

Abstract
Introduction
Succinic semialdehyde dehydrogenase (SSADH) deficiency (γ-hydroxybutyric aciduria) is a rare neurometabolic disorder of γ-aminobutyric acid degradation. While neurological manifestations, such as developmental delay, are typical during infancy, limited data are available on adolescent and adult symptomatology.
Methods
We overview the phenotype of 33 adolescents and adults (10.1–39.5 years of age, mean: 17.1 years, 48% females) with SSADH deficiency. For this purpose, we applied a database with systematic questionnaire-based follow-up data.
Results
Two thirds of patients (n=21) presented by 6 months of age, 14% from 6–12 months of age, 5% from 1–2 years of age, and 14% from 2–4 years of age, mean age at first symptoms was 11±12 months. However, mean age at diagnosis was 6.6±6.4 years of age. Presenting symptoms encompassed motor delay, hypotonia, speech delay, autistic features, seizures, and ataxia. Eighty-two percent demonstrated behavioral problems, such as attention deficit, hyperactivity, anxiety, or aggression, and 33% had ≥3 behavior problems. Electroencephalograms showed background slowing or epileptiform discharges in 40% of patients. Treatment approaches are then summarized.
Conclusion
The variable phenotype in SSADH deficiency suggests the likelihood that this disease may be under-diagnosed. Families of patients with SSADH deficiency should be counseled and supported regarding the anticipated persistence of various neuropsychiatric symptoms into adulthood.
FOCUS POINTS.
Succinic semialdehyde dehydrogenase deficiency is a rare disorder of γ-aminobutyric acid metabolism with a high neuropsychiatric morbidity.
Presenting symptoms in younger children are motor delay, hypotonia, speech delay, autistic features, seizures, and ataxia.
The phenotype in older patient comprises various neuropsychiatric symptoms, such as mental deficits, seizures, and various behavior problems.
Patients with complex neuropsychiatric problems in the setting of a suspected organic etiology should be screened via urine organic acid analysis.
INTRODUCTION
Succinic semialdehyde dehydrogenase (SSADH) deficiency is a raredisorder of γ-aminobutyric acid (GABA) metabolism. We summarize the neuropsychiatric symptomatology in a cohort of 33 adolescents and adults. The variable phenotype suggests the likelihood that SSADH deficiency may be under-diagnosed. Our findings indicate the occurrence of various neuropsychiatric symptoms in adolescence and adulthood.
SSADH deficiency (γ-hydroxybutyric aciduria; OMIM 271980, 610045) is an autosomal-recessive inherited disorder of GABA metabolism. The phenotype is variable and non-specific, including a variety of neurological and psychiatric symptoms. 1-7 The diagnosis requires a high degree of clinical suspicion. It may be unrecognized, as it has the course of a nonprogressive encephalopathy or even a psychiatric disorder, and lacks the cardinal features associated with most metabolic encephalopathies, such as hypoglycemia, hyper-ammonemia, or intermittent lethargy. Patients are often assigned diagnoses such as global developmental delay, pervasive developmental delay, or cerebral palsy. Approximately 50% of SSADH deficiency patients have epilepsy, usually of the generalized type.6 Magnetic resonance imaging may show increased T2-weighted signal abnormalities usually involving the globi pallidi, cerebellar dentate nucleus, white matter, or brainstem. A normal magnetic resonance imaging is reported in almost 40% of patients.6
Approximately 10% of patients present with a more severe phenotype characterized by developmental regression; this group appears to have extrapyramidal manifestations.5 It is likely that multiple neurotransmitter disturbances, in association with profound glial and neuronal dysfunction, underlie the pathophysiology of SSADH deficiency. A number of potential pathomechanisms have been identified in the Aldh5a1−/− mouse model. 8-13
The underlying enzyme deficiency impairs the oxidation of succinic semialdehyde (SSA) to succinic acid (Figure), leading to the accumulation of SSA and its downstream metabolite γ-hydroxybutyric acid (GHB). Detection of GHB, on urine organic acid testing, is performed with gas chromatography-mass spectrometry.14 Diagnostic pitfalls are the variable excretion of GHB in urine, its potential volatilization from acidified urine using organic solvent extraction and the exogenous application of GHB as a drug.15 The diagnosis may be confirmed using enzyme analysis in leukocytes,16,17 which can be augmented with molecular genetic analysis of the Aldh5a1 gene at chromosome 6p22.8,19
The first case of SSADH deficiency was described over 25 years ago.20 The same group also identified the first adult case.21 There are only a few reports on adolescent and adult patients in the literature; the major manifestations reported in this age group are expressive language dysfunction, sleep anomalies, and psychiatric symptoms.1-3,7,22 Owing to the non-specific clinical features and specific diagnostic requirements in physiological fluids, SSADH deficiency is likely under-diagnosed in adolescent and adult patients. In this report, we focus on neuropsychiatric morbidity reported in confirmed patients >10 years of age.
METHODS
The SSADH-deficient patient database maintained in the Department of Neurology at Children's National Medical Center in Washington, DC, contains systematic questionnaire-based, anonymized data of 63 patients (retrospective analysis with longitudinal follow-up). This study was approved by the Children's National Medical Center institutional review board. The cohort's mean age at the current time is 12.1±7.6 years of age (median age: 11.8 years, age range: 2.2–39.6 years).
There were 33 patients (48% females) identified >10 years of age. Mean age of this group is 17.1±6.4 years of age (median age: 15.2. years, age range: 10.1–39.6 years). Parental consanguinity was noted in six (18%) patients. Two-thirds of patients were white and the remainder of varying ethnicity. There were affected siblings (two children) in three families.
Language skills, cognitive performance, as well as gross and fine motor skills were stratified by a physician into five levels (1=severe deficit, 2=moderate, 3=mild, 4=minor partial restrictions, 5=normal performance.) Hypotonia was classified according to severity into four levels (0=none, 1=mild, 2=moderate, 3=severe). Ataxia was classified into six categories (decreased balance, wide-based gait, uncoordinated walking, uncoordinated movements, hand tremors, and excessive movements during fine motor tasks). Behavioral problems were classified into inattention, hyperactivity, anxiety, obsessive-compulsive disorder, aggression, hallucinations, pervasive developmental disorder, and autistic behavior. Sleep disturbances were categorized into four categories (difficulty falling asleep, difficulty maintaining sleep, daytime somnolence, and disrupted sleep).
Data were analyzed using Graph Pad Prism software 4.0 (San Diego, Calif.) and SPSS version 14.0. Values are given as mean ± standard deviation. Spearman correlation and multivariate regression analysis were used to evaluate possible associations, if applicable, and differences between male and female patients were tested with Mann-Whitney U test. A P value <.05 was considered significant.
RESULTS
Presenting Symptoms and Developmental History
In 21 patients, age at onset could be tracked in detail. Fourteen patients presented from birth to 6 months of age (67%), 3 patients (14%) from 6–12 months of age, one patient (5%) between 12–24 months of age, and three patients (14%) between 2 and 4 years of age. Mean age at first symptoms was 11±12 months (median age: 6 months, age range 0-44 months). Mean age at diagnosis was 6.6±6.4 years (median age: 5 years of age, range: 0–25 years). One patient was diagnosed soon after birth in relation to an affected sibling. The two patients in whom diagnosis was established as late as 21 and 25 years of age, respectively, presented early in life with global delays associated with absences or generalized tonic-clonic convulsions (GTC). Generally, there was a strong correlation between age at disease onset and age at diagnosis (r=0.4509, P<.05).
Developmental history revealed global delays of milestones for the entire cohort. In detail, mean age for sitting up was 10.0±2.6 months, for walking 21.8±15.6 months, and for first words 27.3±13.1 months. Two males and two females did not develop expressive language, and one male manifested regression of language skills at 30 months of age. Presenting symptoms were gross motor delay (64%), hypotonia (58%), speech delay (55%), fine motor delay (45%), global delay (48%), autistic features (12%), seizures (12%), and ataxia (9%). Normal early infantile development was noted in two patients, who presented at 18–24 months of age with speech delay and ataxia or seizures. Statistical analysis using Spearman's correlation demonstrated a link between later age at presentation and earlier age of independent walking (r=−0.5614, P=.0081). Age at onset, age at diagnosis, P and number of presenting symptoms did not differ between both sexes.
Ataxia
At the first presentation or during the disease course, 20 patients (61%) showed various atactic features (Table 1). Twenty-one percent of patients exhibited at least three features. Two patients who manifested ataxia as the main symptom when diagnosed at 2 and 3 years of age, respectively, showed persistent anomalies in at least four categories relating to ataxia.
TABLE 1.
Clinical Phenotype, Including Behavior Problems, Ataxia, and Seizures, in 33 Adolescent and Adult SSADH Deficiency Patients (mean age: 12.1±7.6 years, range: 2.2-39.6 years; 48% females)
| Clinical Phenotype in Adolescent and Adult Patients with SSADH Deficiency (N=33) | ||
| Symptoms | n | Percentage |
| Developmental delay | 33 | 100 |
| Behavior problems | 27 | 82 |
| Ataxia | 20 | 61 |
| Seizures | 19 | 58 |
| Hypotonia | 15 | 45 |
| Sleep disturbances | 15 | 45 |
| Behavior Problems in Patients with SSADH Deficiency | ||
| Symptoms | n | Percentage |
| Attention-deficit | 18 | 55 |
| Hyperactivity | 13 | 39 |
| Anxiety | 12 | 36 |
| Obsessive-compulsive | 11 | 33 |
| Aggressive behavior | 6 | 18 |
| Hallucinatory episodes | 5 | 15 |
| Autistic features | 4 | 12 |
| Ataxia in Patients with SSADH Deficiency | ||
| Symptoms | n | Percentage |
| Decreased balance | 8 | 24 |
| Uncoordinated movements | 8 | 24 |
| Wide-based gait | 7 | 21 |
| Uncoordinated walking | 7 | 21 |
| Hand tremors | 6 | 18 |
| Excessive movements | 4 | 12 |
| Tripping | 2 | 6 |
| Seizures in Patients with SSADH Deficiency | ||
| Symptoms | n | Percentage |
| Generalized tonic-clonic | 14 | 42 |
| Absence | 12 | 36 |
| Myoclonic | 3 | 9 |
| Others (febrile, unspecified) | 9 | 27 |
SSADH=succinic semialdehyde dehydrogenase.
Behavior Problems
Twenty-seven (82%) of patients had various behavior problems (Table 1). Eleven patients (33%) demonstrated at least three behavior problems. Moreover, we found a significant correlation between occurrence of ataxic features and number of behavioral problems (r=0.3726, P=0.0327).
Sleep Disturbances
Sleep disturbances were classified into four categories as previously mentioned. Fifteen patients (45%) had at least one major sleep problem, primarily difficulties in sleep maintenance. One patient had daytime somnolence.
Follow-Up
Of the 33 patients currently ≥10 years of age, 24 were available for follow-up assessment of language, cognition, and gross and fine motor performance (Table 2). Language delay was a cardinal symptom, as none of the patients had normal language skills. Fifteen patients had reduced muscle tone and strength in relation to initial presentation, three patients had improved muscular tone, and one boy had persistent severe hypotonia.
TABLE 2.
Follow-up Assessment of Language, Cognition, and Gross and Fine Motor Performance in 24 Adolescent and Adult SSADH Deficiency Patients (mean age: 18.1±7.0 years, age range: 10.6–40.0 years; 50% females)
| Items |
Relative Units (mean) |
Relative Units (Range) |
|---|---|---|
| Language skills | 2.0±1.1 | 1–4 |
| Cognitive performances | 2.3±1.1 | 1–4 |
| Gross motor performances | 3.2±0.8 | 3–4 |
| Fine motor performances | 2.5±0.9 | 2–4 |
The mean follow-up period was 11. ±4.6 years. (range: 5.3-21.4 years.)
All performances were classified by an experienced physician in relative units from 1 (severe deficit) to 5 (normal).
SSADH=succinic semialdehyde dehydrogenase.
Seizures
Nineteen patients (58%) developed seizures during the course of the disease, primarily GTC convulsions or absences (Table 1). Simple and complex partial seizures were not independently reported, but the possibility remains that some of the patients for whom unspecified seizures were described may have had focal seizures. In the nine patients suffering from absences and GTC, significant behavioral problems were found in eight (89%), developmental deficits in seven (78%), and ataxia in six patients (67%).
Electroencephalography Recordings
For 25 patients, electroencephalography (EEG) follow-up data were available. Forty percent showed persistent anomalies, such as pathological slowing or epileptiform discharges. As expected, there was a positive correlation between epileptiform discharges and clinical seizure activity (r=0.6226, P<.0001). Statistically, there was a significant association between EEG anomalies and the presence of behavior problems, seizures, and ataxic features in the course of the disease (P<.0001), which suggests that an abnormal EEG is consistent with a more severe encephalopathy.
Anticonvulsants
Seventeen patients (52% of cohort) were at least treated transiently with anticonvulsants. For 13 out of the 19 patients with at least one seizure, long-term seizure medications were used. Fourteen patients received vigabatrin [AU: DOSAGE?], which is theoretically a logical choice because this irreversible inhibitor of GABA-transaminase prevents the formation of GHB (although GABA levels will not be decreased). However, the vigabatrin was discontinued due to side effects, such as heightened hypotonia with drop attacks, drowsiness, or absence of therapeutic benefit, in seven patients (50%). “Poor” vigabatrin responders, and those with adverse effects, were often older with earlier presentation compared with those considered vigabatrin responders, whereas neuropsychiatric morbidity did not differ between these subjective groups. No patient had sustained seizure control with vigabatrin. Three patients received valproate 30 mg per kg/day divided BID or 1,500 mg BID in adults, and one of them reported improved seizure control. No other neurodevelopmental benefit (or deterioration) was noted. Carbamazepine 20 mg per kg/day divided BID or 1,000 mg BID in adults was employed in five patients. Of these, two patients showed a good seizure response and one patient had reported improvement in concentration and sleep. Two patients received topiramate 5 mg/kg/day divided BID or 250 mg BID in adults, phenytoim 5 mg/kg/day divided BID or 200 mg BID in adults, or phenobarbital 5 mg/kg/day divided BID or 100 mg BID in adults without obvious clinical benefits. Of the five patients who received lamotrigine 5 mg/kg/day divided BID or 200 mg BID in adults, one showed a good clinical response. Overall, seven patients were treated with more than one antiepileptic.
Behavioral Medicines
Eight of 33 patients (24%) were treated with behavioral medications, including methylphenidate, risperidone, fluoxetine, and fluvoxamine. Seven of 33 patients (21%) received methylphenidate. In one, methylphenidate 10–20 mg TID improved attention, balance, and coordination based on the parental questionnaire. Two patients were treated with risperidone 2 mg BID, resulting in behavioral improvement in one. Fluoxetine 20 mg/day was administered to one patient and was effective for anxiety and obsessive- compulsive symptoms. This patient was concomitantly on carbamazepine 20 mg/kg/day divided BID or 1,000 mg BID in adults, methylphenidate 10-20 mg TID and risperidone 2 mg BID. One patient received fluvoxamine 50 mg BID without therapeutic benefit.
Taurine And Dietary Approaches
Because of efficacy in the SSADH-deficient (Aldh5a1−/−) mouse model, taurine 500 mg BID was given to two patients without clear benefit. None of the patients with epilepsy were treated with the ketogenic diet during the observational period.
DISCUSSION
There are few reports describing the clinical spectrum of SSADH deficiency in adolescents and adults.1,3,7,21 In our cohort, the most prevalent symptoms were neuropsychiatric, including behavior problems and sleep disturbances. Almost two thirds of patients experienced seizures, but irreversible status epilepticus or worsening of overall symptomatology with age was uncommon. Sleep disturbances were observed in 45% of patients. A single published case report of polysomnography in SSADH deficiency22 revealed subtle sleep abnormalities and, during a second consecutive night of monitoring, increased slow-wave sleep following an epileptic convulsion. Moreover, patients with SSADH deficiency demonstrate EEG recordings that may show background slowing, disorganization, or epileptiform discharges that are usually 2–3 Hz generalized spike-and-wave complexes.23
Human SSADH deficiency typically has the temporal course of a static encephalopathy, in contrast with the corresponding animal model. SSADH-deficient knockout mice (Aldh5a1−/− mice) present with progressive neurologic deterioration and essentially uniform mortality by 1 month of age.10,11 Differences between the human and murine disorders could relate to species-specific features, enzymatic variations or different compensatory mechanisms. Pathophysiology is being characterized in Aldh5a1−/− mice. High levels of GHB and GABA in the mouse model eventually lead to receptor dysfunctions and other probable neurotoxic effects.8,11,24-28 In the murine model, downregulation of GABA(B) and GABA(A) receptor function, likely the result of high circulating GHB and GABA, may contribute to the progression of generalized convulsive seizures.27,29-31 Of interest, Mehta and colleagues32 demonstrated that most pathophysiological alterations are likely at GABAergic systems in the Aldh5a1−/− mice, as there were no detectable alterations of GHBergic binding or activity in the animal model. Other pathogenetic mechanisms in Aldh5a1−/− mice include dysregulated glutamine metabolism and functional glutamate receptor abnormalities,2,28 altered dopaminergic neurotransmission, oxidative stress, and myelin alterations.9,12,33-37
At least two pharmacologically active species, GABA and GHB, accumulate in heritable SSADH deficiency (Figure). The pharmacology of GABA is well described, and at least one third of brain synapses utilize it as an inhibitory neurotransmitter. 38 Of interest, in the developing embryo GABA is excitatory and critically important in the development and patterning of the synapse.34 Conversely, the exact roles of GHB in the central nervous system are incompletely defined. GHB is capable of rapidly passing the blood-brain barrier into the central nervous system; it may function as a mood-affecting sedative drug and treatment option for alcohol addiction or even narcolepsy. 39,40 Patients acutely intoxicated with GHB as a selfadministered euphoric agent display sedation, amnesia, or even coma, but they may also present with paradoxical agitation and ataxia compared with some of the symptoms seen in our older patient cohort.41 In baboons, chronic administration of GHB leads to physical dependence and withdrawal phenomena.42,43
Therapeutic approaches in SSADH deficiency have been challenging, perhaps related to the complex disease pathophysiology. Vigabatrin, an irreversible inhibitor of GABA transaminase (Figure), is the most widely used therapy, but it may exacerbate the hyper-GABAergic status.1,6,44-46 Thus, it is not surprising that clinical results with vigabatrin are not uniform, ranging from partial efficacy to neurological decline.1,47-49 In our cohort, vigabatrin-poor responders, and those patients with severe adverse effects, were generally older and had presented earlier in life compared with those considered vigabatrin responders. Application of vigabatrin enhances survival of the Aldh5a1−/− mice at high doses.25,50 vigabatrin may be considered in individual cases, but its overall beneficial effect in patients with SSADH deficiency remains in question.
Vigabatrin augments GABAergic functions in some brain areas and modulates N-methyl-Daspartate receptor function.51,52 GHB concentrations increased during vigabatrin therapy in a patient with SSADH deficiency.53 vigabatrin may also down-regulate SSADH activity in vitro,54 suggesting that it should be used with caution in this disease. Carbamazepine and lamotrigine showed some efficacy in individual cases, whereas topiramate, phenytoin, or phenobarbital were not clinically beneficial. In Aldh5a1−/− mice, ethosuximide was effective in ameliorating absence seizures.1,55 Phenytoin and phenobarbital were unsuccessful in rescue from status epilepticus.25 Benzodiazepines enhance GABAergic effects by binding to the GABA(A) receptor. There are limited data showing that benzodiazepines may lead to decreased aggression and agitation in patients with SSADH deficiency,2 but it may exacerbate ataxia and hypotonia.
To produce its stimulant effect, methylphenidate activates the brainstem arousal system and cortex, whereas risperidone affects primarily dopaminergic and serotoninergic neurotransmission. Conversely, fluoxetine and fluvoxamine are selective serotonin reuptake inhibitors. Outcome data for methylphenidate, risperidone, fluoxetine, or fluvoxamine in SSADH-deficient patients are anecdotal. Taurine is a sulfur-containing amino acid that exhibits neuroprotective and neuromodulating properties and is a candidate inhibitory neurotransmitter.56 Its potential therapeutic value in SSADH deficiency has been reviewed.35 In Aldh5a1−/− mice, taurine application attenuated early lethality.50 Although taurine has not been investigated in controlled trials (K.M. Gibson, PhD, personal communications [AU: WAS THE COMMUNICATIONS WRITTEN OR ORAL? PLEASE PROVIDE YEAR OF COMMUNICATION.], it has been administered to two patients without clear beneficial effects. However, controlled trials with taurine are needed, before solid conclusions can be made. Finally, another potential approach in the treatment of epilepsy associated with SSADH deficiency, particularly in view of the questionable efficacy of various anticonvulsants, is the ketogenic diet. Fasting or ketogenic diet treatment produces ketone bodies, such as β-hydroxybutyric acid, which serve as major brain fuels and spare glucose consumption.57 Ketogenic diet is often applied in children and adolescents with severe epilepsy,58,59 but it is not generally used in adults. Although its mode of action is not totally characterized, there is evidence that Ketogenic diet improves bioenergetics and manifests neuroprotective effects.57 While data on the efficacy of Ketogenic diet in patients with SSADH deficiency are not yet available, there are promising results that Ketogenic diet improves synaptic function and survival rate in Aldh5a1−/− mice.60
Although the number of patients in our study was small, it includes ~10% of all patients diagnosed with SSADH deficiency worldwide. Variation in phenotype and time of presentation, and the non-specific clinical features, suggest that SSADH deficiency may be frequently overlooked in the clinic. The pathophysiology of SSADH deficiency seems complex, involving various metabolic pathways and systems. Further studies in the Aldh5a1−/− mouse model will be beneficial in this arena. Moreover, other model systems could have utility, such as a tissue-specific knockout (eg, neural tissue only) or a conditional knockout manipulated at particular developmental periods. Ablation of the Aldh5a1 gene in other species (eg, zebrafish, yeast) could provide new insight into the pathophysiology of the human disorder. For the future, therapeutic intervention in the SSADH-deficient state will likely need to target multiple neural systems and probably comprise a multi-drug therapy.
CONCLUSION
Neuropsychiatric morbidity is prevalent in older children and adults with SSADH deficiency. Patients presenting with behavior problems, attentional deficits, sleep disturbances, hyperactivity, anxiety, obsessive-compulsive symptoms, and aggression in the setting of a suspected organic etiology should be screened utilizing urine organic acid analysis. Additionally, families of diagnosed patients should be counseled regarding the expected occurrence of neuropsychiatric symptoms into adulthood. CNS
FIGURE. GABA metabolic interrelationships and conversion to succinate in the mitochondria.
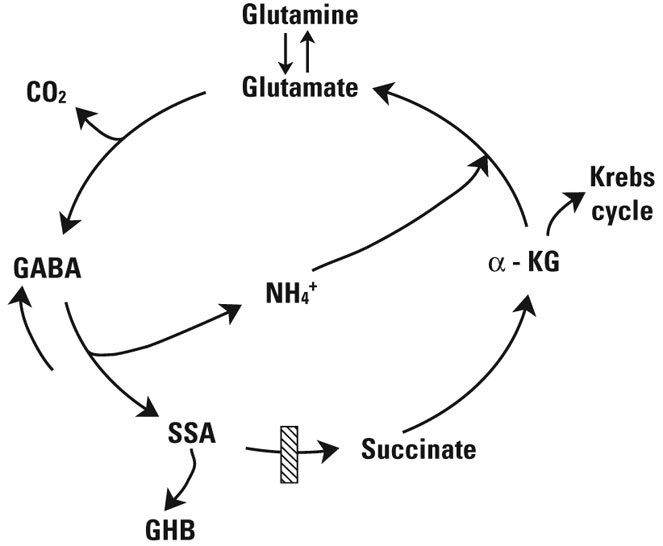
The site of the defect in patients with SSADH deficiency (and Aldh5a1−/− mice) is depicted by the hatched box. GABA is synthesized primarily from glutamate via glutamic acid decarboxylase, an irreversible reaction that releases CO2. GABA is transaminated to SSA by the action of GABA-transaminase. The NH4+ of GABA condenses with α-KG from the Krebs cycle to generate glutamate for each GABA molecule consumed (the “GABA shunt”). In the central nervous system, this ensures balance between production of inhibitory and excitatory neurotransmitters. SSA that accumulates is most likely the metabolic precursor of GHB, produced by AKR7a5. Another key glutamate precursor is glutamine (synthesized in glial cells via glutamine synthase). In neurons, glutamine is converted to glutamate by the action of glutaminase.
GABA=TK; CO2=carbon dioxide; α-KG=α-ketoglutarate; NH4+=nitrogen; SSA=succinic semialdehyde; GHB=γ-hydroxybutyrate; SSADH=succinic semialdehyde dehydrogenase; AKR7a5=aldo-keto reductase 7a5.
Acknowledgments
Funding/Support: This study was supported in part by the National Institutes of Health (R01 NS40270, Dr. Gibson), the Pediatric Neurotransmitter Disease Association (Drs. Pearl and Gibson), SHS International (Dr. Knerr), and the University of Erlangen-Nuremberg.
Footnotes
Faculty Disclosures: The authors do not have an affiliation with or financial interest in any organization that might pose a conflict of interest.
REFERENCES
- 1.Gibson KM, Christensen E, Jakobs C, et al. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): case reports of 23 new patients. Pediatrics. 1997;99:567–574. doi: 10.1542/peds.99.4.567. [DOI] [PubMed] [Google Scholar]
- 2.Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria) Biol Psychiatry. 2003;54:763–768. doi: 10.1016/s0006-3223(03)00113-6. [DOI] [PubMed] [Google Scholar]
- 3.Pearl PL, Gibson KM. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology. 2003;60:1413–1417. doi: 10.1212/01.wnl.0000059549.70717.80. [DOI] [PubMed] [Google Scholar]
- 4.Pearl PL, Gibson KM. Clinical aspects of the disorders of GABA metabolism in children. Curr Opin Neurol. 2004;17:107–113. doi: 10.1097/00019052-200404000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Pearl PL, Acosta MT, Wallis DD, Bottiglieri T, Miotto K, Jakobs C. Dyskinetic features of succinate semialdehyde dehyrdrogenase deficiency, a GABA degradative defect. In: Fernandez-Alvarez E, Arzimanoglu A, Tolosa E, editors. Paediatric Movement Disorders: Progress in Understanding. 1st. Surrey, United Kingdom: John Libbey Eurotext; 2005. pp. 203–212. [Google Scholar]
- 6.Pearl PL, Capp PK, Novotny EJ, Gibson KM. Inherited disorders of neurotransmitters in children and adults. Clin Biochem. 2005;38:1051–1058. doi: 10.1016/j.clinbiochem.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Philippe A, Deron J, Geneviève D, et al. Neurodevelopmental pattern of succinic semialdehyde dehydrogenase deficiency (gamma-hydroxybutyric aciduria) Dev Med Child Neurol. 2004;46:564–568. doi: 10.1017/s0012162204000933. [DOI] [PubMed] [Google Scholar]
- 8.Gibson KM, Schor DS, Gupta M, et al. Focal neurometabolic alterations in mice deficient for succinate semialdehyde dehydrogenase. J Neurochem. 2002;81:71–79. doi: 10.1046/j.1471-4159.2002.00784.x. [DOI] [PubMed] [Google Scholar]
- 9.Gupta M, Hogema BM, Grompe M, et al. Murine succinate semialdehyde dehydrogenase deficiency. Ann Neurol. 2003;54(suppl 6):S81–S90. doi: 10.1002/ana.10625. [DOI] [PubMed] [Google Scholar]
- 10.Gupta M, Polinsky M, Senephansiri H, et al. Seizure evolution and amino acid imbalances in murine succinate semialdehyde dehydrogenase (SSADH) deficiency. Neurobiol Dis. 2004;16:556–562. doi: 10.1016/j.nbd.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Gibson KM, Jakobs C, Pearl PL, Snead OC. Murine succinate semialdehyde dehydrogenase (SSADH) deficiency, a heritable disorder of GABA metabolism with epileptic phenotype. IUBMB Life. 2005;57:639–644. doi: 10.1080/15216540500264588. [DOI] [PubMed] [Google Scholar]
- 12.Gibson KM, Gupta M, Senephansiri H, et al. Oxidant stress and neurodegeneration in murine succinic semialdehyde dehydrogenase (SSADH) deficiency. In: Hoffman GF, editor. Diseases of Neurotransmission: From Bench to Bed. 1st. Heilbronn, Germany: SPS Publication; 2006. pp. 199–212. [Google Scholar]
- 13.Chowdhury GM, Gupta M, Gibson KM, Patel AB, Behar KL. Altered cerebral glucose and acetate metabolism in succinic semialdehyde dehydrogenase-deficient mice: evidence for glial dysfunction and reduced glutamate/glutamine cycling. J Neurochem. 2007;103:2077–2091. doi: 10.1111/j.1471-4159.2007.04887.x. [DOI] [PubMed] [Google Scholar]
- 14.Gibson KM, Aramaki S, Sweetman L, et al. Stable isotope dilution analysis of 4- hydroxybutyric acid: an accurate method for quantification in physiological fluids and the prenatal diagnosis of 4-hydroxybutyric aciduria. Biomed Environ Mass Spectrom. 1990;19:89–93. doi: 10.1002/bms.1200190207. [DOI] [PubMed] [Google Scholar]
- 15.Wolf NI, Haas D, Hoffmann GF, et al. Sedation with 4-hydroxybutyric acid: a potential pitfall in the diagnosis of SSADH deficiency. J Inherit Metab Dis. 2004;27:291–293. doi: 10.1023/b:boli.0000028842.15981.6e. [DOI] [PubMed] [Google Scholar]
- 16.Gibson KM, Sweetman L, Jansen I, et al. Properties of succinic semialdehyde dehydrogenase in cultured human lymphoblasts. J Neurogenet. 1985;2:111–122. doi: 10.3109/01677068509100146. [DOI] [PubMed] [Google Scholar]
- 17.Gibson KM, Lee CF, Chambliss KL, et al. 4-Hydroxybutyric aciduria: application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin Chim Acta. 1991;196:219–221. doi: 10.1016/0009-8981(91)90076-o. [DOI] [PubMed] [Google Scholar]
- 18.Akaboshi S, Hogema BM, Novelletto A, et al. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum Mutat. 2003;22:442–450. doi: 10.1002/humu.10288. [DOI] [PubMed] [Google Scholar]
- 19.Chambliss KL, Hinson DD, Trettel F, et al. Two exon-skipping mutations as the molecular basis of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria) Am J Hum Genet. 1998;63:399–408. doi: 10.1086/301964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakobs C, Bojasch M, Mönch E, Rating D, Siemes H, Hanefeld F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin Chim Acta. 1981;111:169–178. doi: 10.1016/0009-8981(81)90184-4. [DOI] [PubMed] [Google Scholar]
- 21.Jakobs C, Smit LM, Kneer J, Michael T, Gibson KM. The first adult case with 4-hydroxybutyric aciduria. J Inherit Metab Dis. 1990;13:341–344. doi: 10.1007/BF01799390. [DOI] [PubMed] [Google Scholar]
- 22.Arnulf I, Konofal E, Gibson KM, et al. Effect of genetically caused excess of brain gamma-hydroxybutyric acid and GABA on sleep. Sleep. 2005;28:418–424. doi: 10.1093/sleep/28.4.418. [DOI] [PubMed] [Google Scholar]
- 23.Pearl PL, Acosta MT, Theodore WH, et al. Human SSADH deficiency-phenotype and treatment strategies. In: Hoffman GF, editor. Diseases of Neurotransmission: From Bench to Bed. 1st. Heilbronn, Germany: SPS Publication; 2006. pp. 187–198. [Google Scholar]
- 24.Chan KF, Burnham WM, Jia Z, Cortez MA, Snead OC., 3rd GABAB receptor antagonism abolishes the learning impairments in rats with chronic atypical absence seizures. Eur J Pharmacol. 2006;541:64–72. doi: 10.1016/j.ejphar.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Hogema BM, Gupta M, Senephansiri H, et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29:212–216. doi: 10.1038/ng727. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Ali S, Ahmadian G, et al. Gamma-hydroxybutyric acid (GHB) and gamma-aminobutyric acidB receptor (GABABR) binding sites are distinctive from one another: molecular evidence. Neuropharmacology. 2004;47:1146–1156. doi: 10.1016/j.neuropharm.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 27.Wu Y, Buzzi A, Frantseva M, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor-mediated mechanisms. Ann Neurol. 2006;59:42–52. doi: 10.1002/ana.20686. [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Buzzi A, Shen L, et al. 2004 Abstract Viewer/Itinerary Planner. Washington, DC: Society for Neuroscience; 2004. Differential expression of ampa-type glutamate receptors in the brain of mice deficient for succinate semialdehyde dehydrogenase. Program No. 952.10, Online. [AU: YOU MAY EITHER PROVIDE URL AND ACCESS DATE FOR THIS REFERENCE OR DELETE “ONLINE”.] [Google Scholar]
- 29.Buzzi A, Wu Y, Frantseva MV, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res. 2006;1090:15–22. doi: 10.1016/j.brainres.2006.02.131. [DOI] [PubMed] [Google Scholar]
- 30.Pearl PL, Taylor JL, Trzcinski S, et al. C-Flumazenil PET imaging in patients with SSADH deficiency. J Inherit Metab Dis. 2007;30(suppl 1):43. [Google Scholar]
- 31.Reis J, Cohen LG, Pearl PL, Gibson KM, Dustin I, Theodore WH.Transcranial magnetic stimulation reveals altered cortical excitability in succinic semialdehyde dehydrogenase deficiency Abstract Viewer Program No. 3.029 Philadelphia, Penn: American Epilepsy Society; 2007 [Google Scholar]
- 32.Mehta AK, Gould GG, Gupta M, Carter LP, Gibson KM, Ticku MK. Succinate semialdehyde dehydrogenase deficiency does not down-regulate gamma-hydroxybutyric acid binding sites in the mouse brain. Mol Genet Metab. 2006;88:86–89. doi: 10.1016/j.ymgme.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Barcelo-Coblijn G, Murphy EJ, Mills K, et al. Lipid abnormalities in succinate semialdehyde dehydrogenase (Aldh5a1−/−) deficient mouse brain provide additional evidence for myelin alterations. Biochim Biophys Acta. 2007;1772:556–562. doi: 10.1016/j.bbadis.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donarum EA, Stephan DA, Larkin K, et al. Expression profiling reveals multiple myelin alterations in murine succinate semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2006;29:143–156. doi: 10.1007/s10545-006-0247-6. [DOI] [PubMed] [Google Scholar]
- 35.Knerr I, Pearl PL, Bottiglieri T, Snead OC, Jakobs C, Gibson KM. Therapeutic concepts in succinate semialdehyde dehydrogenase (SSADH; ALDH5a1) deficiency (gamma-hydroxybutyric aciduria). Hypotheses evolved from 25 years of patient evaluation, studies in Aldh5a1−/− mice and characterization of gamma-hydroxybutyric acid pharmacology. J Inherit Metab Dis. 2007;30:279–294. doi: 10.1007/s10545-007-0574-2. [DOI] [PubMed] [Google Scholar]
- 36.Latini A, Scussiato K, Leipnitz G, Gibson KM, Wajner M. Evidence for oxidative stress in tissues derived from succinate semialdehyde dehydrogenase-deficient mice. J Inherit Metab Dis. 2007;30:800–810. doi: 10.1007/s10545-007-0599-6. [DOI] [PubMed] [Google Scholar]
- 37.Sauer SW, Kölker S, Hoffmann GF, et al. Enzymatic and metabolic evidence for a region specific mitochondrial dysfunction in brains of murine succinic semialdehyde dehydrogenase deficiency (Aldh5a1−/− mice) Neurochem Int. 2007;50:653–659. doi: 10.1016/j.neuint.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 38.Gibson KM, Hoffmann GF, Hodson AK, Bottiglieri T, Jakobs C. 4-Hydroxybutyric acid and the clinical phenotype of succinic semialdehyde dehydrogenase deficiency, an inborn error of GABA metabolism. Neuropediatrics. 1998;29:14–22. doi: 10.1055/s-2007-973527. [DOI] [PubMed] [Google Scholar]
- 39.Scharf MB, Brown D, Woods M, Brown L, Hirschowitz J. The effects and effectiveness of gamma-hydroxybutyrate in patients with narcolepsy. J Clin Psychiatry. 1985;46:222–225. [PubMed] [Google Scholar]
- 40.Shannon M, Quang LS. Gamma-hydroxybutyrate, gamma-butyrolactone, and 1,4-butanediol: a case report and review of the literature. Pediatr Emerg Care. 2000;16:435–440. doi: 10.1097/00006565-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 41.Drasbek KR, Christensen J, Jensen K. Gamma-hydroxybutyrate–a drug of abuse. Acta Neurol Scand. 2006;114:145–156. doi: 10.1111/j.1600-0404.2006.00712.x. [DOI] [PubMed] [Google Scholar]
- 42.Goodwin AK, Griffiths RR, Brown PR, et al. Chronic intragastric administration of gamma-butyrolactone produces physical dependence in baboons. Psychopharmacology (Berl) 2006;189:71–82. doi: 10.1007/s00213-006-0534-9. [DOI] [PubMed] [Google Scholar]
- 43.Weerts EM, Goodwin AK, Griffiths RR, et al. Spontaneous and precipitated withdrawal after chronic intragastric administration of gamma-hydroxybutyrate (GHB) in baboons. Psychopharmacology (Berl) 2005;179:678–687. doi: 10.1007/s00213-004-2079-0. [DOI] [PubMed] [Google Scholar]
- 44.Gibson KM, DeVivo DC, Jakobs C. Vigabatrin therapy in patient with succinic semialdehyde dehydrogenase deficiency. Lancet. 1989;2:1105–1106. doi: 10.1016/s0140-6736(89)91126-4. [DOI] [PubMed] [Google Scholar]
- 45.Gibson KM, Jakobs C, Ogier H, et al. Vigabatrin therapy in six patients with succinic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 1995;18:143–146. doi: 10.1007/BF00711750. [DOI] [PubMed] [Google Scholar]
- 46.Gropman A. Vigabatrin and newer interventions in succinic semialdehyde dehydrogenase deficiency. Ann Neurol. 2003;54(suppl 6):S66–S72. doi: 10.1002/ana.10626. [DOI] [PubMed] [Google Scholar]
- 47.Ergezinger K, Jeschke R, Frauendienst-Egger G, Korall H, Gibson KM, Schuster VH. Monitoring of 4-hydroxybutyric acid levels in body fluids during vigabatrin treatment in succinic semialdehyde dehydrogenase deficiency. Ann Neurol. 2003;54:686–689. doi: 10.1002/ana.10752. [DOI] [PubMed] [Google Scholar]
- 48.Gordon N. Succinic semialdehyde dehydrogenase deficiency (SSADH) (4-hydroxybutyric aciduria, gamma-hydroxybutyric aciduria) Eur J Paediatr Neurol. 2004;8:261–265. doi: 10.1016/j.ejpn.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Matern D, Lehnert W, Gibson KM, Korinthenberg R. Seizures in a boy with succinic semialdehyde dehydrogenase deficiency treated with vigabatrin (gamma-vinyl-GABA) J Inherit Metab Dis. 1996;19:313–318. doi: 10.1007/BF01799261. [DOI] [PubMed] [Google Scholar]
- 50.Gupta M, Greven R, Jansen EE, et al. Therapeutic intervention in mice deficient for succinate semialdehyde dehydrogenase (gamma-hydroxybutyric aciduria) J Pharmacol Exp Ther. 2002;302:180–187. doi: 10.1124/jpet.302.1.180. [DOI] [PubMed] [Google Scholar]
- 51.Löscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16:669–694. doi: 10.2165/00023210-200216100-00003. [DOI] [PubMed] [Google Scholar]
- 52.Löscher W. Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Prog Neurobiol. 1999;58:31–59. doi: 10.1016/s0301-0082(98)00075-6. [DOI] [PubMed] [Google Scholar]
- 53.Shinka T, Ohfu M, Hirose S, Kuhara T. Effect of valproic acid on the urinary metabolic profile of a patient with succinic semialdehyde dehydrogenase deficiency. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;792:99–106. doi: 10.1016/s1570-0232(03)00276-9. [DOI] [PubMed] [Google Scholar]
- 54.Pattarelli PP, Nyhan WL, Gibson KM. Oxidation of [U-14C]succinic semialdehyde in cultured human lymphoblasts: measurement of residual succinic semialdehyde dehydrogenase activity in 11 patients with 4-hydroxybutyric aciduria. Pediatr Res. 1988;24:455–460. doi: 10.1203/00006450-198810000-00007. [DOI] [PubMed] [Google Scholar]
- 55.Cortez MA, Wu Y, Gibson KM, Snead O., 3rd Absence seizures in succinic semialdehyde dehydrogenase deficient mice: a model of juvenile absence epilepsy. Pharmacol Biochem Behav. 2004;79:547–553. doi: 10.1016/j.pbb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Saransaari P, Oja SS. Taurine and neural cell damage. Amino Acids. 2000;19:509–526. doi: 10.1007/s007260070003. [DOI] [PubMed] [Google Scholar]
- 57.Freeman J, Veggiotti P, Lanzi G, et al. The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy Res. 2006;68:145–180. doi: 10.1016/j.eplepsyres.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Groesbeck DK, Bluml RM, Kossoff EH. Long-term use of the ketogenic diet in the treatment of epilepsy. Dev Med Child Neurol. 2006;48:978–981. doi: 10.1017/S0012162206002143. [DOI] [PubMed] [Google Scholar]
- 59.Henderson CB, Filloux FM, Alder SC, Lyon JL, Caplin DA. Efficacy of the ketogenic diet as a treatment option for epilepsy: meta-analysis. J Child Neurol. 2006;21:193–198. doi: 10.2310/7010.2006.00044. [DOI] [PubMed] [Google Scholar]
- 60.Nylen K, Likhodii S, Perez Velasquez JL, Burnham WM, Gibson KM, Snead O. The ketogenic diet rescues the lethal phenotype and restores synaptic activity in succinic semialdehyde dehydrogenase deficient mice. Clin Neurophysiol. 2007;118:e187. [Google Scholar]