

Abstract
Brain-derived neurotrophic factor (BDNF) acting through the tyrosine kinase B receptor (TrkB) is thought to be a critical mediator of learning. As there are no available selective antagonists of TrkB, we used a lentivirus encoding a dominant-negative TrkB (TrkB.t1) to antagonize BDNF signaling during extinction of conditioned fear. Whereas TrkB.t1-infected rats showed normal within-session extinction, their retention of extinction was impaired, suggesting that amygdala TrkB activation is required for the consolidation of stable extinction memories.
The persistence of fear memories is thought to be a major contributor to the morbidity associated with a number of psychiatric disorders, including post-traumatic stress disorder, panic disorder, and specific and social phobia1. Although currently available anxiolytic medication can be helpful in decreasing symptoms, the most effective means of specific treatment for these disorders includes exposure-based psychotherapy that targets the specific fear. This form of treatment relies on the underlying process of extinction of fear. Our group has recently had success in using agents that augment extinction in rodent models to successfully enhance the treatment of specific phobias in humans2. By understanding the mechanisms of encoding and consolidation of extinction, new and powerful treatment modalities may become available.
The extinction of fear involves new learning of an inhibitory signal that competes with the previously learned fear memory. Extinction is dependent upon the basolateral nucleus of the amygdala (BLA)3,4 and the infralimbic cortex5,6. Within the BLA, it is probably dependent on the NMDA receptor3,4 and on voltage-gated calcium channels (VGCCs)7. Notably, recent evidence suggests that the extinction of fear and the extinction of drug use may involve similar mechanisms within the BLA (ref. 8). However, despite a wealth of behavioral data, there is limited understanding of the molecular mechanisms mediating the acquisition and consolidation of extinction learning.
BDNF acting on the TrkB receptor within the amygdala is required for normal learning of conditioned fear9. To examine its role in extinction, we first examined whether expression of the gene encoding BDNF is altered following the extinction of conditioned fear. Rats were fear-conditioned with 2 d of 10 light-shock pairings (Supplementary Methods and Supplementary Fig. 1 online). One day later, they were given a pre-extinction test so that they could be matched into two groups with similar levels of conditioned fear. Four days later, the rats were extinction-trained by exposure to 90 lights without shocks; they were then killed after 30 min, 2 h or 4 h. Other rats were fear-conditioned but not extinction-trained, and were killed from their homecage or after exposure to the extinction-training context (Fig. 1a). We found significantly increased expression of Bdnf mRNA within the BLA (Fig. 1a–b) of the 2-h postextinction group compared to the homecage group and the 30-min and 4-h postextinction groups (P < 0.05). This difference was not attributable to context exposure alone as an additional replication study showed no increase in Bdnf mRNA in a context-only exposure group compared to the extinction-trained groups (Fig. 1a). These data suggest that increased Bdnf mRNA transcription or stability is associated with the extinction of fear memory. The level of Bdnf mRNA expression is related to the level of BDNF release and TrkB activation9. The increase in Bdnf mRNA in the 2-h postextinction group, but not in the 30-min or 4-h postextinction groups, is consistent with findings that there are several ‘molecular peaks’ during the consolidation of memory (for example, see ref. 10). We do not know with certainty whether the increase in mRNA at this time point represents a homeostatic increase in transcription in order to replace BDNF peptide that had been previously released from vesicle pools or, alternatively, whether Bdnf mRNA is increased concomitantly with increasing BDNF release to enable greater TrkB activation during the late consolidation phase of memory formation.
Figure 1.
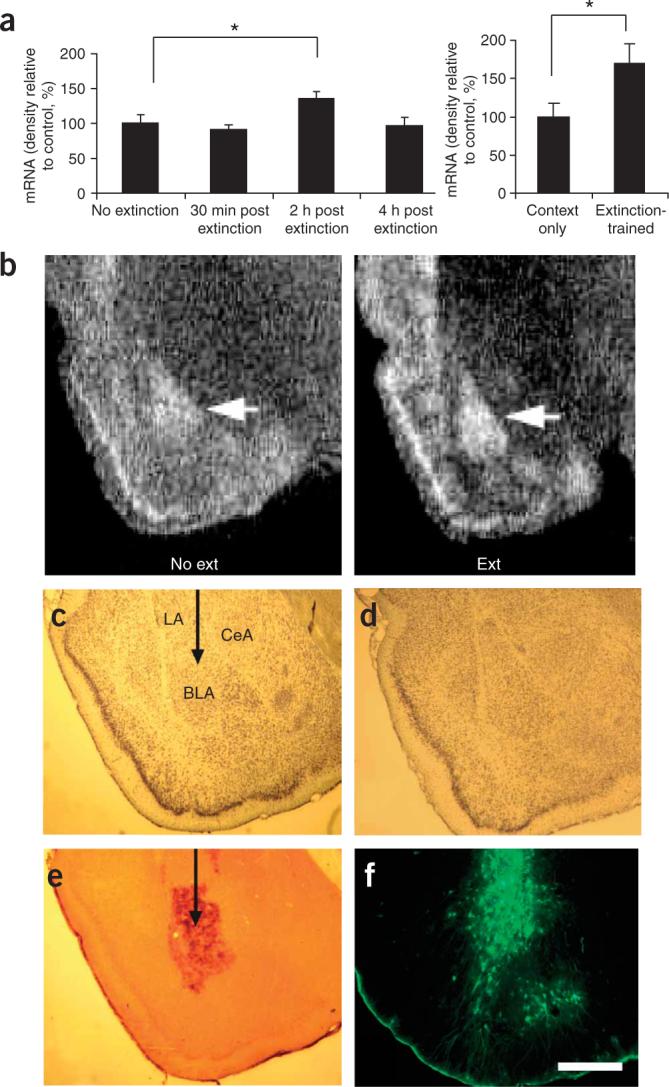
Bdnf mRNA expression and assessment of lentiviral gene expression within the BLA. (a) Regulation of Bdnf mRNA following extinction. In situ hybridization was used to assess the amount of Bdnf mRNA (exon V) present at three time points following extinction. n = 6 per group. At right, a replication experiment in which some rats underwent the same extinction protocol as on the left, whereas others were simply placed in the extinction context without light presentations. Both groups were killed 2 h after the exposure. n = 8 per group. (b) At the 2-h time point, significant increases in Bdnf mRNA were found within the basolateral amygdala (BLA, arrow), as shown with dark-field optics. (c,d) Histological examination of viral infection in the BLA following behavioral studies. No amygdala damage was seen following infection (visualized with cresyl violet staining). LA, lateral amygdala. CeA, central amygdala. (e) Expression of TrkB.t1 was assessed using immunocytochemistry (ICC) for a hemagglutinin (HA) epitope tag incorporated into the TrkB.t1 coding sequence. (f) GFP expression was directly assessed by visualizing tissue under an epifluorescence microscope. *P < 0.05. Error bars represent s.e.m. Scale bar, 1 mm.
To more directly investigate the role of the activation of the receptor for BDNF in the formation of extinction memories, we examined the effects of manipulating the TrkB receptor. As there are no available pharmacological antagonists to block TrkB, we used a viral gene-delivery system to express a dominant-negative truncated TrkB receptor (TrkB.t1)9,11 in BLA neurons. This protocol allowed us to examine whether genetic antagonism of TrkB-mediated signaling in BLA neurons prevents the initial acquisition of extinction and/or the retention of extinction memory.
Rats were fear-conditioned and matched into two groups with equivalent levels of conditioned fear, before virus infusion. One day later, a lentivirus encoding either green fluorescent protein (GFP) or TrkB.t1 was infused into the BLA bilaterally. Extinction training (90 lights with no shocks) was performed 12−14 d after the viral infections, and rats were tested for conditioned fear 1 h and 2 d after extinction. The rats were then killed, and their brains were sectioned and immunostained to visualize the site of injection and the amount of TrkB.t1 and GFP expression (Fig. 1c–f). Only data from rats that received infusions directly within the BLA were analyzed in the behavioral studies. TrkB.t1 infection within the amygdala does not affect the expression of conditioned fear9. Therefore, any differences between the experimental and control groups would be due to the acquisition or consolidation of the extinction memory. We found that rats infected with the TrkB.t1 dominant-negative virus demonstrated more fear than those with the GFP virus (Fig. 2a) when tested 2 d after extinction training, suggesting that genetic blockade of the TrkB receptor prevents the retention of extinction memories.
Figure 2.

Lentivirus expression of TrkB.t1 in BLA blocks the consolidation but not the acquisition of extinction. (a) Level of fear-potentiated startle (FPS) following extinction training, when tested at 1 h or 48 h. TrkB.t1-infected rats showed significantly higher levels of fear 2 d after extinction, as compared to GFP-infected rats (n = 9 and 7 for TrkB.t1 and GFP groups, respectively). (b) TrkB.t1 impairs extinction retention. Averaging across all trials, TrkB.t1-infected rats showed a deficit in extinction as compared to GFP-infected rats. (c) Examining extinction within the testing session suggested that the TrkB.t1-infected rats had normal within-session extinction, but lacked extinction retention across the 2-d interval between tests (n = 9 and 10 for TrkB.t1 and GFP groups, respectively). Note that GFP-infected rats showed complete extinction by test 3 and were not tested thereafter. TrkB.t1-infected rats showed poor extinction retention even on day 4, as evidenced by high FPS during the first 5 trials of the test. (d) Normalization of FPS across the first 3 d (100% = mean of first 5 trials per rat per day) demonstrated that there was no difference in the rates of within-session acquisition of extinction between the two groups. *P < 0.05, **P < 0.01. Error bars represent s.e.m.
We wondered whether the blockade of extinction with TrkB.t1 prevented the initial encoding of the extinction process or the consolidation of extinction memories. To address this, we altered our extinction protocol so that we could examine both within-session and between-session extinction. Rats were trained and injected as above. In this experiment, however, extinction occurred over multiple days of testing; tests were administered every 2 d and each involved 30 light-startle trials intermixed with 30 startle-only trials (to measure fear-potentiated startle). This ‘slow extinction’ approach provided extinction training while also allowing us to measure fear within each extinction session.
Rats expressing TrkB.t1 demonstrated significantly (P < 0.05) decreased extinction (Fig. 2b) across several days of extinction training and testing, as compared to rats expressing GFP (which showed marked extinction across days). The impairment of extinction in TrkB.t1-infected rats was not complete, however, as there appeared to be some level of extinction occurring over the first 2 d, perhaps due to incomplete TrkB.t1 infection within the amygdala. An alternative explanation for this partial deficit is that other non-BDNF mechanisms are also involved in extinction (for example, the NMDA receptor4 and the VGCC (ref. 7)), and these may allow for partial consolidation in the absence of normal TrkB activation.
To compare rates of within-session extinction between the two groups (Fig. 2c), the first 3 d of repeated extinction testing were combined, and the level of fear for each rat at the beginning of testing was normalized to 100%. We observed that there was no difference between the groups in the rate of within-session extinction (Fig. 2c,d). However, whereas the GFP-infected rats showed progressive reductions in fear during the first block of trials across testing days (a measure of retention), rats infected with TrkB.t1 manifested fairly similar levels of fear at the beginning of each extinction session, even after 4 d of extinction training (Fig. 2c), in contrast to their normal rates of within-session extinction. This suggests that these rats may initially encode the extinction memory within each session but are not able to consolidate it, making it unavailable for recall during subsequent sessions. This is similar to the effect seen following lesions of the ventromedial prefrontal cortex5 or the systemic administration of an NMDA antagonist12.
Together, these data suggest that the TrkB receptor is not involved in the process of within-session encoding of fear extinction, but instead is required for the normal consolidation of extinction as measured during the retention test. These results are also consistent with findings from in vitro slice physiology showing that BDNF may be primarily involved in ‘synaptic consolidation’ of late-phase long-term potentiation (LTP)13,14.
The dissociation of within-session extinction from extinction retention in the present study suggests that these are two separable phenomena. The role of the BDNF/TrkB system in extinction contrasts with the apparent role of the cannabinoid receptor type 1 (CB1) in mediating the extinction of fear, as it has been shown that CB1 antagonism prevents both within-session extinction (Supplementary Fig. 2 online) and the retention of extinction15. Together with previous work5,12, it seems that within-session extinction may be a necessary but not sufficient condition for the formation of stable extinction memories.
Clinically, this idea may have important implications, as different treatment strategies may be appropriate for patients with deficits in within-session extinction versus the retention of extinction. Moreover, these results imply that enhancing TrkB activation at the time of extinction learning, or shortly thereafter during the consolidation period, could enhance the process of consolidation and extinction retention. Furthermore, because we previously found that BDNF also is required for the consolidation but perhaps not the acquisition of fear learning9, a BDNF antagonist given after the experience of trauma may decrease trauma memory consolidation and thus block the later onset of post-traumatic stress disorder.
ACKNOWLEDGMENTS
Support was provided by the US National Institutes of Health (MH47840 to M.D., MH069884 to K.J.R. and MH070218 to J.P.C.), the National Alliance for Research on Schizophrenia and Depression (NARSAD; K.J.R.), NIH/NCRR base grant (P51RR000165) to Yerkes National Primates Research Center and the Center for Behavioral Neuroscience (National Science Foundation agreement IBN-987675).
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
Note: Supplementary information is available on the Nature Neuroscience website.
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
Supplementary Material
References
- 1.Cannistraro PA, Rauch SL. Psychopharmacol. Bull. 2003;37:8–25. [PubMed] [Google Scholar]
- 2.Ressler KJ, et al. Arch. Gen. Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- 3.Falls WA, Miserendino MJ, Davis M. J. Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker DL, Ressler KJ, Lu KT, Davis M. J. Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quirk GJ, Russo GK, Barron JL, Lebron K. J. Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milad MR, Quirk GJ. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 7.Cain CK, Blouin AM, Barad M. J. Neurosci. 2002;22:9113–9121. doi: 10.1523/JNEUROSCI.22-20-09113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroeder JP, Packard MG. Learn. Mem. 2004;11:641–647. doi: 10.1101/lm.78504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rattiner LM, Davis M, French CT, Ressler KJ. J. Neurosci. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ressler KJ, Paschall G, Zhou XL, Davis M. J. Neurosci. 2002;22:7892–7902. doi: 10.1523/JNEUROSCI.22-18-07892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Middlemas D, Lindberg R, Hunter T. Mol. Cell. Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santini E, Muller RU, Quirk GJ. J. Neurosci. 2001;21:9009–9017. doi: 10.1523/JNEUROSCI.21-22-09009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang H, Welcher AA, Shelton D, Schuman EM. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 14.Bramham CR, Messaoudi E. Prog. Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Marsicano G, et al. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.