

Abstract
The Listeria monocytogenes protein listeriolysin-O (LLO) is a pore-forming protein essential for virulence. Although LLO's major role is to allow L. monocytogenes entry into the cytosol, it also induces apoptosis of activated lymphocytes, an obligatory cellular response that modulates the infection. Induction of apoptosis by LLO proceeds through a fast, caspase-dependent pathway, and a slow, caspase-independent pathway. Polyclonal T cell lines were generated from either normal mice or mice deficient in granzyme and perforin proteins, and then treated with apoptogenic doses of LLO. Here we show that apoptosis of lymphocytes induced by LLO was characterized by activation of caspases as quickly as 30 minutes that was dependent on the expression of granzymes. In the absence of granzymes, all parameters of apoptosis such as caspase activation, phosphatidylserine exposure, mitochondrial depolarization, and DNA fragmentation, were dramatically reduced in magnitude. Removal of perforin inhibited the apoptotic effect of LLO on cells by about 50%. Neutralization of intracellular acidification using chloroquine inhibited the rapid apoptotic death. In agreement with these findings granzyme deficient mice harbored lower bacterial titers and decrease splenic pathology compared to normal mice following L. monocytogenes infection. Thus, LLO exploits apoptotic enzymes of the adaptive immune response to eliminate immune cells and increase its virulence.
Keywords: apoptosis, T Cells, bacterial
Introduction
Infection with the gram-positive bacterium Listeria monocytogenes is a powerful model to examine bacterial virulence and immune regulation after infection. Infection of mice with L. monocytogenes causes marked apoptosis of lymphocytes, hepatocytes, and neurons (1-3). Indeed there is a growing list of bacterial pathogens that induce apoptosis making it important to understand the molecular mechanisms behind it (4, 5).
L. monocytogenes expresses a virulence cluster dedicated to invasiveness in mammalian species (6). One of the virulence factors in the cluster is the pore-forming toxin LLO, a member of the cholesterol dependent cytolysin family (CDC) (7-12). CDC are expressed by a number of gram-positive bacteria, and have various functions, from delivery of toxins (streptolysin O)(13), to compromising phagosomes of infected cells (LLO). The main role attributed to LLO is to allow L. monocytogenes to escape from the phagosome into the permissive environment of the host cell cytosol (14, 15). Bacteria deficient in LLO are avirulent in vivo and in vitro. Treatment of mice with a monoclonal antibody that neutralizes LLO also renders L. monocytogenes avirulent in vivo and in vitro (16, 17).
Lymphocyte apoptosis takes place in infective foci at the time of exponential growth of the microbe (1). Phagocytes also die after infection, but the mechanism of death is not understood. Mice deficient for the type I interferon receptor have decreased lymphocyte apoptosis and increased survival of a subset of macrophages (18-20). The apoptotic lesion is immuno-modulatory, leading to decreased host-resistance and increased bacterial proliferation (4, 21). We have postulated that during the exponential growth of L. monocytogenes, LLO is released to the extracellular fluids, leading to lymphocyte apoptosis. We favor this hypothesis based on the findings that, i) antibodies to LLO blocked apoptosis in vivo (16), ii) free L. monocytogenes was found in the inflamed lesions surrounding apoptotic cells; iii) lymphocytes were never infected with L. monocytogenes; and iv) nanomolar doses of purified LLO induced apoptosis of dividing T cells with fast kinetics, and activation of caspase-3, noted as early as 30 minutes after treatment (22).
In examining the mechanism of action of LLO in causing lymphocyte apoptosis, we considered the role of granzymes. We reasoned that due to the rapid kinetics, a membrane proximal event should be the inductive event. Of all the apoptotic signals studied to date, granzyme-mediated induction of cellular death has kinetics most similar to LLO induced apoptosis. LLO has a pH optimum that enhances its activity in the phagosomal environment and could either lyse or permeabilize the acidic vesicles/granules that contain granzymes releasing them into the cytosol (23, 24). Hence, LLO may act as an endosomolytic agent. In fact, extracellular LLO has been used to deliver large amounts of purified recombinant granzyme B to target cells (25). Alternatively, LLO could be inducing signaling cascades inside cells that lead to granzyme dependent apoptosis. To our knowledge this is the only example of a protein that induces apoptosis through cell autonomous granzyme activity. We prove that granzyme is the major executor of the fast cellular death seen in activated T cells treated with LLO and, importantly, we also indicate an effect in the infection.
Materials and Methods
Mice and Infections
129/SvJ mice were purchased from The Jackson Laboratories (Bar Harbor, ME). Granzyme A−/−×B−/−, granzyme B−/−, and granzyme A−/−×B cluster−/− mice on a 129/SvJ background were a kind gift from Dr. Timothy J. Ley (Washington University School of Medicine, St. Louis, MO) A detailed description of how all the granzyme deficient strains of mice were generated can be found in (26). In vivo infections were performed as described previously (21). Hematoxylin and eosin and TdT-mediated dUTP nick-end labeling (TUNEL) staining was performed as described previously (1). All mice were bred and maintained at our animal facility and used according to the protocols defined by the Division of Comparative Medicine of Washington University School of Medicine.
Cell Culture and LLO treatment
T cell lines were generated by immunizing mice in the hind-footpad with 10 nmoles of ovalbumin (Sigma Chemical Co, St. Louis, MO) in Complete Freund's Adjuvant (Difco brand, Sigma Chemical Co.). Lymph nodes were isolated and lines were generated using conventional techniques. The initial T cell line was passaged as follows: 1×106 T cells and 2×107 dispersed irradiated (3000 Rad) splenocytes were cultured in 20 mL of Dulbecco's Modified Eagles Medium supplemented with 10% defined fetal calf serum, 50 U/mL interleukin-2, and 10 μM ovalbumin. T Cell lines were passaged every 7 days. At every passage and before any experiment T cell lines were extensively purified by centrifugation over a Histopaque 1119 gradient (Sigma) to remove dead cells. This process also removes the irradiated splenocytes. T cells purified this way were 100% CD4+, CD69+, and CD62L− by flow cytometric analysis (22). Wild-type 129/SvJ T cells were ∼30-40%+ for granzyme B expression by intracellular flow cytometry, versus ∼0% for the granzyme A−/−× granzyme B−/−, granzyme B−/− and granzyme A−/−× B cluster−/− T cells lines. T cell lines were treated with 250 ng/mL of purified recombinant LLO as described previously (22). For chloroquine treatments, cells were incubated in the indicated concentrations of chloroquine (Sigma, St. Louis, MO) for 10 minutes before being treated with LLO for the indicated times.
RNA was isolated from cultured 129/SvJ or granzyme AC T cells using the RNeasy Mini Kit (Qiagen, Valencia, CA). First strand cDNA was made using the Superscript III First Strand Synthesis Kit (Invitrogen, Carlsbad, CA). PCR was carried out with the primers indicated in Supplementary Table I using GoTaq Green Master Mix (Promega, Madison, WI). All protocols were performed using the manufacturer's protocols.
Concanavalin A (ConA, Sigma, St. Louis, MO) was used at 25 μg/mL. Whole splenocytes were incubated in ConA for 16 hours, then purified by density gradient, and treated with LLO. For experiments involving calcium-free media, MEM base (Sigma) was supplemented with all the components of complete DMEM except calcium.
Conventional DMEM containing calcium was made following the same protocol. T cells were washed out of conventional DMEM and resuspended in DMEM with or without calcium and 1% fetal calf serum for the duration of the experiment.
Antibodies and immunoblotting
Whole protein was isolated from 1 to 6 million cells by standard techniques, resolved by SDS-PAGE and transferred to Hybond-P membranes (Amersham Biosciences, Piscataway, NJ). Caspase-3 (8G10), caspase-6 (9762), and caspase-9 (9504) antibodies were obtained from Cell Signaling Technologies (Beverley, MA) and used at 1:4000, 1:2000 and 1:2000 dilutions, respectively. Goat-anti-rabbit horseradish peroxidase was obtained from Cell Signaling Technologies and used at 1:8000 (caspase-3) or 1:4000 (caspase-6 and -9) dilution. Detection was performed with ECL Plus (Amersham Biosciences).
Flow cytometry
The cationic dye JC-1 was purchased from Molecular Probes (Eugene, OR). For JC-1 incorporation assays, cells were washed twice in PBS and resuspended in DMEM containing 10 μg/mL JC-1. Cells were incubated for 30 minutes at 37°C, washed twice in DMEM, and analyzed by flow cytometry. Anti active-caspase-3 antibody, annexin V-PE and 7-AAD were obtained from BD Biosciences (Valencia, CA) and used as per the manufacturer's protocol. All flow cytometry was carried out on a FACSCalibur (BD Biosciences) and data was collected using CellQuest Software (BD Biosciences) and subsequently analyzed using FlowJo (FlowJo, Ashland, OR). CFDA-SE was used by the manufacturer's instructions at a final concentration of 10 μM dye (Eugene, OR).
Results
Granzymes are responsible for most of the rapid T cell apoptosis induced by LLO
CD4 T cells from normal mice and from mice deficient in granzyme expression were examined for LLO-induced apoptosis. We have chosen to mainly examine CD4 T cell lines because they are easily cultured, polyclonal, and are well propagated in culture. Limited experiments were made with CD8 T cells which are more fastidious in long term culture.
Antigen-specific T cell lines were generated against ovalbumin by immunizing either 129/SvJ or granzyme A−/− × B cluster deficient mice (GzmAC3). The GzmAC mice are a cross between granzyme A−/− (Gzma−/−) and granzyme B−/− (Gzmb−/−) cluster mice. The Gzmb−/− cluster mouse is deficient for granzyme B and has reduced expression of granzymes C, D, F, and G due to a positional effect of neomycin insertion into the Gzmb gene locus (27, 28). The GzmAC T cell lines are completely deficient in granzymes A and B and have reduced expression of the B-cluster granzymes C, D, F, and G. This observation was confirmed by RT-PCR. Our wild-type T cell lines expressed granzyme A and B at the RNA level (Fig. 1A). We also detected granzyme C, D and G. In the GzmAC T cells, we did not detect granzymes A, B, or C, but still detected RNA for granzymes D and G. The reduction in granzyme A, B, and C mRNA in the GzmAC T cells is consistent with previous data (27). There is little data on the function of granzymes D and G. Based on sequence analysis they appear to cleave hydrophobic residues, unlike granzymes A and B which are a tryptase and aspase, respectively. We were unable to detect granzymes E, F, K, M, or N in either cellular preparation (29).
Figure 1.

Rapid LLO-induced T cell apoptosis is dependent on expression of granzyme. (A) Wild type T cell lines express granzymes A, B, C, D and G at levels detectable by RT-PCR. Granzyme A × B cluster−/− T cell lines do not express detectable levels of granzymes A, B or C. Granzyme D and G are expressed like the wild-type L = ladder, Ctrl = no RNA. Letters A-G, K, M, N over the lanes refers to the granzyme being amplified. (B-D) 1×106 purified T cells were treated with or without (UT) 250 ng/mL LLO for the indicated times. Cells were then stained for annexin V and 7-AAD. For panel (B) the UT cells were in culture for 8 h before being stained. For panel (C) the UT cells were in culture for 24 h before being stained. The flow cytometry plots are representative of at least three independent experiments performed in duplicate or triplicate. The percentages given in each of the four quadrants in (B-C) was plotted in the graph in panel (D). The green part of the bars represent the annexin V+/7-AAD− (lower right) quadrant, the red part of the bars indicates the annexin V+/7-AAD+ (upper right) quadrant, and the grey part of the bars represents the annexin V−/7-AAD− (lower left) quadrant of cells. All bars are the mean +/− S.E.M. for at least three independent experiments.
A time-course study examined if the kinetics of LLO-induced apoptosis differed between cells derived from 129/SvJ and GzmAC mice. Fig. 1B-C are flow cytometry plots for annexin V and 7-aminoactinomycin D (7-AAD) demonstrating a dramatic reduction of apoptosis following LLO treatment of GzmAC T cells. Fig. 1D shows the compiled data of all the flow cytometry plots of annexin V/7-AAD staining following LLO treatment. Apoptotic cells are annexin V+ (a marker of phosphatidylserine) and 7-AAD− (a marker of DNA), depicted in green in Fig. 1D. Late apoptotic/necrotic cells are positive for 7-AAD (depicted in red in Fig. 1D).
LLO caused approximately 44.8%, 57.1%, and 76.7% T cell apoptosis at 2, 4 and 8 hours post-treatment respectively in the 129/SvJ T cells with a background apoptosis in untreated cells of 9.61% (Fig. 1B,D). In contrast, GzmAC T cells showed a reduction, only 13.2%, 21.5% and 28.6% apoptotic cells at 2, 4, and 8 hours, respectively over a background of 15.2% (Fig. 1B, D).
Despite the dramatic reduction of apoptosis and total cell death within the first 8 hours following LLO treatment, a slower death was found in the GzmAC T cells, at 24 hours after treatment. By 24 hours of treatment with LLO there was a total of 74.7% cell death in the 129/SvJ T cells, and 48.8% in the GzmAC T cells (annexin V+ cells). The cell death seen in GzmAC cells at 24 hours was distinct from the rapid granzyme-mediated death, the annexin V+/7-AAD− cells were not found but instead annexin V+/7-AAD dull to bright cells and annexin V−/7-AAD+ cells dominated the culture (Fig. 1C-D).
CD4+ T lymphocytes are not conventionally thought to be cytotoxic cells that express granzyme and perforin. However, recent evidence has shown that human CD4+ T cells activated with anti-CD3 and anti-CD46 express granzyme B (30). Also, granzyme B is important in the regulation of activation induced cell death in different T helper subsets (31). Granzyme B RNA was detected by qRT-PCR in the wild-type but not the GzmAC T cell lines. Levels of message were >212 higher in the 129/SvJ T cells than the GzmAC T cells. This result was confirmed with a cross-reactive mouse anti-human granzyme B antibody. Intracellular staining for granzyme B protein, although weak, disclosed 29.6% of the T cells positive for granzyme B by flow cytometry in the wild type and 0.61% granzyme positive cells in the GzmAC T cells. Examination of lymphocytes by immunofluorescent microscopy revealed that granzyme B showed a punctate distribution compatible with compartmentalization in secretory granules.
Caspase activation, mitochondrial depolarization and DNA fragmentation following LLO treatment was attenuated in GzmAC T cells
Caspase immunoblots performed on whole cell lysates of T cells treated with LLO detected both the pro- and active forms of the caspases (32). Activated caspases-3, 6, and 9 were detected as early as 30 minutes after treatment with LLO but were not found in GzmAC T cells (Fig. 2A). Fig. 2B shows representative flow cytometry plots for activated caspase-3 on either untreated or LLO treated T cells. By 2 hours after treatment with LLO, 41% of 129/SvJ T cells were caspase-3+ over a background of 6.09%. In contrast, only 12.1% of the GzmAC T cells were caspase-3+ after two hours of LLO treatment compared to a background of 9.32%.
Figure 2.

Treatment of T cells with LLO induces granzyme-dependent rapid caspase activation. 1×106 T cells were treated with 250 ng/mL of LLO for the indicated times and then tested for caspase-3, -6, and -9 activation by immunoblot (A). The antibodies used in these assays detect both the inactive pro-form of the enzyme (the high molecular weight species) and the cleaved, active form of the enzyme. Expected pro- and active-form band sizes are labeled on the gel. The gel pictures shown are representative of 2-6 independent experiments. (B) Caspase-3 activation was measured by intracellular flow cytometry after treatment of 1×106 T cells with 250 ng/mL of LLO for 2 or 24 h. Untreated cells were tested at either 2 or 24 h after being put in culture. Cells were permeabilized and stained using an antibody that detects only the active-form of caspase-3. The plots are representative of 3 independent experiments, each performed in duplicate or triplicate.
After 24 hours of treatment there were 70.1% active caspase-3+ T cells, but the background cell death had risen to 24.1% (Fig. 2B). The background levels were approximately the same in GzmAC T cells after 24 hours in culture, but after treatment with LLO only 46.1% active caspase-3 positive cells were found. This result indicates that most of the caspase activation seen within the first two hours of treatment with LLO is granzyme dependent, but that there is a slower pathway to caspase activation that operates in the absence of granzyme.
Fig. 3A shows the data obtained after staining LLO treated T cell lines with the mitochondrial dye JC-1, a cationic lipophilic dye that binds preferentially to mitochondrial membranes and undergoes a green to red color transition when the intermembrane space depolarizes due to apoptosis. A rapid increase in mitochondrial permeability was detected, with approximately 66.1%, 72.5% and 81.4% of the 129/SvJ T cells becoming JC-1 red after 2, 4, and 8 hours compared to about 21.9% background. This magnitude of mitochondrial depolarization was not seen in GzmAC T cells, which displayed 27.8%, 32.7% and 42.4% JC-1 red at 2, 4, and 8 hours after LLO treatment versus a background of about 21.3%.
Figure 3.

Rapid mitochondrial depolarization and DNA degradation in T cells caused by LLO is granzyme dependent. (A) 1×106 T cells were treated with 250 ng/mL of LLO for the indicated times and then stained using the mitochondrial dye JC-1. The bars are the percent of total cells that became JC-1 red, indicating loss of mitochondrial permeability. (B) T cells were treated with 250 ng/mL of LLO for either 2 or 24 h and then cells were permeabilized and stained by TUNEL. Cells under the marker are indicated as % of TUNEL+. Untreated cells were assayed 8 h after being put in culture.
The final parameter examined was DNA fragmentation as measured by terminal transferase dUTP nick end labeling (TUNEL) reaction. As early as 2 hours after treatment with LLO, 57.2% of the 129/SvJ T cells showed fragmented DNA, compared to 11% for the untreated (Fig. 3B). The GzmAC T cells, showed 24.8% TUNEL+ after LLO treatment compared to 13.8% for the control. By 24 hours of treatment with LLO, 86.4% of the 129/SvJ T cells were TUNEL+ over a 26.1% background. Even in the absence of granzyme we still detected DNA fragmentation, with 48.9% of the GzmAC T cells TUNEL+ over a 17.1% untreated background.
Role of perforin, extracellular calcium and mitogenic stimulation on granzyme mediated LLO induced apoptosis
Delivery of granzymes from cytotoxic lymphocytes to their targets is dependent on the expression of perforin. Perforin allows granzyme to translocate across membranes through an unknown mechanism. Both perforin and the CDCs contain a conserved membrane attack complex/perforin (MACPF) domain that participates in the oligomerization and pore formation of these proteins (33). To determine the extent of perforin's contribution to the granzyme-dependent death seen after treatment with LLO, we treated perforin deficient (Prf−/−) T cells with LLO and tested annexin V/7-ADD as describe previously. Figure 4A shows representative flow cytometry plots of either untreated or LLO treated T cell lines. We detected approximately the same level of background cell death in all three cell lines. After treatment with LLO, we detected a 22%, 16%, and 5% increase in the number of annexin V+/7-AAD− cells in 129/SvJ, GzmAC and Prf−/− respectively. Analysis of three independent experiments revealed that Prf−/− cells died early after LLO treatment but the incidence dropped approximately 50% compared to the 129/SvJ T cells (Fig 4B). Perforin appears to enhance the ability of LLO to induce apoptosis of treated lymphocytes but the mechanism for this enhancement inside cells is unknown.
Figure 4.
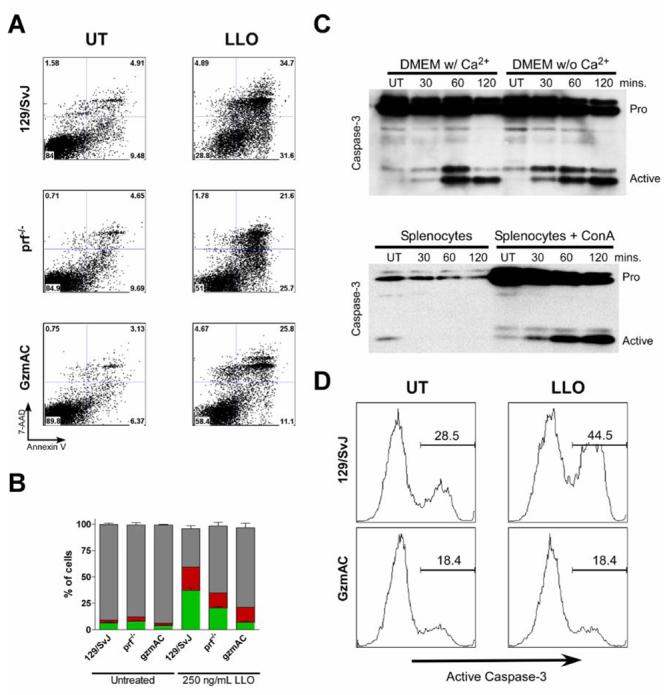
Participation of perforin and calcium and testing of ConA activated splenocytes treated with LLO. (A) T cell lines from perforin−/− mice were treated with LLO for 4 hours and then annexin V/ 7-AAD straining was performed. Panel B summarizes the data of three independent experiments as performed in Panel A. UT cells were untreated and cultured for 8 h (C, Top Blot) 129/SvJ T cells were incubated in either DMEM with or without calcium and then treated with 250 ng/mL LLO for the indicated times. Cells were then immunoblotted for caspase-3. (C, Bottom B, blot) Whole splenocytes were either left untreated or activated using concanavalin A (ConA) and then treated with 250 ng/mL LLO for the indicated times. Immunoblots were performed for total caspase-3. UT cells were assayed at 2 h after culture. (D) ConA activated splenocytes were fractionated to purify CD8+ T cells. Cells were treated with 250 ng/mL LLO for 2 hour and then caspase-3 activation was measured by intracellular stain and flow cytometry. UT cells were assayed after 2 h in culture.
To examine a possible role of extracellular calcium, T cells were cultured in complete media in the presence or absence of calcium and then treated with LLO for different times. We found equivalent caspase-3 activation after treatment with LLO regardless of the presence of extracellular calcium (Fig. 4A). We also tested the involvement of the calcium dependent protease calpain by treating T cells with the calpain inhibitor ALLM, but this had no effect on LLO-induced apoptosis (data not shown). Perforin activity is calcium dependent (34). The fact that depletion of calcium had no effect on LLO activity in T cells suggests to us that there is no secretion of lytic granules that are causing neighboring T cells to die. This data would suggest that perforin potentiates the release of granzyme inside the LLO treated cell.
One potential caveat to this work is that bulk antigen specific T cells may reflect altered responses to stimuli after culture. We wanted to validate that the result obtained with bulk T cell lines in culture could be replicated with previously uncultured lymphocytes. Whole spleen cells were treated with ConA for 16 h to induce them to proliferate. The lymphocytes were then purified by density gradient centrifugation and treated with LLO for different times. Control splenocytes were treated in the same way but did not receive ConA. Fig. 4C shows the result of immunoblot against active caspase-3 following LLO treatment. We observed that treatment with ConA was sufficient to induce upregulation of pro-caspase-3 in the splenocytes. This is consistent with previous observations that showed upregulation of caspase-3 in T cells following activation (35). Treatment with ConA also sensitized the cells to the killing effects of LLO. There was activation of caspase-3 only in ConA-treated, but not resting splenocytes. We fractionated the ConA treated T cells to determine if CD8+ T cells would also undergo apoptosis in response to LLO treatment. Fig. 4C shows that activated CD8+ spleen T cells also activated caspase-3 in response to LLO exposure. Activation of caspase-3 in ConA activated CD8+ T cells was also dependent on expression of granzymes (Fig 4D).
Cellular acidification was required for LLO induced rapid apoptosis
LLO is unique in the CDC family of toxins in its pH dependence of its pore-forming activity (23, 24). At neutral pH and physiological temperatures LLO becomes inactivated due to an irreversible conformational change in the domain responsible for penetrating lipid bilayers (36). This dependence on pH limits its activity in the cytosol of infected cells; at pH 6.5 LLO would only be very weakly active (24). Accordingly, we tested the pH dependence of apoptosis induced by LLO by using the phagosomal neutralizing agent chloroquine (37).
Treatment of the T cell lines with chloroquine alone did not cause cell death as measured by annexin V/7-AAD staining (Fig. 5A). Chloroquine reduced the number of 129/SvJ T cells that became annexin V+/7-AAD− after 2 hours of LLO treatment from 42.1% to 10.9%. The effect of chloroquine was titrateable, as lowering the dose from 10 μM to 1 μM ablated its inhibitory ability on LLO (Fig. 5B-C). Despite the effect of chloroquine on the wild-type cells, there was no effect on the 2 h LLO treated GzmAC T cells. We found 9.72% annexin V+/7-AAD− GzmAC T cells after 2 h of LLO treatment and 8.38% if the cells were incubated in 10 μM chloroquine. Chloroquine reduced the number of annexin V+/7-AAD− 129/SvJ T cells found 24 h after treatment of LLO from 55.9% to 25.6% (30.3% reduction) (Fig. 5A,C). However, this was the same difference found at 2 h (42.1% to 10.9% is 32.1% reduction). Interestingly, chloroquine reduced the number of dead GzmAC T cells at 24 h from 59.9% to 32.5%. This result suggests that part of the slow-death seen in the absence of granzymes also depends on phagolysosomal acidification. Neutralization of cellular acidity would have no effect on granzyme B activity in our assays because granzyme is more active at neutral than acidic pH (38). Also, perforin activity is also more active at neutral pH, therefore the entire perforin/granzyme pathway would be increased in activity by neutralization of acidity, arguing that the effect of choloroquine on LLO-induced apoptosis is at the level of LLO activity inside cells.
Figure 5.

Neutralization of lysosomal acidity by chloroquine blocks rapid granzyme-dependent LLO killing of T cells. 1×106 129/SvJ or GzmAC T cells were either treated with 250 ng/mL LLO or left untreated (UT) for either 2 or 24 h. Cells were stained for annexin V/7-AAD. (A) Flow cytometry plots for annexin V/7-AAD staining. (B-C) Graphs of the flow cytometry data at 2 h (B) or 24 h (C) after treatment with LLO. All the UT cells were in culture for the indicated times (2 or 24 h). The green part of the bars represent the annexin V+/7-AAD− (lower right) quadrant, the red part of the bars indicates the annexin V+/7-AAD+ (upper right) quadrant, and the grey part of the bars represents the annexin V−/7-AAD− (lower left) quadrant of cells. Bars are the mean+/−SEM for two independent experiments performed in triplicate.
Granzyme B accounted for most of the rapid LLO-induced apoptosis
We wanted to determine the contribution of specific granzymes to the rapid LLO induced apoptosis. By immunoblot, we did not find much activation of either caspase-3 or caspase-9 in either a Gzma−/−×b−/− or a Gzmb−/− T cell (Fig. 6A). We detected a very small amount of caspase-3 and -9 activation in Gzmb−/− T cells by immunoblotting at 2 hours after treatment. This would suggest that a minor component of the caspase activation seen after LLO treatment is dependent on granzyme A expression. The result was confirmed by intracellular staining for activated caspase-3: only the 129/SvJ T cells had activated caspase-3 over background after 2 hour of treatment (Fig. 6B). By 24 hours after treatment with LLO some level of activated caspase-3 was found in all granzyme deficient T cell lines; however, none of them reached the level of the 129/SvJ (Fig. 6C). Similar results were obtained with annexin V/7-AAD staining. The annexin V/7-AAD data for the Gzmb−/− and Gzma−/−×b−/− T cells was similar to the data obtained with the GzmAC T cells. The granzyme deficient T cells displayed cell death, again not to the same level as the wild-type (Fig. 6D-E). Removal of granzyme B alone eliminated most of the caspase-3 activation and diminished the rapid cell death seen after LLO treatment. In the absence of granzyme B there was still a small amount of caspase-3 activation that was dependent on granzyme A expression. It did not appear that this low level of caspase-3 activation was sufficient to induce extensive cell death, arguing that granzyme B was the dominant enzyme in the fast cell death induced by LLO.
Figure 6.

Granzyme B is responsible for all of the rapid apoptosis following LLO treatment of T cells. (A) 1×106 activated Gzmb−/−, and Gzma−/− × Gzmb−/− T cells were treated with 250 ng/mL of LLO for the indicated times. Immunoblots were performed against caspase-3 and -9. (B-C) 1×106 Gzmb−/−, Gzma−/− × Gzmb−/−, and GzmAC T cells were treated with 250 ng/mL LLO or left untreated for either 2 hours (B) or 24 h (C) and then assayed for activated caspase-3 by flow cytometry. The bars indicated the mean+/−SEM percent of active caspase-3+ cells from at least 3 independent experiments performed in duplicate or triplicate. (D-E) 1×106 Gzmb−/−, Gzma−/− × Gzmb−/−, and GzmAC activated T cells were treated with 250 ng/mL of LLO for either 2 (D) or 24 (E) h and then stained for annexin V/7-AAD. The green part of the bars represent the annexin V+/7-AAD− (lower right) quadrant, the red part of the bars indicates the annexin V+/7-AAD+ (upper right) quadrant, and the grey part of the bars represents the annexin V−/7-AAD− (lower left) quadrant of cells.
It is possible that LLO is causing the release of lytic granules from the treated lymphocyte to adjacent lymphocytes. A variety of assays, including removal of calcium from the medium, treatment with monoclonal antibodies against LLO, treatment with chloroquine, and using conditioned medium from LLO treated cells suggests that this is not the case. To directly examine this possibility the following experiment was done. We labeled either 129/SvJ or GzmAC T cells with CFDA-SE and then treated them with LLO to see if we could detect caspase-3 activation. We mixed CFDA-SE labeled 129/SvJ cells with unlabeled GzmAC cells or vice versa at approximately 1:1 ratios then determined which population of cells was dying after treatment with LLO. In unlabeled wild-type cells treated with LLO we detected 52.6% caspase-3+ cells (Fig. 7). When we mixed labeled 129/SvJ cells with unlabeled GzmAC cells we found that only the wild-type 129/SvJ T cells activated caspase-3 following treatment with LLO. Approximately 38% of the CFDA-SE labeled cells were positive for active caspase-3 versus 6% for the unlabeled GzmAC T cells. In the reciprocal experiment, we found that 44% of the unlabeled 129/SvJ cells activated caspase-3 versus 7% for the labeled GzmAC cells. We also performed control experiments without mixing the cells and found approximately 36% of the labeled 129/SvJ cells activated caspase-3 after treatment with LLO versus 3% for the labeled GzmAC T cells treated with LLO (Data not shown). This demonstrates that granzyme is not being transferred to another cell after treatment with LLO.
Figure 7.

The activity of LLO is cell autonomous. T Cells were either left untreated for 2 h (UT) or treated with LLO at 250 ng/mL for 2 h (LLO). The top two plots show unlabeled T cells. The two middle plots are a 1:1 mix between 129/SvJ T cells stained with CFDA-SE (green) and unlabeled GzmAC T cells. The two bottom plots are a 1:1 mix of unlabeled 129/SvJ T cells and GzmAC cells labeled with CFDA-SE.
Decreased apoptosis and increased resistance to infection in granzyme AC mice
129/SvJ and granzyme AC mice were infected with ∼2.5×104 CFU intraperitoneally and bacterial burden and histology was determined. At day 2 of the infection granzyme AC mice contained ∼15 fold fewer bacteria than the wild-type mice in both the spleens and livers (Fig. 8A-B). At day 4 of the infection there was a ∼10-fold decrease in the spleens and livers of the granzyme AC mice compared to 129/SvJ (Fig. 8C-D). The difference in colony counts was significant, with p-values ranging from 0.0313 to 0.0015 when comparing the difference between the 129/SvJ and the granzyme AC mice (Fig. 8). The results of one experiment showed a drop in L. monocytogenes burden in both strains after 6 and 8 days of infection, an indication that T cell immunity was taking place. Examination of spleen histology at day 2 following infection, the peak time of lymphocyte apoptosis (1), revealed infectious foci in the periarteriolar lymphoid sheath containing L. monocytogenes: however there was a noticeable decrease in apoptotic lesions in the spleens of granzyme AC mice compared to the 129/SvJ (Fig. 9). Fig. 9 A, C shows an apoptotic lesion found in the 129/SvJ mice at day 2 of infection. The 129/SvJ mice showed the typical lesions in the periarteriolar lymphoid sheath with depletion of the lymphocytes and TUNEL+ cells. About 1/3 of the white pulp profiles were TUNEL+. In contrast the profiles found in the granzyme AC mice showed no apoptotic lesions as examined by TUNEL (Fig. 9 B, D). Similar results were obtained in two independent experiments each examining 4 and 6 mice. Thus, our data demonstrates that one of the potential mechanisms of induction of lymphocyte apoptosis by L. monocytogenes infection is dependent on granzyme. Experiments of others have not shown such a protective effect with Prf−/− mice. Perforin deficiency leads to impairment in sterilization of the splenic compartment, but the overall bacterial growth is unaffected (39). This result has been confirmed by our laboratory (data not shown). The major phenotype of perforin during L. monocytogenes infection is in the secondary response, where it is required in the CD8+ T cell compartment to mount an effective recall response. Additionally, we have examined the splenic pathology in Prf−/− mice following infection with L. monocytogenes, and did not see a major difference in the extent or presence of apoptotic lesion.
Figure 8.

Increased resistance to infection in granzyme AC mice. A-D) Mice were infected with ∼2.5×104 CFU intraperitoneally. After the indicated days (2 or 4 d) mice were sacrificed and colony counts were determined as described previously (21). Each bar represents the mean+/− S.E.M. for a total of 10 mice analyzed in two independent experiments. p-values were determined using the Mann-Whitney unpaired test with two-tailed p-values and 95% confidence intervals.
Figure 9.

Lymphocyte apoptosis is decreased in granzyme AC mice following infection with L. monocytogenes. Mice were infected with 2.5×104 CFU of L. monocytogenes and spleens were stained by either hematoxylin and eosin (A-B), or TUNEL (C-D). A and C show the results obtained from 129/SvJ mice. B and D show the results obtained from granzyme AC mice. All micrographs were taken at 400x magnification. Black bars represent 50 micrometers. Results are representative of two independent experiments containing 10 total mice per strain.
Discussion
This study shows that granzymes are a major participant in lymphocyte apoptosis mediated by LLO, in vitro, as well as in vivo, during the first days of infection. The activation of caspases, kinetics of apoptosis and the magnitude were all dependent on granzyme expression.
The results are best explained by an increase in permeability of vesicular compartments of the lymphocytes induced by LLO. When LLO is initially internalized it triggers a response by the cell which causes translocation of granzyme from the lytic granule into the cytosol. This effect is enhanced by the presence of perforin. Breakdown of the lytic granule requires that the phagolysosomal compartments acidify; suggesting that increased LLO activity in the endocytic compartment is required for inducing granzyme release into the cytosol. Indeed, chloroquine, which increases lysosomal pH, protected the lymphocyte from the rapid apoptotic death. We do not favor the interpretation that LLO allows transfer of granzyme from one cell to the next: neither cells treated with LLO nor the supernatant following LLO treatment induced apoptosis in fresh cells (4). Labeling with CFDA-SE demonstrated that only the 129/SvJ cells and not GzmAC cells could activate caspase-3 following LLO activation, further excluding the possibility of cell to cell transfer as the mechanism of LLOs proapoptotic activities. Along this line, a previous report from Trapani's laboratory used LLO to deliver extracellular granzyme into target cells as a proof that LLO could act similarly to perforin in serving as a conduit to the cytosol. Thus the precedent is there that LLO can permit the passage of an enzyme into cytosol, and our studies place these findings into a pathophysiological context. Others have examined the mechanisms whereby granzyme induces apoptosis and envision a scenario in which granzyme induces maximal apoptosis through cleavage of bid to promote release of mitochondrial factors, apoptosome formation, and caspase-9 activation. Granzyme B can also directly cleave procaspase-3 into the active form, but this pathway is inefficient in the absence of bid activity (38).
The effects of LLO on T cells are akin to a model proposed for perforin in delivering granzyme to target cells (34). In the hybrid model proposed by Lieberman, perforin triggers coendocytosis of granzyme through calcium signaling and then disrupts the endocytic vesicle to allow entry of granzyme into the cytosol (34, 40, 41). It is believed that perforin and granzyme enter the target cells through calcium dependent endocytosis and that the perforin allows granzyme egress from the endosome to the cytosol (25, 34, 42). In the case of LLO, its effects by way of granzyme were also enhanced by the presence of perforin.
The effects of LLO may vary depending on the cell type and its content of vesicular enzymes. In the case of L. monocytogenes infected macrophages and dendritic cells the cell death has slower kinetics than the death process seen in lymphocytes following LLO treatment (43-45). There is no immediate caspase activation or phosphatidylserine exposure in macrophages or dendritic cells either infected with L. monocytogenes or treated with LLO (Carrero and Unanue, unpublished observations, (45). We speculate that it would be disadvantageous to the microbe to kill its cellular host too quickly, but important to cause rapid death of adaptive immune cells. In fact, LLO is degraded very quickly in the cytosol of infected phagocytic cells and mutants of LLO that are not degraded or properly translationally regulated kill the host cell and decrease L. monocytogenes virulence (11, 12, 46). It remains to be determined if other cell types that have high granzyme or protease content such as NK cells or neutrophils are also rapidly killed by LLO.
We still detected a slow cell death in limited cells, in the absence of granzymes and after blocking proteolytic activity. Examination of this slow death program is in progress but it appears not to depend on extracellular calcium signaling, cathepsins, granzymes, or autophagy. Some good candidates for slow death triggering of lymphocyte following LLO treatment are apoptosis inducing factor, reactive oxygen intermediates, or reactive nitrogen moieties. To note is that pneumolysin triggers the release of intracellular calcium stores which leads to release of apoptosis inducing factor from the mitochondria and subsequent apoptosis in a peroxide-dependent manner (47).
In vivo, the peak of apoptosis was detected in wild-type mice at 48 hours following infection. This coincides with the earliest detectable non-specific activation of lymphocytes following infection with L. monocytogenes (19, 48). The apoptotic lesion resolves by 96 hours following infection, presumably because of the initiation of a robust adaptive immune response that controls the infection (1). Removal of granzyme led to decreased apoptosis and bacterial titer. However, the difference was smaller compared to mice lacking lymphocytes (scid or Rag−/−) or mice lacking the type I interferon receptor (18-21). The difference in the former (i.e. normal vs. granzyme AC) was ∼10-15 fold compared to the latter (i.e. wild-type vs. scid/Rag−/− or type I interferon receptor−/−) which was ∼100-1000 fold. We propose that there are multiple cell death pathways operating during L. monocytogenes infection. Some, like granzyme dependent LLO cell death, are driven directly by the bacterial toxin(s). But in contrast, the perforin deficient mice moreover, have a slight defect in clearance of the infection (39, 49). Other host molecules, like TRAIL enhance apoptosis after infection with L. monocytogenes, although the mechanism of induction of this pathway remains unresolved (50). In addition to a role for granzymes in the death of lymphocytes following infection, it is possible that other granzyme/protease expressing cell types, such as NK cells and neutrophils may be dying by L. monocytogenes-induced apoptosis during the early phase of the infection. This remains to be determined.
Acknowledgements
We wish to thank Dr. Boris Calderon for assistance with staining of tissue sections. We thank Kathy Frederick for her animal husbandry. We thank Dr. Timothy J. Ley for his kind gift of mice.
Footnotes
Disclosures
The authors have no financial conflict of interest.
This work was funded by the National Institutes of Health.
Abbreviations used in this paper: GzmAC, granzyme A−/− x B cluster deficient mice; Gzma−/−, granzyme A−/− mice; Gzmb−/−, granzyme B−/− mice; Gzma−/− x b−/−, Gzma−/− crossed to Gzmb−/− mice; ConA, Concanavalin A; Prf, Perforin.
References
- 1.Merrick JC, Edelson BT, Bhardwaj V, Swanson PE, Unanue ER. Lymphocyte apoptosis during early phase of Listeria infection in mice. Am J Pathol. 1997;151:785–792. [PMC free article] [PubMed] [Google Scholar]
- 2.Rogers HW, Callery MP, Deck B, Unanue ER. Listeria monocytogenes induces apoptosis of infected hepatocytes. J Immunol. 1996;156:679–684. [PubMed] [Google Scholar]
- 3.Schluter D, Domann E, Buck C, Hain T, Hof H, Chakraborty T, Deckert-Schluter M. Phosphatidylcholine-specific phospholipase C from Listeria monocytogenes is an important virulence factor in murine cerebral listeriosis. Infect Immun. 1998;66:5930–5938. doi: 10.1128/iai.66.12.5930-5938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrero JA, Unanue ER. Lymphocyte apoptosis as an immune subversion strategy of microbial pathogens. Trends Immunol. 2006;27:497–503. doi: 10.1016/j.it.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Ulett GC, Adderson EE. Regulation of Apoptosis by Gram-Positive Bacteria: Mechanistic Diversity and Consequences for Immunity. Curr Immunol Rev. 2006;2:119–141. doi: 10.2174/157339506776843033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couve E, de Daruvar A, Dehoux P, Domann E, Dominguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, Garcia-del Portillo F, Garrido P, Gautier L, Goebel W, Gomez-Lopez N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, de Pablos B, Perez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, Simoes N, Tierrez A, Vazquez-Boland JA, Voss H, Wehland J, Cossart P. Comparative genomics of Listeria species. Science. 2001;294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- 7.Palmer M. The family of thiol-activated, cholesterol-binding cytolysins. Toxicon. 2001;39:1681–1689. doi: 10.1016/s0041-0101(01)00155-6. [DOI] [PubMed] [Google Scholar]
- 8.Kayal S, Charbit A. Listeriolysin O: a key protein of Listeria monocytogenes with multiple functions. FEMS Microbiol Rev. 2006;30:514–529. doi: 10.1111/j.1574-6976.2006.00021.x. [DOI] [PubMed] [Google Scholar]
- 9.Rossjohn J, Polekhina G, Feil SC, Morton CJ, Tweten RK, Parker MW. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J Mol Biol. 2007;367:1227–1236. doi: 10.1016/j.jmb.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tweten RK. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnupf P, Portnoy DA, Decatur AL. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST-like sequence. Cell Microbiol. 2006;8:353–364. doi: 10.1111/j.1462-5822.2005.00631.x. [DOI] [PubMed] [Google Scholar]
- 12.Schnupf P, Zhou J, Varshavsky A, Portnoy DA. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect Immun. 2007 doi: 10.1128/IAI.00164-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madden JC, Ruiz N, Caparon M. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell. 2001;104:143–152. doi: 10.1016/s0092-8674(01)00198-2. [DOI] [PubMed] [Google Scholar]
- 14.Gaillard JL, Berche P, Mounier J, Richard S, Sansonetti P. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect Immun. 1987;55:2822–2829. doi: 10.1128/iai.55.11.2822-2829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Portnoy DA, Jacks PS, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edelson BT, Cossart P, Unanue ER. Cutting edge: paradigm revisited: antibody provides resistance to Listeria infection. J Immunol. 1999;163:4087–4090. [PubMed] [Google Scholar]
- 17.Edelson BT, Unanue ER. Intracellular antibody neutralizes Listeria growth. Immunity. 2001;14:503–512. doi: 10.1016/s1074-7613(01)00139-x. [DOI] [PubMed] [Google Scholar]
- 18.Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med. 2006;203:933–940. doi: 10.1084/jem.20060045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrero JA, Calderon B, Unanue ER. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J Immunol. 2004;172:4866–4874. doi: 10.4049/jimmunol.172.8.4866. [DOI] [PubMed] [Google Scholar]
- 23.Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J Cell Biol. 2002;156:1029–1038. doi: 10.1083/jcb.200201081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beauregard KE, Lee KD, Collier RJ, Swanson JA. pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J Exp Med. 1997;186:1159–1163. doi: 10.1084/jem.186.7.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Browne KA, Blink E, Sutton VR, Froelich CJ, Jans DA, Trapani JA. Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol Cell Biol. 1999;19:8604–8615. doi: 10.1128/mcb.19.12.8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, Ley TJ. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–646. doi: 10.1016/j.immuni.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 27.Pham CT, MacIvor DM, Hug BA, Heusel JW, Ley TJ. Long-range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci U S A. 1996;93:13090–13095. doi: 10.1073/pnas.93.23.13090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shresta S, Graubert TA, Thomas DA, Raptis SZ, Ley TJ. Granzyme A initiates an alternative pathway for granule-mediated apoptosis. Immunity. 1999;10:595–605. doi: 10.1016/s1074-7613(00)80059-x. [DOI] [PubMed] [Google Scholar]
- 29.Trapani JA. Granzymes: a family of lymphocyte granule serine proteases. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-12-reviews3014. REVIEWS3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104:2840–2848. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- 31.Devadas S, Das J, Liu C, Zhang L, Roberts AI, Pan Z, Moore PA, Das G, Shi Y. Granzyme B is critical for T cell receptor-induced cell death of type 2 helper T cells. Immunity. 2006;25:237–247. doi: 10.1016/j.immuni.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A. 1999;96:10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosado CJ, Buckle AM, Law RH, Butcher RE, Kan WT, Bird CH, Ung K, Browne KA, Baran K, Bashtannyk-Puhalovich TA, Faux NG, Wong W, Porter CJ, Pike RN, Ellisdon AM, Pearce MC, Bottomley SP, Emsley J, Smith AI, Rossjohn J, Hartland EL, Voskoboinik I, Trapani JA, Bird PI, Dunstone MA, Whisstock JC. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317:1548–1551. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 34.Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol. 2007;19:301–308. doi: 10.1016/j.coi.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabbagh L, Kaech SM, Bourbonniere M, Woo M, Cohen LY, Haddad EK, Labrecque N, Ahmed R, Sekaly RP. The selective increase in caspase-3 expression in effector but not memory T cells allows susceptibility to apoptosis. J Immunol. 2004;173:5425–5433. doi: 10.4049/jimmunol.173.9.5425. [DOI] [PubMed] [Google Scholar]
- 36.Schuerch DW, Wilson-Kubalek EM, Tweten RK. Molecular basis of listeriolysin O pH dependence. Proc Natl Acad Sci U S A. 2005;102:12537–12542. doi: 10.1073/pnas.0500558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wowk ME, Trapani JA. Cytotoxic activity of the lymphocyte toxin granzyme B. Microbes Infect. 2004;6:752–758. doi: 10.1016/j.micinf.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 39.Kagi D, Ledermann B, Burki K, Hengartner H, Zinkernagel RM. CD8+ T cell-mediated protection against an intracellular bacterium by perforin-dependent cytotoxicity. Eur J Immunol. 1994;24:3068–3072. doi: 10.1002/eji.1830241223. [DOI] [PubMed] [Google Scholar]
- 40.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 41.Bolitho P, Voskoboinik I, Trapani JA, Smyth MJ. Apoptosis induced by the lymphocyte effector molecule perforin. Curr Opin Immunol. 2007;19:339–347. doi: 10.1016/j.coi.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Froelich CJ, Orth K, Turbov J, Seth P, Gottlieb R, Babior B, Shah GM, Bleackley RC, Dixit VM, Hanna W. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J Biol Chem. 1996;271:29073–29079. doi: 10.1074/jbc.271.46.29073. [DOI] [PubMed] [Google Scholar]
- 43.Guzman CA, Domann E, Rohde M, Bruder D, Darji A, Weiss S, Wehland J, Chakraborty T, Timmis KN. Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol Microbiol. 1996;20:119–126. doi: 10.1111/j.1365-2958.1996.tb02494.x. [DOI] [PubMed] [Google Scholar]
- 44.Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, Unger H, Chakraborty T, Levy DE, Muller M, Decker T. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol. 2002;169:6522–6529. doi: 10.4049/jimmunol.169.11.6522. [DOI] [PubMed] [Google Scholar]
- 45.Barsig J, Kaufmann SH. The mechanism of cell death in Listeria monocytogenes-infected murine macrophages is distinct from apoptosis. Infect Immun. 1997;65:4075–4081. doi: 10.1128/iai.65.10.4075-4081.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Villanueva MS, Sijts AJ, Pamer EG. Listeriolysin is processed efficiently into an MHC class I-associated epitope in Listeria monocytogenes-infected cells. J Immunol. 1995;155:5227–5233. [PubMed] [Google Scholar]
- 47.Braun JS, Sublett JE, Freyer D, Mitchell TJ, Cleveland JL, Tuomanen EI, Weber JR. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J Clin Invest. 2002;109:19–27. doi: 10.1172/JCI12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang J, Lau LL, Shen H. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. Journal of Immunology. 2003;171:4352–4358. doi: 10.4049/jimmunol.171.8.4352. [DOI] [PubMed] [Google Scholar]
- 49.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 50.Zheng SJ, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol. 2004;173:5652–5658. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]