, Horowitz JM. A single amino acid substitution results in a retinoblastoma protein defective in phosphorylation and oncoprotein binding. Proc Natl Acad Sci U S A. 1990;87:6922–6. doi: 10.1073/pnas.87.17.6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–24. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- 21.De Falco G, Comes F, Simone C. pRb: master of differentiation. Coupling irreversible cell cycle withdrawal with induction of muscle-specific transcription. Oncogene. 2006;25:5244–9. doi: 10.1038/sj.onc.1209623. [DOI] [PubMed] [Google Scholar]
- 22.Kitzmann M, Fernandez A. Crosstalk between cell cycle regulators and the myogenic factor MyoD in skeletal myoblasts. Cell Mol Life Sci. 2001;58:571–9. doi: 10.1007/PL00000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen TT, Wang JY. Establishment of irreversible growth arrest in myogenic differentiation requires the RB LXCXE-binding function. Mol Cell Biol. 2000;20:5571–80. doi: 10.1128/mcb.20.15.5571-5580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kratzke RA, Otterson GA, Hogg A, Coxon AB, Geradts J, Cowell JK, Kaye FJ. Partial inactivation of the RB product in a family with incomplete penetrance of familial retinoblastoma and benign retinal tumors. Oncogene. 1994;9:1321–6. [PubMed] [Google Scholar]
- 25.Qin XQ, Chittenden T, Livingston DM, Kaelin W., Jr Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev. 1992;6:953–64. doi: 10.1101/gad.6.6.953. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu E, Coxon A, Otterson GA, Steinberg SM, Kratzke RA, Kim YW, Fedorko J, Oie H, Johnson BE, Mulshine JL, Minna JD, Gazdar AF, Kaye FJ. RB protein status and clinical correlation from 171 cell lines respresenting lung cancer, extrapulmonary small cell carcinoma, and mesothelioma. Oncogene. 1994;9:2441–8. [PubMed] [Google Scholar]
- 27.Fujiwara T, Motoyama T, Ishihara N, Watanabe H, Kumanishi T, Kato K, Ichinose H, Nagatsu T. Characterization of four new cell lines derived from small-cell gastrointestinal carcinoma. Int J Cancer. 1993;54:965–71. doi: 10.1002/ijc.2910540617. [DOI] [PubMed] [Google Scholar]
- 28.Dick FA, Dyson N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol Cell. 2003;12:639–49. doi: 10.1016/s1097-2765(03)00344-7. [DOI] [PubMed] [Google Scholar]
- 29.Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–16. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 30.Helt AM, Galloway DA. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75:6737–47. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sano T, Oyama T, Kashiwabara K, Fukuda T, Nakajima T. Expression status of p16 protein is associated with human papillomavirus oncogenic potential in cervical and genital lesions. Am J Pathol. 1998;153:1741–8. doi: 10.1016/S0002-9440(10)65689-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams JP, Stewart T, Li B, Mulloy R, Dimova D, Classon M. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Mol Cell Biol. 2006;26:1170–82. doi: 10.1128/MCB.26.4.1170-1182.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valverde JR, Alonso J, Palacios I, Pestana A. RB1 gene mutation up-date, a meta-analysis based on 932 reported mutations available in a searchable database. BMC Genet. 2005;6:53. doi: 10.1186/1471-2156-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brichard B, Heusterspreute M, De Potter P, Chantrain C, Vermylen C, Sibille C, Gala JL. Unilateral retinoblastoma, lack of familial history and older age does not exclude germline RB1 gene mutation. Eur J Cancer. 2006;42:65–72. doi: 10.1016/j.ejca.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Richter S, Vandezande K, Chen N, Zhang K, Sutherland J, Anderson J, Han L, Panton R, Branco P, Gallie B. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72:253–69. doi: 10.1086/345651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dryja TP, Rapaport J, McGee TL, Nork TM, Schwartz TL. Molecular etiology of low-penetrance retinoblastoma in two pedigrees. Am J Hum Genet. 1993;52:1122–8. [PMC free article] [PubMed] [Google Scholar]
- 37.Bremner R, Du DC, Connolly-Wilson MJ, Bridge P, Ahmad KF, Mostachfi H, Rushlow D, Dunn JM, Gallie BL. Deletion of RB exons 24 and 25 causes low-penetrance retinoblastoma. Am J Hum Genet. 1997;61:556–70. doi: 10.1086/515499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Notis CM, Niksarli K, Abramson DH, DeLillo AR, Ellsworth RM. Parents with unilateral retinoblastoma: their affected children. Br J Ophthalmol. 1996;80:197–9. doi: 10.1136/bjo.80.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46:617–34. doi: 10.1002/gcc.20457. [DOI] [PubMed] [Google Scholar]
- 40.Meyer JL, Hanlon DW, Andersen BT, Rasmussen OF, Bisgaard K. Evaluation of p16INK4a expression in ThinPrep cervical specimens with the CINtec p16INK4a assay: correlation with biopsy follow-up results. Cancer. 2007;111:83–92. doi: 10.1002/cncr.22580. [DOI] [PubMed] [Google Scholar]
- 41.Serrano M, Gomez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995;267:249–52. doi: 10.1126/science.7809631. [DOI] [PubMed] [Google Scholar]
- 42.Brookes S, Rowe J, Ruas M, Llanos S, Clark PA, Lomax M, James MC, Vatcheva R, Bates S, Vousden KH, Parry D, Gruis N, Smit N, Bergman W, Peters G. INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. EMBO J. 2002;21:2936–45. doi: 10.1093/emboj/cdf289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bautista D, Emanuel JR, Granville C, Howard R, Costa J. Identification of mutations in the Ki-ras gene in human retinoblastoma. Invest Ophthalmol Vis Sci. 1996;37:2313–7. [PubMed] [Google Scholar]
- 44.Sun H, Chang Y, Schweers B, Dyer MA, Zhang X, Hayward SW, Goodrich DW. An E2F binding-deficient Rb1 protein partially rescues developmental defects associated with Rb1 nullizygosity. Mol Cell Biol. 2006;26:1527–37. doi: 10.1128/MCB.26.4.1527-1537.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onadim Z, Hogg A, Baird PN, Cowell JK. Oncogenic point mutations in exon 20 of the RB1 gene in families showing incomplete penetrance and mild expression of the retinoblastoma phenotype. Proc Natl Acad Sci U S A. 1992;89:6177–81. doi: 10.1073/pnas.89.13.6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seminara SB, Dryja TP. Unbiased transmission of mutant alleles at the human retinoblastoma locus. Hum Genet. 1994;93:629–34. doi: 10.1007/BF00201561. [DOI] [PubMed] [Google Scholar]
- 47.Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94:349–54. doi: 10.1007/BF00201591. [DOI] [PubMed] [Google Scholar]
- 48.Huang Q, Dryja TP, Yandell DW. Distinct Rb gene point mutations in families showing low penetrance of hereditary retinoblastoma. Chung Hua I Hsueh I Chuan Hsueh Tsa Chih. 1998;15:139–42. [PubMed] [Google Scholar]
- 49.Cowell JK, Bia B. A novel missense mutation in patients from a retinoblastoma pedigree showing only mild expression of the tumor phenotype. Oncogene. 1998;16:3211–3. doi: 10.1038/sj.onc.1201833. [DOI] [PubMed] [Google Scholar]
- 50.Gallie BL, Hei YJ, Mostachfi H, M DJ. Retinoblastoma: for the next generation. In: Cowell JK, editor. Molecular genetics of cancer. Oxford: Bios Scientific Publishers, Limited; 1995. pp. 1–30. [Google Scholar]
- 51.Yilmaz S, Horsthemke B, Lohmann DR. Twelve novel RB1 gene mutations in patients with hereditary retinoblastoma. Mutations in brief no. 206. Online. Hum Mutat. 1998;12:434. doi: 10.1002/(SICI)1098-1004(1998)12:6<434::AID-HUMU16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]