

Abstract
Studies in cell-culture systems and in postmortem tissue from human disease have suggested a connection between cell-cycle activation and neurodegeneration. The fruit fly Drosophila melanogaster has recently emerged as a powerful model system in which to model neurodegenerative diseases. Here we review work in the fly that has begun to address some of the important questions regarding the relationship between cell-cycle activation and neurodegeneration in vivo, including recent data implicating cell-cycle activation as a downstream effector of tau-induced neurodegeneration. We suggest how methods in the fly might be utilized to approach fundamental questions that remain.
1. Introduction
In 1992, a report by Feddersen, Orr and colleagues showed that SV40-mediated disruption of the tumor suppressor pRb leads to Purkinje cell degeneration [1]. In an insightful perspective piece that followed this study, Nathaniel Heintz postulated that there may be a fundamental relationship between neurodegeneration and neoplastic transformation and that “genes that can cause transformation in dividing cell populations (and thus result in clonal expansion and neoplastic disease) can cause programmed cell death of terminally differentiated neurons (and thus result in clonal elimination and neurodegenerative disease)” ([2]; Figure 1). In the last decade, an important body of work in cell-culture systems and in postmortem tissue from human disease has further raised the possibility of a mechanistic connection between transformation and neurodegeneration [3]. There have, for example, been several studies indicating that posmitotic neurons abnormally re-express numerous cell-cycle markers and replicate their DNA in neurodegenerative disorders, including tauopathies and Alzheimer’s disease (AD) [4, 5]. In various neurotoxic paradigms in cell-culture systems, neuronal apoptosis can be prevented by blocking various components of the cell-cycle machinery [3]. Finally, certain cell-cycle regulators, including E2F and Cdc2, have been shown to directly activate the neuronal apoptotic machinery [6–9].
Figure 1. Schematic diagram contrasting different consequences of cell-cycle activation for proliferating and postmitotic cells.
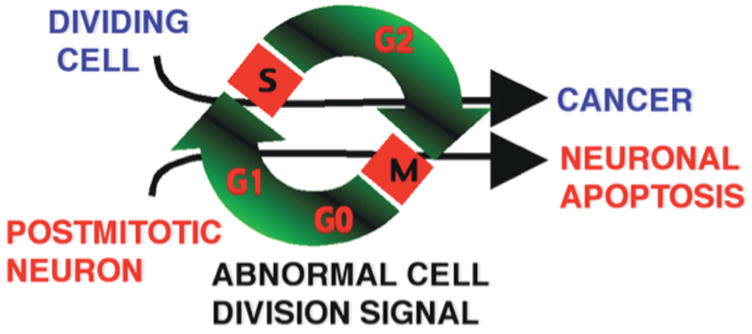
In this scheme, postmitotic neurons forced to re-enter a cell cycle undergo apoptosis rather than cell division.
For all their strengths, studies associating aberrant cell-cycle marker expression with neurodegeneration in postmortem tissue have been largely correlative. For example, immunostaining in these studies has often only been in abnormal structures such as neurofibrillary tangles (NFTs), proteinaceous aggregates of the microtubule-associated protein tau that could potentially bind antibodies non-specifically. Also, proteins expected to be nuclear during cell-cycle progression have sometimes been found to be cytoplasmic in dying neurons [10, 11]. Thirdly, both cell-cycle activating and inhibitory proteins are upregulated in AD and animal models [11, 12], making it difficult to conclude whether activation or inhibition of the cell cycle, or neither, contribute to neuronal demise. These findings raise the concerns that either aberrant cell-cycle marker expression indicates nothing more than dysregulated protein synthesis in dying neurons or, alternatively, that the upregulated proteins may be subserving non cell-cycle roles. In this regard, Herrup and colleagues made a significant advance by showing that DNA is actually replicated in susceptible and dying neuronal populations in AD, supporting the idea that neurons re-enter a coordinated cell cycle in this disease. However, to add complexity, DNA replication and abnormal cell-cycle marker re-expression in postmitotic neurons has been described in several animal models in which neurons do not appreciably degenerate, including rodent models of ATM [13] and AD [14], and in Retinoblastoma Protein-1 (pRb-1)-deficient mice. For example, while widespread neurodegeneration occurs in the central nervous system (CNS) of pRb-1-deficient mice [15, 16], this effect is non cell-autonomous because in chimeric mice, the pRb-1-/-clones in the CNS exhibit tetraploidy in the absence of appreciable neuronal death [17].
Bearing these data in mind, we wondered first whether there was a causal relationship between cell-cycle activation and neurodegeneration in animal models of tauopathy and second, whether cell-cycle activation was sufficient to cause neurodegeneration. Third, if cell-cycle activation did mediate neuronal death, it remained to be seen which phase of the cell cycle was lethal for a neuron. The absence of any description of mitotic figures or cytokinesis of a postmitotic neuron certainly implied that adult neurons could not progress through mitosis, although studies in cell-culture systems also implicated early G1/S mediators, such as E2F and Cyclin D, in causing neuronal apoptosis. Finally, the question of the triggers for postmitotic neuron to re-enter the cell cycle remained unknown. In particular, in postmortem tissue from AD, numerous mitogenic signaling molecules had been shown to be upregulated [18–22], but the consequences of these pathways being activated in postmitotic neurons, whether apoptotic or pro-mitotic, were unclear.
In this context, we felt the genetically tractable fruit fly, in which neurodegenerative diseases are now modeled, had a unique role to play. In this review, we highlight the utility of this model organism to investigate the connection between cell-cycle activation and neurodegeneration. We analyze recent experimental data that have begun to address some of the important questions in the field, and discuss how methods in the fly might be utilized to approach fundamental questions that remain.
2. The utility of Drosophila in investigating the cell-cycle/neurodegeneration connection
Drosophila is ideally suited to investigate the relationship between neurodegeneration and cell-cycle activation in vivo for several reasons. First, the cell-cycle machinery is substantially conserved between Drosophila and mammalian systems [23]. Indeed, considerable advances in our understanding of the role of cell-cycle mediators in vivo have been afforded by investigations in flies. Second, there are now several well-established Drosophila models of neurodegeneration, including tauopathies, Parkinson’s disease (PD) and polyglutamine disorders (reviewed in [24]). As noted above, postmortem analyses associate cell-cycle activation with tauopathies including AD. Of primary importance, the relative low level of redundancy in the fly genome (that is, a single gene in Drosophila is often the only homolog for an entire family of mammalian genes) and the availability of many sophisticated genetic tools make genetic analysis in Drosophila well suited to address issues of causality. Finally, the short lifespan and reproductive cycle of flies allow for the analysis of many potential genetic modifiers in a relatively short amount of time.
2.1 Cell-cycle and mitogenic signaling machineries are well conserved in Drosophila
In the cell cycle, DNA synthesis occurs during S-phase and mitosis occurs in M-phase. Two gap phases (G1 and G2) separate the S- and M-phases. Between division cycles, cells are in G0. Neurons are generally considered postmitotic and permanently in G0 (Figure 2). Immunohistochemical markers can be used to identify cell-cycle phases. For example, the proliferating cell nuclear antigen (PCNA), thought to function as a “sliding clamp” for DNA polymerase-δ [25], is upregulated during G1, S and G2. Phosphorylation at Ser10 of histone-3 (PH3) occurs during condensation of chromosomes in M-phase [26] and is thus considered an M-phase-specific marker. It should be noted, however, that these markers are upregulated in situations other than cell division – for example, PCNA is involved in DNA repair [27] and H3 phosphorylation at Ser10 occurs during immediate-early gene transcription [28]. 5-bromo-2’-deoxyuridine (BrdU) incorporation into actively synthesized DNA is a more definitive S-phase marker.
Figure 2. Simplified representation of cell-cycle regulators in mammalian (blue) and Drosophila (black) cells.
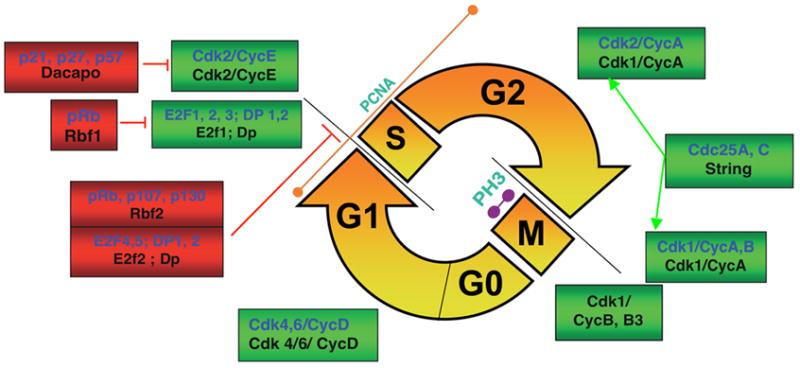
Positive regulators that facilitate progression are boxed in green and negative regulators that inhibit progression are indicated in red. The cell cycle consists of an S-phase (in which DNA is replicated) and an M-phase (in which mitosis occurs), separated by two gap phases G1 and G2. G0 is a resting phase and is the phase which postmitotic neurons are considered to occupy after differentiation. Cyclin-dependent kinases (Cdks) form complexes with cyclins and regulate progression through the G1, S, G2 and M-phases of the cell-division cycle. The E2F transcription factors and pocket proteins of the retinoblastoma family (pRb) help coordinate the G1/S transition. The Cip inhibitor Dacapo is the fly homolog of the Cip proteins p21/p27 and inhibits the G1/S transition. In Drosophila, Cdk1/CyclinA catalyzes the G2/M transition and Cdk1/CyclinB and B3 catalyze progression through mitosis. The phosphatase Cdc25 is a positive cell-cycle regulator, dephosphorylating and activating key enzymes including Cdk1 and Cdk2 [23, 34]. In vertebrate and Drosophila cell cycles, PCNA is predominantly an S-phase marker and phosphorylation at Ser-10 of Histone 3 (PH3) accompanies condensed chromosomes during M-phase.
Cyclin-dependent kinases (Cdks) catalyze reactions essential for progression through the cell cycle. Key regulators of the G1/S checkpoint include the E2F family of transcription factors, the pocket proteins (including pRb) and the Cdk inhibitors p21 and p27 (Figure 2). E2F factors 1 and 2 can directly activate the transcription of many S-phase genes, including DNA polymerase-α and Cyclin E [29]. Pocket proteins of the pRb family can directly bind and inhibit E2F-mediated transactivation [30]. This inhibition is relieved by pocket protein phosphorylation by Cdks. Alternatively, E2F 4 and 5 family members can bind pocket proteins and form complexes that repress transcription. p21 and p27 regulate the G1/S transition by directly binding and inhibiting Cdk2/Cyclin E. These inhibitors can also inhibit DNA synthesis by binding PCNA [31].
In Drosophila, the essential features of the cell cycle are conserved [23]. Homologs of Cdks, Cyclins, Cip inhibitors, E2F transcription factors and pocket proteins have been described [32]. Drosophila homologs are indicated in Figure 2 (black). Cdk1 and Cdk2 are often referred to in the literature as cdc2 and cdc2c, respectively. Note that Drosophila Cyclin A complexes with Cdk1 but does not complex with Cdk2. Cyclin E, however, does complex with Cdk2 to facilitate the G1/S transition. The G2/M transition is dependent upon Cdk1/Cyclin A in flies, with Cdk1/Cyclin B complexes thought to be important in progression through the various stages of mitosis. The fly homolog for p27/p21 is Dacapo (Dap)[33]. There are two E2f family members in Drosophila [34]. E2f1 directly transactivates gene transcription, like the vertebrate E2Fs 1/2/3, and is inhibited by Rbf, the homolog of pRb-1. E2f2, like the vertebrate E2Fs 4/5, forms repressor complexes with Rbf2. In studies of Drosophila development, inhibition of the G1/S transition is achieved by overexpressing Rbf1, dE2f2, Rbf2, Dacapo or human p21 [33, 34]. The G1/S transition appears to be most effectively inhibited in the developing larval eye disc by co-expressing both Dap and Rbf1 [33].
The primary function of Cdk4/Cyclin D during fly development appears to be as a growth activator, although Cdk4/Cyclin D drives the G1/S transition indirectly in some tissues [35, 36]. Similarly, classic mitogenic and oncogenic signaling pathways, including Myc [37], Ras [38] and TOR [39], are highly conserved in flies and appear to drive the G1/S transition indirectly by promoting cellular growth. Interestingly, several papers have shown that TOR activity is abnormally upregulated in AD brain [18, 40].
2.2 A Drosophila model of tauopathy
In postmortem studies, early cell-cycle markers including PCNA are upregulated in a number of acute and chronic neurodegenerative conditions [6]. In contrast, markers of the G2/M transition appear to be more selectively upregulated in tauopathies [4]. Tauopathies are neurodegenerative diseases characterized by intraneuronal accumulations of hyperphosphorylated forms of the microtubule-associated protein tau. The term encompasses hereditary frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), and sporadic tauopathies including AD, progressive supranuclear palsy and Pick’s disease. We, and others, have developed Drosophila models of tauopathies [41, 42]. Transgenic flies are created by expressing human wild-type or FTDP-17-associated mutant tau constructs. Tissue-specific expression of the transgenes is achieved using the bipartite UAS/GAL4 system derived from yeast [43]. For example, GMR-GAL4 drives expression within retinal tissue, in both neuronal (photoreceptors) and non-neuronal postmitotic cells. elav-GAL4 drives transgene expression within postmitotic neurons, including photoreceptors of the retina and the brain (Figure 3H).
Figure 3. Drosophila model of tauopathy.
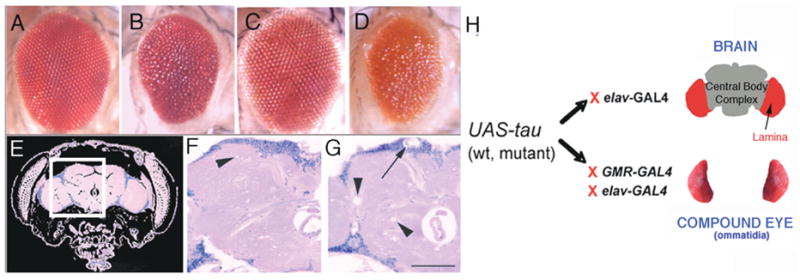
(A–D) The tau-induced rough eye phenotype consists of a small irregular eye with fused ommatidia and small focal areas of degeneration. Compare the elav-GAL4/+ (A) and GMR-GAL4/+ (C) control eyes with eyes of transgenic flies expressing mutant forms of tau, tauR406W (B; genotype: elav-GAL4/+; UAS-tauR406W6/+) or tauV337M (D; genotype: UAS-tauV337M1/+; GMR-GAL4/+), This phenotype is visible in adults at eclosion. (E) Hematoxylin and eosin-stained frontal section through the head and brain of a wild-type fly. (F–G) Areas of the fly brain similar to that boxed in (G) are shown for control elav-GAL4/+ (F) and mutant tau-expressing (G) flies. Vacuolization is observed in the neuropil (N, arrowheads) and cortex (Cx, arrow) [42]. (H) Bipartite UAS/GAL4 system: This system allows for specific expression of tau either in postmitotic neurons, including the fly brain, with elav-GAL4, or in the Drosophila eye with elav-GAL4 (photoreceptors only) or GMR-GAL4 (photoreceptors and support cells). F and G have been reproduced with permission from [42].
Tau expression induces retinal and brain phenotypes that have been well characterized in our laboratory and by other investigators [41, 42, 44]. Retinal toxicity is observed in adult flies as a “rough” eye phenotype. The eye is reduced in size, loses its regular ommatidial structure and is irregularly contoured (Figures 3A–D). The rough eye phenotype is useful because it is easily scored and amenable to genetic modification, making it a valuable tool for both forward (unbiased) and candidate genetic screens. Recently, this phenotype was used to conduct a P element-based forward modifier screen for tau-induced neurotoxicity [45].
When tau is expressed with elav-GAL4, adult flies eclose with morphologically normal brains. However, in adult life, accumulation of disease-associated phospho- and conformational tau epitopes accompanies progressive neurodegeneration, histologically characterized by neuronal nuclear fragmentation and vacuolization of the cortex and neuropil (Figures 3F–G) [42, 44], and TUNEL (terminal transferase-mediated deoxy-UTP biotin nick end-labeling) staining of apoptotic neurons.
3. Investigating cell-cycle activation in a Drosophila tauopathy model
3.1 Establishing a causal connection between tau-induced neurodegeneration and cell-cycle activation in vivo
Recently, we found that, as in tauopathies, the cell-cycle markers PCNA and PH3 were abnormally expressed in neurons in the Drosophila tauopathy model [46]. These could always be found in transgneic tau fly brains, but were completely absent in age-matched control flies. PCNA staining was both cytoplasmic and nuclear, and PH3 staining was entirely nuclear, consistent with the subcellular localization patterns during cell-cycle activation. In agreement with previous postmortem and cell-culture studies, we did not see abnormal cytokinesis of postmitotic neurons in the brains of transgenic tau flies.
A comprehensive genetic analysis in the retina and brain (Table 1) established a clear causal relationship between cell-cycle activation and neurodegeneration. In other words, the use of mutant alleles or transgene combination to inhibit cell-cycle progression substantially reduced tau-induced neuronal apoptosis. In contrast, co-expressing genetic activators of the cell-cycle synergistically enhanced tau-induced neurodegeneration. Cdk1 and Cdk2 enhanced tau-induced neurodegeneration when co-expressed with their respective Cyclins. Since multiple reagents at both the G1/S and G2/M transition fitted this pattern, our data strongly implicated the cell-cycle pathway as a whole in mediating tau-induced neurodegeneration. There were rare exceptions to the rule. For example, two separate Cyclin E mutants did not suppress tau-induced retinal degeneration. This may be trivially related to haplosufficiency of Cyclin E. Notably, however, these alleles do exert dominant effects in developmental contexts [47] and, overall, the effects of modulating the G2/M Cyclins A and B were certainly more striking in modifying tau-induced toxicity than the effects of modulating the G1/S Cyclins D and E.
Table 1.
Cell-cycle mediators tested for modification of tau-induced neurodegeneration phenotypes
| Cell-cycle mediator | System* | Interaction with tau** |
|---|---|---|
| UAS-Dacapo/UAS-Rbf1 | Ret, Br | Sup |
| UAS-Dacapo/UAS-Cdk1DN | Ret, Br | Sup |
| Cyclin ALOF | Ret | Sup |
| Cyclin BLOF | Ret | Sup |
| Cyclin B3LOF | Ret | Sup |
| E2f1LOF | Ret | Sup |
| Cyclin DLOF | Ret | Sup |
| Cdk4LOF | Ret | Sup |
| Cyclin ELOF | Ret | None |
| UAS-Cyclin A | Ret, Br | Enh |
| UAS-Cyclin B | Ret, Br | Enh,(Ret), Lethal (Br) |
| UAS-Cyclin B3 | Ret, Br | Enh,(Ret), Lethal (Br) |
| UAS-Cdk1, UAS-Cyclin B | Ret, Br | Enh,(Ret), Lethal (Br) |
| UAS-Cdk4, UAS-Cyclin D | Ret, Br | Enh,(Ret), Semilethal (Br) |
| UAS-Cyclin E | Ret, Br | Enh(Ret), Lethal (Br) |
| UAS-Cdk1 | Ret | None |
| UAS-Cdk2 | Ret | None |
Retina (Ret; driver: GMR-GAL4), Brain (Br; driver: elav-GAL4)
Synergistic Enhancement (Enh), Suppression (Sup) of tau-induced neurodegeneration, No effect (None), Lethal/Semilethal combination when co-expressed (Lethal, Semilethal).
Abbreviations: Dominant negative (DN), Loss-of-function allele (LOF); UAS (Upstream Activating Sequence) refers to GAL4-responsive transgenes.
Bold face indicate unexpected interactions.
Information regarding specific genetic reagents is published [46] or is available from the authors by request.
Postmortem analyses of AD and tauopathy tissue have associated cell-cycle marker expression with abnormal tau phosphorylation. Since Cdks are well known to phosphorylate tau [48], the question of whether cell-cycle activation acts up- or downstream of tau phosphorylation remained an important open question amenable to genetic analysis. In the fly tauopathy model, reducing tau phosphorylation genetically, by reducing the level of a known tau kinase glycogen synthase kinase-3 (GSK-3), led to decreased cell-cycle activation. Conversely, a pseudophosphorylated tau construct exhibited dramatically increased toxicity and commensurately increased ectopic cell-cycle marker expression in the brain. Furthermore, the toxicity induced by this pseudophorylated construct was blocked by cell-cycle inhibition. Finally, immunohistochemical data indicated that only a small subset of neurons containing abnormally phosphorylated tau were immunopositive for cell-cycle markers [46]. Taken together, these experiments indicated that cell-cycle activation occurred downstream of abnormal tau phosphorylation in the fly model. In our view, these experiments exemplify the utility of simple genetic models in casting light upon mechanistic questions that remain difficult to resolve in other model systems, or in human postmortem tissue analysis. Of course, the findings in the fly model do not rule out alternative mechanisms operating in mammalian models or in human disease.
Since cell-cycle activation occurred downstream of abnormal tau phosphorylation in the fly tauopathy model, cell-cycle activation might directly precede apoptosis [46]. We tested whether ectopic cell-cycle activation was sufficient to trigger apoptosis by using elav-GAL4 (or the inducible GeneSwitchelav) drivers to express E2f1/Dp, Cdk2/Cyclin E and Cyclin A in neurons. We found that increased TUNEL staining accompanied upregulation of PCNA and PH3, indicating that ectopic cell-cycle activation is sufficient to trigger neuronal apoptosis. Ectopic E2f1/Dp expression, in particular, lead to dramatic neuronal apoptosis and cell loss commensurate with the degree of cell-cycle marker upregulation. Diseases. It should be noted, however, that these experiments do not rule out potential cell-cycle-independent apoptotic effects of ectopic cell-cycle activation. For example, E2f1/Dp is known to directly transactivate proapoptotic genes in cell-culture system and in flies [49]. Interestingly, our preliminary data indicate that cyclins vary in their efficacy to induce apoptosis, and that different neuronal populations within the brain respond differently to the same cell-cycle stimulus.
3.2 Determining the link between tau and cell-cycle reentry
Having established a causal relationship between cell-cycle activators and tau-induced neurodegeneration, we focused on the mechanism of tau-induced cell-cycle activation. Potential mechanisms include those more closely related to tau, both in terms of its role as a microtubule-associated protein and its amino acid sequence, and general mechanisms, including oxidative stress, DNA damage and inhibition of the ubiquitin/proteasome system (Figure 4). The general mechanisms each have precedents in cell-culture systems. Our data indicated that ectopic cell-cycle marker expression was not a feature of the PD and spinocerebellar ataxia-3 (SCA3) fly models, and cell-cycle modification did not modify the SCA3-induced retinal phenotypes, supporting the possibility that cell-cycle activation may be tau-specific [46].
Figure 4. Potential mechanisms for tau-induced cell-cycle activation.
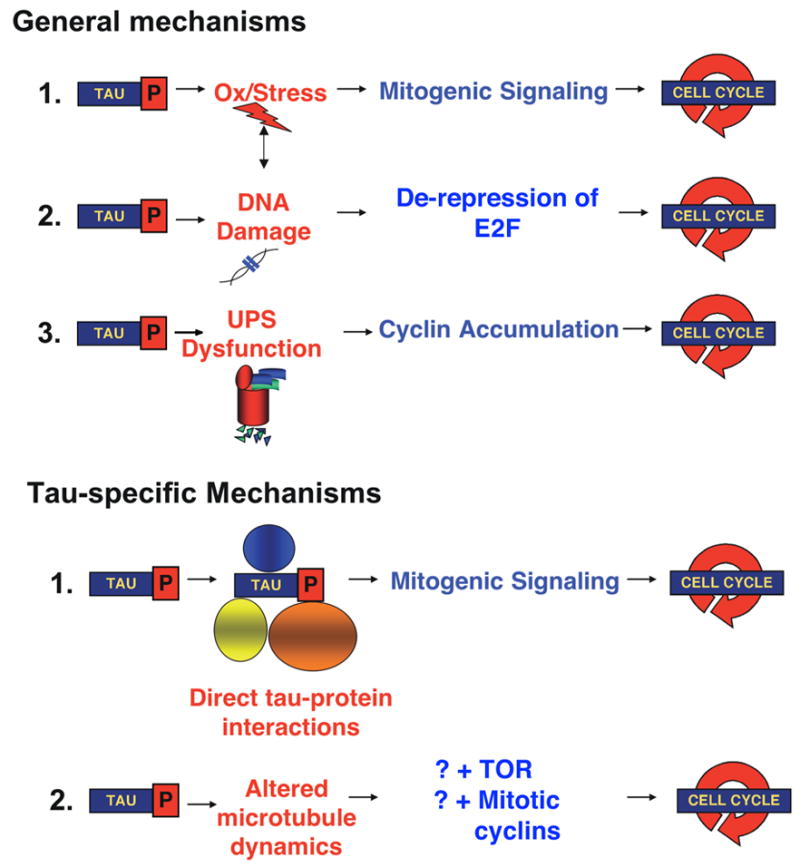
These include general mechanisms, such as DNA damage [67, 88, 89], oxidative stress [66] and dysfunction of the ubiquitin-proteasome system [80]. Other mechanisms may be tau-specific, including direct-tau protein interactions that subserve activation of mitogenic signaling pathways, and indirect activation of the cell cycle through alterations of microtubules.
Intriguing data from cell-culture systems implicate tau as a potential signaling molecule. Tau has been identified at the cell membrane and is enriched in axon growth cones. Furthermore, transfection of tau into neuronal cell lines results in neurite extension, suggesting that tau is capable of activating complex and coordinated intracellular signals [50, 51]. Tau is well known to be phosphorylated at many disease-associated phosphoepitopes in fetal life [52], a period of dynamic growth cone changes, and tau-induced neuritic extension in cell culture studies is modulated by Ser-Pro/Thr-Pro phosphorylation [50]. Phosphorylated tau may thus be particularly effective at transducing growth signals, a finding that may have relevance for disease states in which tau is hyperphosphorylated. As noted above, tau phosphorylation was required for tau-induced cell-cycle activation in the fly tauopathy model.
The amino acid sequence of tau reveals 5 tyrosines, 7 N-terminal PXXP motifs and multiple SP/TP motifs (Figure 5). Since SH2 domains bind phosphotyrosine, SH3 domains bind proline-rich domains that commonly contain a “PXXP” motif and WW domains bind SP/TP motifs, tau could potentially bind proteins containing these modular domains and be capable of transducing complex signals through binding multiple regulatory and adapter proteins. The receptor tyrosine kinases (RTKs), including the platelet-derived growth factor receptor, are classic example of such molecules. RTKs have tyrosine kinase catalytic function but contain multiple tyrosines that, when phosphorylated, bind SH2 domain-containing proteins including the adapter protein PLC-γ and Src family tyrosine kinases. Such interactions serve to amplify mitogenic signals by providing additional docking sites for modular signaling proteins. Hyperactive RTK signals can lead to oncogenesis through multiple mechanisms including increased transcription and translation of necessary components for growth and proliferation [53]. Viral antigens, such as the large and small SV40 T antigens, are other examples of proteins involved in multiple interactions, including with pRb, protein phosphatase 2A (PP2A) and p53, that induce cellular transformation [54]. In fact, the literature reveals that tau also binds several SH2/SH3 and WW domain-containing proteins, including non-receptor tyrosine kinases of the Src-family and c-Abl, the prolyl isomerase Pin-1 and phospholipase C-γ (PLC-γ ). An intriguing association has recently been described between the N-terminal of tau and the pRb-binding protein Che-1 [55]. Che-1 blocks both pRb-1 function in human cell lines by competing for HDAC binding [56]. The direct binding between tau and a negative regulator of the cell cycle is of obvious interest in the context of investigating the connection of tau to the cell cycle. Interestingly, tau is a substrate of the neuronally enriched PP2A and PP2A activity is decreased in AD [57–60].
Figure 5. Drawing analogies between tau and receptor tyrosine kinases.
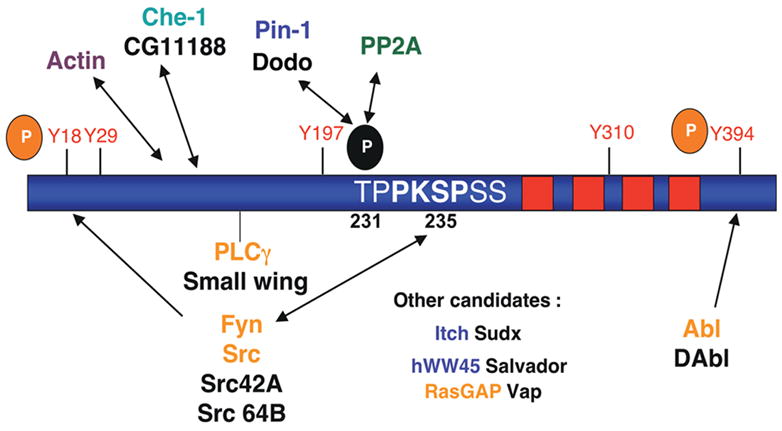
Tau contains 14 SP/TP, 5 tyrosines (at least two of which - Y18 and Y394 - are known to be phosphorylated in PHF-tau), and 7 PXXP-type motifs, making it a potential binding partner for WW, SH2 and SH3 domain-containing proteins, respectively. In cell culture and in vitro systems, tau is known to bind to SH2/3-containing proteins including Src family non-receptor tyrosine kinases [51], c-Abl [90] and PLC-γ [91, 92]. Binding of tau to the Fyn-SH3 domain occurs at the PXPXXP sequence shown (232-236). Tau also binds the WW domain-containing protein Pin-1, which isomerizes P-Thr-231 to render it more readily dephosphorylated by PP2A [93]. Other candidate proteins of interest, with genetic reagents available in flies, include the WW domain-containing proteins hWW-45 and Itch and the SH2/SH3-containing RASGAP. Note that fly homologs are labeled in black.
Since fly homologs for classic oncogenic signaling pathways and known tau interactors exist, we tested many signaling molecules for genetic modification of the tau-induced rough eye. Our candidate analysis is ongoing, but has hitherto strongly implicated signaling pathways known to activate growth as mediators of tau-induced neurotoxicity. Since the TOR pathway is a point of convergence for multiple growth-activating pathways (Figure 6; [61]) and TOR signaling has been linked to aging [62, 63] and AD, we thoroughly investigated the effect of modulating this pathway on tau-induced neurodegeneration [46]. We consistently found that TOR-activated genetic backgrounds synergistically enhanced tau-induced neurotoxocity, and TOR inhibition ameliorated tau-induced neurodegeneration in the retina and brain (Table 2). Our data indicated that TOR-dependent enhancement of tau-induced neurodegeneration could be blocked by concomitant cell-cycle inhibition and that ectopic TOR activation induced aberrant neuronal cell-cycle marker expression and apoptosis. These data strongly implicate TOR as an important mediator of tau-induced cell-cycle activation, although do not rule out parallel pathways existing for tau-induced cell-cycle activation.
Figure 6. TOR signaling in Drosophila [94–96].
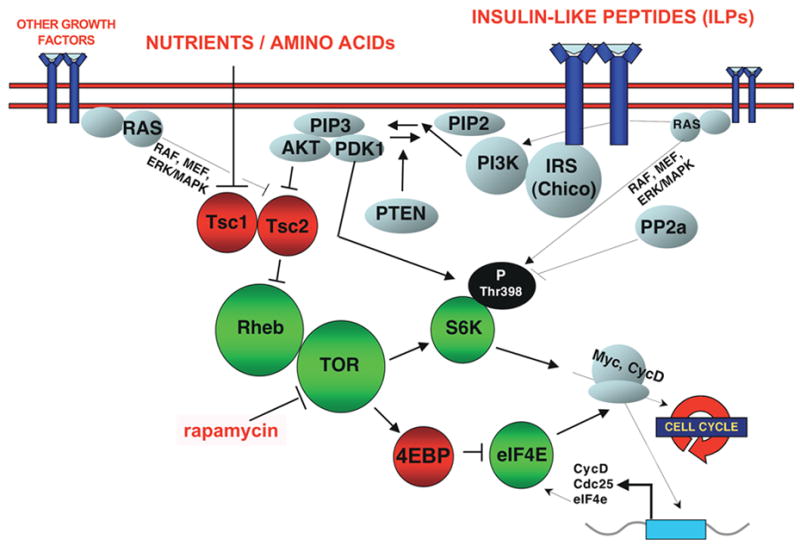
TOR integrates nutrient availability with signals from multiple pathways, including insulin signaling/PI3K. The insulin receptor is a ~400kDa tyrosine kinase receptor. PI3K is activated by direct recruitment to the insulin receptor substrate (IRS; also known as Chico). In mammalian systems, a well-characterized mechanism of PI3K activation is through Ras. PI3K appears to activate MAP kinase pathways in mammalian cells, but this does not appear to be the case in flies [96]. However, signaling from PI3K to Akt, and to TOR is well conserved. Akt phosphorylates and inhibits Tsc2 [97], the GAP protein inhibitory for the Rheb GTPase [98]. Rheb activates TOR and drives growth and indirectly the G1/S transition [99–101]. Over-expressing TOR itself paradoxically downregulates TOR signaling [102]. The two major downstream effectors are S6k and eIF4e. In flies, TOR activates S6k by phosphorylating Thr398. eIF4E is inhibited by 4EBP and TOR-dependent phosphorylation of 4EBP relieves this inhibition, leading to protein translation [103]. Thin arrows indicate links established in other systems. Studies in mammalian cell-culture systems have demonstrated that S6K phosphorylates the ribosomal protein S6 and thus activates ribosome biogenesis. eIF4E activates Cap-dependent protein translation. Targets may include Cyclin D and the Myc oncogene [61]. eIF4e is in turn a transcriptional target of Myc [104].
Table 2.
TOR-signaling mediators tested for modification of tau-induced neurodegeneration phenotypes
| TOR signaling mediator | System* | Interaction with tau** |
|---|---|---|
| TORLOF | Ret | Sup |
| RhebLOF | Ret | Sup |
| S6kLOF | Ret | Sup |
| eIF4eLOF | Ret | Sup |
| UAS-4ebpGOF | Ret | None |
| UAS-Tsc1 | Ret | Sup |
| UAS-Tsc2 | Ret | Sup |
| UAS-Tsc2GOF | Ret | Sup |
| UAS-TOR | Ret | None*** |
| UAS-Rheb | Ret, Br | Enh (Ret), Lethal (Br) |
| UAS-Tsc2LOF | Ret | Enh |
Retina (Ret; driver: GMR-GAL4), Brain (Br; driver: elav-GAL4)
Synergistic Enhancement (Enh), Suppression (Sup) of tau-induced neurodegeneration, No effect (None), Lethal combination when co-expressed (Lethal).
Overexpression of TOR is known to paradoxically cause a loss-of-function phenotype during development [102]. Overexpression of the Rheb GTPase, however, activates the pathway [99, 100].
Abbreviations: Dominant negative (DN), Loss-of-function allele (LOF), Gain-of-function/constitutively active allele (GOF); UAS (Upstream Activating Sequence) refers to GAL4-responsive transgenes.
Bold face indicate unexpected interactions.
Information regarding specific genetic reagents is published [46], or is available from the authors by request.
4. Fundamental questions that remain and how Drosophila might help
The Drosophila tauopathy model has proved useful in resolving three issues to date. First and foremost, a causal connection has been established between cell-cycle mediator activation and tau-induced neurodegeneration in vivo. Second, in the fly model, genetic manipulations and the use of different tau constructs has been useful in determining the epistasis of tau phosphorylation to cell-cycle activation. Our data would indicate that tau phosphorylation is upstream of cell-cycle activation, Third, we have shown that TOR signaling is required for tau-induced neurodegeneration and that TOR can ectopically activate cell cycle-dependent neuronal apoptosis in vivo. Fundamental questions remain more open, however, and these will be discussed in turn.
4.1 “Cell-cycle activation” in neurodegeneration: do we mean a coordinated cell cycle or cell-cycle mediator-dependent apoptosis?
Our genetic analyses in the tauopathy model indicate that activation of cell-cycle mediators is necessary for tau-induced neurodegeneration, since numerous manipulations at both the G1/S and G2/M checkpoints with multiple genetic reagents produce a consistent pattern of modification [46]. The presence of both PCNA and PH3, markers of different cell-cycle phases, supports the possibility that neurons may have re-entered a coordinated cell cycle. Certainly, the absence of any evidence of cytokinesis in our model indicates that neurons cannot successfully progress through mitosis. Our genetics would also generally indicate stronger effects of modulating G2/M cyclins on tau phenotypes than G1/S cyclins (see Section 3.1, above), indirectly supporting neuronal apoptosis being transduced later in the cell cycle. We cannot, however, rule out a second possibility that positive cell-cycle regulators, including Cdks and E2f1/Dp, are involved in transducing tau-induced apoptosis in the absence of a coordinated cell-cycle progression. Studies in cell-culture systems have revealed that E2F family transcription factors can directly transactivate pro-apoptotic genes [9], and that Cdk1 can directly lead to BAD-dependent neuronal apoptosis [8]. In contrast, postmortem tissue analyses in AD and AD mouse models suggest that DNA has been replicated in susceptible postmitotic neurons. Furthermore, in cell-culture AD models, neuronal apoptosis can be blocked by inhibiting DNA polymerase [3]. These latter investigations would support a coordinated cell-cycle progression being potentially responsible for neurodegeneration. The distinction is biologically important because, in the former case, cell-cycle activators are simply upstream components of the neuronal apoptosis mechanism. In the latter case, cell-cycle processes, the replication of DNA or the G2/M transition for example, may be responsible for cell death.
The Drosophila tauopathy model may be useful in resolving this issue, although more definitive measures of cell-cycle phasing are required. Such techniques would include assays for neuronal BrdU incorporation and FISH or fluorescence-activated cell sorting (FACS) to directly assess neuronal ploidy. BrdU incorporation [33, 64] and FACS [35] are standard techniques that have been used to determine the effect of cell-cycle modulators in proliferating fly larval tissues. We have not yet observed BrdU incorporation in the fly brain of transgenic tau flies, although whether this is a true negative result or simply reflects the technical limitations of the experiment remains unclear. It is important to note that relatively few neurons in the brain of transgenic tau flies demonstrate re-expression of cell-cycle markers at any given time, and thus negative results from any of these approaches would be difficult to interpret. An alternative approach would be to express tau in proliferating tissue, such as fly imaginal discs in which ploidy analysis is a well-established technique. Coupled with FACS analysis, this may provide interesting insights into how tau affects cell-cycle phasing. Our preliminary data would suggest that tau expression in imaginal discs does in fact lead to marked increases in cell-cycle marker activation, indicating either hyperproliferation or delayed cell-cycle exit.
4.2 Is neuronal cell-cycle activation sufficient to cause neuronal apoptosis?
The question of sufficiency of cell-cycle activation for neuronal apoptosis has plagued the cell-cycle/neurodegeneration field since its inception with the observation that DNA replication can be observed in the absence of neurodegeneration in a variety of context in postmitotic neurons, including in Rb-/- chimeric mice, Atm-/- mice, APP transgenic mice and in certain populations of neurons in AD brain [5, 13, 14, 17]. Our data would indicate the ectopic expression of multiple cyclins and E2f/Dp in the brain is sufficient to drive apoptosis of postmitotic neurons in vivo [46]. Interpretation of these experiments, however, is complicated by PCNA and PH3 being our only available read-out for cell-cycle activation in the fly brain. Each marker could, for example, represent multiple phases of the cell cycle (PCNA: G1, S, G2; PH3: G2, M), a transcriptional upregulation of cell-cycle regulators in the absence of coordinated cell-cycle activation, an immediate-early gene response (PH3) or a response to DNA damage (PCNA). Again, accurate determination of cell-cycle phasing in different contexts may help resolve these issues because it may be that neuronal apoptosis is transduced at the G2/M and not the G1/S transition in vivo. It is possible that this is context-dependent and dependent upon pathogenesis, or even variable among different populations of neurons exposed to the stimulus. Indeed, in flies, as described in Section 3.1, ectopic expression of different cyclins leads to cell-cycle marker expression and apoptosis in different neuronal populations.
Investigating the issue of sufficiency is important for several reasons. First, understanding the phase at which the apoptotic signal is transduced may have significant implications for designing appropriate therapies. Also, events occurring up- or downstream of tau-induced cell-cycle activation may be critical for an apoptotic response once the cell cycle becomes activated [65]. These events could also potentially be therapeutic targets. Recently, it has been proposed that oxidative stress may be one such critical upstream event [65, 66]. For example, a neuron might be able to enter an S-phase without detriment, unless its DNA has previously been damaged. Thus, an attempt to replicate damaged DNA may be the trigger for apoptosis rather than cell-cycle re-entry itself [67], and oxidative stress may be one important cause of such DNA damage. We have recently showed that genetic and pharmacologic manipulation of oxidative stress modifies tau-induced neurodegeneration in flies (Dias et al., in press), and thus the fly tauopathy model may be very useful in investigating the cell-cycle/oxidative stress connection. Determining critical upstream events may also shed light on the tau-induced phenotype itself because, while ectopic TOR and cell-cycle activation can lead to neuronal apoptosis in the fly brain, in neither situation is the tau phenotype precisely recapitulated, suggesting that events occurring upstream of TOR/cell cycle may be crucial determinants of this phenotype.
4.3 Are there critical non cell-autonomous factors that mediate cell-cycle-dependent apoptosis?
An issue related to sufficiency of cell-cycle activation for neuronal apoptosis is discerning cell-autonomous from non cell-autonomous factors in the mediation of cell-cycle-dependent neuronal death. Cell-cycle-dependent neuronal apoptosis has generally been thought of as a cell-autonomous process, as suggested by investigations in cell culture paradigms. However, there may be critical non cell-autonomous contributors to this process in vivo. The CNS phenotype of mice null for the tumor suppressor pRb (pRb-1 -/-) is a striking example of such a possibility. pRb-1-/- mice die in embryonic life (E13–E15) with significant hematopoietic abnormalities and massive apoptotic neuronal death in the central and peripheral nervous system [15, 16]. The appearance of the neuronal phenotype temporally coincides with the normal neuronal cell-cycle exit and commencement of differentiation. Histologically, proliferation-competent neurons in the ventricular zone of the neural tube are normal but apoptotic and mitotic markers are found in CNS regions usually populated by postmtitotic cells. These findings imply a role for pRb-1 in either establishing or maintaining the postmitotic state, and possibly in neuronal differentiation. Interestingly, more recent studies have indicated that central neuronal apoptosis in pRb-1-/- mice is not cell-autonomous. In chimeric mice, composed of wild-type and pRb-1-/- cells, central (but not peripheral) neuronal apoptosis does not occur despite the failure to achieve cell-cycle exit, indicated by abnormal tetraploid cells. The cell-cycle profile, however, of these neurons appear to be different because, while PH3-staining and FACS analyses indicate that many CNS neurons have entered late G2 or M phase in the germline pRb-1-/- animals, neurons in the chimeric mice do not appear to enter M-phase [17]. Surprisingly, the largely normal development of the CNS in chimeras implies that tetraploid neurons are perhaps even capable of differentiation. Thus, these results indicate that apoptosis in this particular developmental context is non cell-autonomous and that failure to exit the cell cycle is not sufficient to trigger central neuronal apoptosis. The results also raise the possibility that apoptosis is triggered at the G2/M checkpoint and that non cell-autonomous factors are required to drive neurons into late G2/M. Finally, the studies indicate important differences between cell-death signaling in central and peripheral nervous system neurons. An intriguing recent study has indicated that the critical non cell-autonomous factors that trigger apoptosis in this model may be placentally derived since pRb-1-/- mice grown in a wild-type placenta do not develop CNS pathology [68, 69].
Drosophila is an attractive model system in which to search for potential non cell-autonomous factors in cell-cycle-dependent neuronal death. For example, in addition to ectopic neuronal expression of cyclins or E2f/Dp, it would be interesting to introduce other genetic or environmental manipulations, both within and outside the nervous system, to determine factors that might modulate the neuronal response to ectopic cell-cycle activation. Obvious candidates for such manipulations would be oxidative stress, DNA damage and cell-cycle checkpoint pathways, all of which have been extensively characterized in a variety of contexts in flies [70, 71]. The modulation of molecules known to alter lifespan, such as those in the insulin signaling pathway or Sir2, within the brain and fat body might also be very interesting in this context [72–74]. In addition, genetic tools are now available in flies to facilitate creation and analysis of clones in the brain, allowing ectopic activation of the cell cycle in a select group of neurons, or in glial cells. It would be interesting if neuronal cell-cycle activation can lead to non cell-autonomous death of surrounding neurons, potentially analogous to the non cell-autonomous apoptosis that occurs in cells surrounding clones that have been conferred a growth advantage. Several recent papers have been published on this phenomenon of “super-competition” in clonal analyses of developing fly imaginal discs [75, 76].
4.4 How do the TOR/cell-cycle pathways become activated in the Drosophila tauopathy model?
In Section 3.2, we outlined several possibilities for tau-induced cell-cycle activation. As depicted in Figure 4, these included more general mechanisms, including generation of oxidative stress or DNA damage, inhibition of the ubiquitin-proteasome system. Other mechanisms depicted were more specifically related to tau, including direct tau-protein interactions and altered microtubule dynamics. We have made good progress in delineating signaling pathways, including TOR, that mediate cell-cycle activation in our model. An important aim of future studies will be to determine the mechanism whereby tau activates mitogenic signaling pathways. In cell-culture systems, oxidative stress activates numerous mitogenic signaling pathways [77] and fluctuations in redox potential may critically regulate cell-cycle proteins [78]. As noted above (Section 4.2), we have recently found that genetically and pharmacologically modulating the oxidative stress response can modify tau-induced neurodegeneration phenotypes (Dias et al., in press). In fact, decreasing anti-oxidant defences in the fly brain does potentiate tau’s ability to induce cell-cycle marker re-expression, implicating oxidative stress in this process. Interestingly, microtubule stabilization in proliferating can induce TOR-dependent apoptosis [79]. It will be interesting to determine whether direct manipulations of the microtubules, whether genetic or pharmacologic, can recapitulate cell-cycle phenotypes, Likewise, numerous genetic reagents are now available in Drosophila to manipulate the UPS and thus the hypothesis that defective degradation of mitotic cyclins leads to aberrant cell-cycle activation [80] is readily testable in vivo in flies.
As mentioned in Section 3.2, tau is known to bind several molecules involved in mitogenic signaling. Homologs of these interacting proteins exist in flies and, in several cases, have been well characterized. Future studies may help determine whether genetic manipulations of these potential interactors alters tau-induced neurodegeneration and, if so, whether tau physically interacts with these proteins in the fly eye or brain. Furthermore, the expression of tau mutants unable to bind these interactors may provide more definitive evidence of the role of such interacting proteins in mediating tau-induced cell-cycle activation. We, and other groups, have successfully made mutant tau constructs and expressed them in flies to elucidate various aspects of tau pathobiology [44, 46]. While these studies would be significant if fruitful, they do rely on limited candidate-based testing based on in vitro tau-protein interactions, or on predictions of potential tau interactors based on modular domains that may not hold true in vivo. Unbiased genetic screens and protein interaction studies, such as yeast-2-hybrid or immunoprecipitation in conjunction with mass spectrometry, may represent powerful alternative approaches. In addition, libraries for many modular domains now exist. It may be worthwhile to test tau for binding in these libraries and to verify interesting candidates for their ability to mediate tau-induced cell-cycle activation in vivo.
Finally, the ability of multiple and diverse insults to mediate neuronal cell-cycle re-entry would imply that the ability to induce cell-cycle activation is not specific to tau phosphorylation [6]. Indeed, cell-cycle mechanisms have been implicated in PD also [81–83]. While we did not find evidence for cell-cycle activation in tau-independent models of neurodegeneration (MJD and PD), other Drosophila models, notably recent models of AD based on ectopic beta-amyloid expression [84–87], remain to be tested for genetic modification by cell-cycle and mitogenic signaling modulation.
5. Conclusion
The idea, proposed more than a decade ago, that ectopic cell-cycle activation in neurons might lead not to their proliferation, but to their demise, is an intriguing one. A number of investigators subsequently identified aberrant expression of cell-cycle markers in human postmortem tissue from a variety of neurodegenerative diseases, most notably AD and other tauopathies, thus establishing this idea as one of potential clinical relevance. Further work in cell-culture systems provided mechanistic insights into how cell-cycle mediators might transduce apoptotic signals.
Recent investigations in a Drosophila tauopathy model have complemented these studies, principally by enabling a thorough genetic substantiation of the idea that cell-cycle activation mediates neuronal death in vivo. In the tauopathy model, inroads have been made in identifying signaling pathways, including TOR kinase signaling, that may mediate aberrant mitogenic signals in the human disease. These investigations raise the possibility that TOR and the cell cycle are therapeutic targets in human tauopathies, including AD, a hypothesis that is now amenable to testing in mouse tauopathy models. Powerful genetic methods available in flies, including the ability to express mutant tau constructs, have enabled mechanistic insights to be made. For example, the experiments investigating the epistasis of tau phosphorylation to cell-cycle activation suggest that tau phosphorylation is upstream of cell-cycle activation [46].
For all the progress that has been made in human tissue, mammalian cell-culture and Drosophila model systems, fundamental and important questions remain. These include whether or not a coordinated cell-cycle activation, or simply certain cell-cycle activators, are required to mediate neuronal death; whether ectopic neuronal cell-cycle activation is always sufficient to mediate neuronal apoptosis; and whether cell-autonomous versus non cell-autonomous factors play a role in cell-cycle-mediated neurodegeneration. Finally, the relationship of cell-cycle activation to other signaling pathways, including those related to cell-cycle checkpoints and aging, remain unclear in vivo.
It is intriguing that there may be a significant mechanistic connection between oncogenesis and neurodegeneration, two major pathologies associated with aging. With versatile and powerful tools available, Drosophila models may be superbly suited to gauge the depth of this connection, and its basis.
Acknowledgments
The authors would like to thank Ilan Elson-Schwab for useful discussions and his thoughtful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT. Neuron. 1992;9:955–66. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- 2.Heintz N. Trends Biochem Sci. 1993;18:157–9. doi: 10.1016/0968-0004(93)90103-t. [DOI] [PubMed] [Google Scholar]
- 3.Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M. Trends Neurosci. 2001;24:25–31. doi: 10.1016/s0166-2236(00)01663-5. [DOI] [PubMed] [Google Scholar]
- 4.Husseman JW, Nochlin D, Vincent I. Neurobiol Aging. 2000;21:815–28. doi: 10.1016/s0197-4580(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Geldmacher DS, Herrup K. J Neurosci. 2001;21:2661–8. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker EB, Bonni A. Prog Neurobiol. 2004;72:1–25. doi: 10.1016/j.pneurobio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Konishi Y, Bonni A. J Neurosci. 2003;23:1649–58. doi: 10.1523/JNEUROSCI.23-05-01649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konishi Y, Lehtinen M, Donovan N, Bonni A. Mol Cell. 2002;9:1005–16. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- 9.Liu DX, Greene LA. Cell Tissue Res. 2001;305:217–28. doi: 10.1007/s004410100396. [DOI] [PubMed] [Google Scholar]
- 10.Busser J, Geldmacher DS, Herrup K. J Neurosci. 1998;18:2801–7. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, Castellani RJ, Perry G, Smith MA. Aging Cell. 2003;2:105–10. doi: 10.1046/j.1474-9728.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- 12.Delobel P, Lavenir I, Ghetti B, Holzer M, Goedert M. Am J Pathol. 2006;168:878–87. doi: 10.2353/ajpath.2006.050540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Herrup K. J Neurosci. 2005;25:2522–9. doi: 10.1523/JNEUROSCI.4946-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, Varvel NH, Lamb BT, Herrup K. J Neurosci. 2006;26:775–84. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 16.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 17.Lipinski MM, Macleod KF, Williams BO, Mullaney TL, Crowley D, Jacks T. Embo J. 2001;20:3402–13. doi: 10.1093/emboj/20.13.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrer I, Blanco R, Carmona M, Puig B. Neuropathol Appl Neurobiol. 2001;27:343–51. doi: 10.1046/j.1365-2990.2001.00348.x. [DOI] [PubMed] [Google Scholar]
- 20.Ferrer I, Blanco R, Carmona M, Puig B. J Neural Transm. 2001;108:1397–415. doi: 10.1007/s007020100016. [DOI] [PubMed] [Google Scholar]
- 21.Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Vinals F, Ribalta T. Brain Pathol. 2001;11:144–58. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X, Lee HG, Raina AK, Perry G, Smith MA. Neurosignals. 2002;11:270–81. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- 23.Edgar BA, Lehner CF. Science. 1996;274:1646–52. doi: 10.1126/science.274.5293.1646. [DOI] [PubMed] [Google Scholar]
- 24.Muqit MM, Feany MB. Nat Rev Neurosci. 2002;3:237–43. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- 25.Bravo R, Frank R, Blundell PA, Macdonald-Bravo H. Nature. 1987;326:515–7. doi: 10.1038/326515a0. [DOI] [PubMed] [Google Scholar]
- 26.Hans F, Dimitrov S. Oncogene. 2001;20:3021–7. doi: 10.1038/sj.onc.1204326. [DOI] [PubMed] [Google Scholar]
- 27.Majka J, Burgers PM. Prog Nucleic Acid Res Mol Biol. 2004;78:227–60. doi: 10.1016/S0079-6603(04)78006-X. [DOI] [PubMed] [Google Scholar]
- 28.Nowak SJ, Corces VG. Trends Genet. 2004;20:214–20. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Stevaux O, Dyson NJ. Curr Opin Cell Biol. 2002;14:684–91. doi: 10.1016/s0955-0674(02)00388-5. [DOI] [PubMed] [Google Scholar]
- 30.Dyson N. Genes Dev. 1998;12:2245–62. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 31.Dotto GP. Biochim Biophys Acta. 2000;1471:M43–56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 32.Adams MD, et al. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 33.de Nooij JC, Letendre MA, Hariharan IK. Cell. 1996;87:1237–47. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- 34.Stevaux O, Dimova D, Frolov MV, Taylor-Harding B, Morris E, Dyson N. Embo J. 2002;21:4927–37. doi: 10.1093/emboj/cdf501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Datar SA, Jacobs HW, de la Cruz AF, Lehner CF, Edgar BA. Embo J. 2000;19:4543–54. doi: 10.1093/emboj/19.17.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer CA, Jacobs HW, Datar SA, Du W, Edgar BA, Lehner CF. Embo J. 2000;19:4533–42. doi: 10.1093/emboj/19.17.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallant P. Curr Top Microbiol Immunol. 2006;302:235–53. doi: 10.1007/3-540-32952-8_9. [DOI] [PubMed] [Google Scholar]
- 38.Prober DA, Edgar BA. Cell. 2000;100:435–46. doi: 10.1016/s0092-8674(00)80679-0. [DOI] [PubMed] [Google Scholar]
- 39.Oldham S, Hafen E. Trends Cell Biol. 2003;13:79–85. doi: 10.1016/s0962-8924(02)00042-9. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Febs J. 2005;272:4211–20. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 41.Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Neuron. 2002;34:509–19. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 42.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Science. 2001;293:711–4. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 43.Brand AH, Perrimon N. Development. 1993;118:401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 44.Nishimura I, Yang Y, Lu B. Cell. 2004;116:671–82. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- 45.Shulman JM, Feany MB. Genetics. 2003;165:1233–42. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. Curr Biol. 2006;16:230–41. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 47.Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cell. 1994;77:107–20. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 48.Billingsley ML, Kincaid RL. Biochem J. 1997;323(Pt 3):577–91. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dimova DK, Dyson NJ. Oncogene. 2005;24:2810–26. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 50.Biernat J, Mandelkow EM. Mol Biol Cell. 1999;10:727–40. doi: 10.1091/mbc.10.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee G. Biochim Biophys Acta. 2005;1739:323–30. doi: 10.1016/j.bbadis.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Johnson GV, Jenkins SM. J Alzheimers Dis. 1999;1:307–328. doi: 10.3233/jad-1999-14-511. [DOI] [PubMed] [Google Scholar]
- 53.Hunter T. Cell. 1997;88:333–46. doi: 10.1016/s0092-8674(00)81872-3. [DOI] [PubMed] [Google Scholar]
- 54.Ahuja D, Saenz-Robles MT, Pipas JM. Oncogene. 2005;24:7729–45. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 55.Barbato C, Corbi N, Canu N, Fanciulli M, Serafino A, Ciotti M, Libri V, Bruno T, Amadoro G, De Angelis R, Calissano P, Passananti C. Mol Cell Neurosci. 2003;24:1038–50. doi: 10.1016/j.mcn.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 56.Bruno T, De Angelis R, De Nicola F, Barbato C, Di Padova M, Corbi N, Libri V, Benassi B, Mattei E, Chersi A, Soddu S, Floridi A, Passananti C, Fanciulli M. Cancer Cell. 2002;2:387–99. doi: 10.1016/s1535-6108(02)00182-4. [DOI] [PubMed] [Google Scholar]
- 57.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Eur J Neurosci. 2005;22:1942–50. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 58.Tanimukai H, Grundke-Iqbal I, Iqbal K. Am J Pathol. 2005;166:1761–71. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL., 3rd J Neuropathol Exp Neurol. 2004;63:1080–91. doi: 10.1093/jnen/63.10.1080. [DOI] [PubMed] [Google Scholar]
- 60.Lim J, Lu KP. Biochim Biophys Acta. 2005;1739:311–22. doi: 10.1016/j.bbadis.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 61.Bjornsti MA, Houghton PJ. Nat Rev Cancer. 2004;4:335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 62.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Curr Biol. 2004;14:885–90. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 64.Duronio RJ, Bonnette PC, O'Farrell PH. Mol Cell Biol. 1998;18:141–51. doi: 10.1128/mcb.18.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu X, Raina AK, Perry G, Smith MA. Lancet Neurol. 2004;3:219–26. doi: 10.1016/S1474-4422(04)00707-0. [DOI] [PubMed] [Google Scholar]
- 66.Klein JA, Ackerman SL. J Clin Invest. 2003;111:785–93. doi: 10.1172/JCI18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nouspikel T, Hanawalt PC. Bioessays. 2003;25:168–73. doi: 10.1002/bies.10227. [DOI] [PubMed] [Google Scholar]
- 68.Dyson N. Nature. 2003;421:903–4. doi: 10.1038/421903a. [DOI] [PubMed] [Google Scholar]
- 69.Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, Rosol TJ, Woollett LA, Weinstein M, Cross JC, Robinson ML, Leone G. Nature. 2003;421:942–7. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- 70.Song YH. Mol Cells. 2005;19:167–79. [PubMed] [Google Scholar]
- 71.Fleming JE, Reveillaud I, Niedzwiecki A. Mutat Res. 1992;275:267–79. doi: 10.1016/0921-8734(92)90031-j. [DOI] [PubMed] [Google Scholar]
- 72.Rogina B, Helfand SL. Proc Natl Acad Sci U S A. 2004;101:15998–6003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Nature. 2004;430:686–9. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 74.Tatar M. Exp Gerontol. 2004;39:1745–50. doi: 10.1016/j.exger.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 75.Gallant P. Cancer Res. 2005;65:6485–7. doi: 10.1158/0008-5472.CAN-05-1101. [DOI] [PubMed] [Google Scholar]
- 76.Secombe J, Pierce SB, Eisenman RN. Cell. 2004;117:153–6. doi: 10.1016/s0092-8674(04)00336-8. [DOI] [PubMed] [Google Scholar]
- 77.Martindale JL, Holbrook NJ. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 78.Menon SG, Goswami PC. Oncogene. 2006 doi: 10.1038/sj.onc.1209895. [DOI] [PubMed] [Google Scholar]
- 79.Asnaghi L, Bruno P. M Priulla and A Nicolin, Pharmacol Res. 2004;50:545–9. doi: 10.1016/j.phrs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 80.Rideout HJ, Wang Q, Park DS, Stefanis L. J Neurosci. 2003;23:1237–45. doi: 10.1523/JNEUROSCI.23-04-01237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S, Slack RS, Przedborski S, Anisman H, Park DS. Proc Natl Acad Sci U S A. 2003;100:13650–5. doi: 10.1073/pnas.2232515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.West AB, Dawson VL, Dawson TM. Trends Neurosci. 2005;28:348–52. doi: 10.1016/j.tins.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 83.Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Neuron. 2003;37:735–49. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- 84.Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, Lomas DA. Neuroscience. 2005;132:123–35. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 85.Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. Mol Cell Neurosci. 2004;26:365–75. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 86.Greeve I, Kretzschmar D, Tschape JA, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R. J Neurosci. 2004;24:3899–906. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Proc Natl Acad Sci U S A. 2004;101:6623–8. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu DX, Nath N, Chellappan SP, Greene LA. Genes Dev. 2005;19:719–32. doi: 10.1101/gad.1296405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. J Neurosci. 1998;18:830–40. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA, Lenz C, Price C, Bird IN, Perera T, Kellie S, Williamson R, Noble W, Van Etten RA, Leroy K, Brion JP, Reynolds CH, Anderton BH. J Neurosci. 2005;25:6584–93. doi: 10.1523/JNEUROSCI.1487-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jenkins SM, Johnson GV. Neuroreport. 1998;9:67–71. doi: 10.1097/00001756-199801050-00014. [DOI] [PubMed] [Google Scholar]
- 92.Hwang SC, Jhon DY, Bae YS, Kim JH, Rhee SG. J Biol Chem. 1996;271:18342–9. doi: 10.1074/jbc.271.31.18342. [DOI] [PubMed] [Google Scholar]
- 93.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. Nature. 1999;399:784–8. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 94.Coleman ML, Marshall CJ, Olson MF. Nat Rev Mol Cell Biol. 2004;5:355–66. doi: 10.1038/nrm1365. [DOI] [PubMed] [Google Scholar]
- 95.Jacinto E, Hall MN. Nat Rev Mol Cell Biol. 2003;4:117–26. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- 96.Wu Q, Brown MR. Annu Rev Entomol. 2006;51:1–24. doi: 10.1146/annurev.ento.51.110104.151011. [DOI] [PubMed] [Google Scholar]
- 97.Potter CJ, Pedraza LG, Xu T. Nat Cell Biol. 2002;4:658–65. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Nat Cell Biol. 2003;5:578–81. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 99.Patel PH, Thapar N, Guo L, Martinez M, Maris J, Gau CL, Lengyel JA, Tamanoi F. J Cell Sci. 2003;116:3601–10. doi: 10.1242/jcs.00661. [DOI] [PubMed] [Google Scholar]
- 100.Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Nat Cell Biol. 2003;5:566–71. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 101.Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E. Nat Cell Biol. 2003;5:559–65. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 102.Hennig KM, Neufeld TP. Genesis. 2002;34:107–10. doi: 10.1002/gene.10139. [DOI] [PubMed] [Google Scholar]
- 103.Miron M, Lasko P, Sonenberg N. Mol Cell Biol. 2003;23:9117–26. doi: 10.1128/MCB.23.24.9117-9126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schmidt EV. Oncogene. 1999;18:2988–96. doi: 10.1038/sj.onc.1202751. [DOI] [PubMed] [Google Scholar]