

Abstract
Prostaglandin E2 (PGE2) is a prototypical inflammatory mediator that excites and sensitizes cell bodies (Kwong and Lee, 2002; 2005) and peripheral nerve terminals (Ho et al. 2000) of primary vagal sensory neurons. Nearly all central nerve terminals of vagal afferents are in the nucleus tractus solitarius (NTS), where they operate with a high probability of release (Doyle and Andresen, 2001). We studied the effect of PGE2 on synaptic transmission between tractus solitarius afferent nerve terminals and the second order NTS neurons in brain stem slices of Sprague Dawley rats. Whole-cell patch recording in voltage clamp mode was used to study evoked excitatory postsynaptic glutamatergic currents (evEPSCs) from NTS neurons elicited by electrical stimulation of the solitary tract (ST). In 34 neurons, bath applied PGE2 (200 nM) decreased the evEPSC amplitude by 49 ± 5 %. In 22 neurons, however, PGE2 had no effect. We also tested 15 NTS neurons for capsaicin sensitivity. Seven neurons generated evEPSCs that were equally unaffected by PGE2 and capsaicin. Conversely, evEPSCs of the other 8 neurons, which were PGE2-responsive, were abolished by 200 nM capsaicin. Furthermore, the PGE2-induced depression of evEPSCs was associated with an increase in the paired pulse ratio and a decrease in both the frequency and amplitude of the spontaneous excitatory postsynaptic currents (sEPSCs) and TTX-independent spontaneous miniature excitatory postsynaptic currents (mEPSCs). These results suggest that PGE2 acts both presynaptically on nerve terminals and postsynaptically on NTS neurons to reduce glutamatergic responses.
Keywords: Glutamate, AMPA receptors, Presynaptic, Postsynaptic
Many neurons in the nucleus of the tractus solitarius (NTS) receive nerve terminals whose axons arise from cell bodies of vagal primary afferent neurons located in the nodose and jugular vagal ganglia. These primary vagal afferents convey diverse sensory information from a variety of receptors located in cardiovascular, respiratory, gastrointestinal and other organs to the NTS neurons in the medulla (Contreras, 1982). Thus, the first monosynaptic contact for myelinated A-type and unmyelinated C-type axons of primary vagal afferent fibers are the second-order neurons in the NTS. These fibers reach second-order NTS neurons via the solitary tract (Doyle et al. 2002) where they evoke ionotropic glutamatergic and/or peptidergic synaptic potentials (Gatti et al. 1995; Aylwin et al. 1997).
For many decades, prostaglandins have been well known to be major mediators in the response to immune challenges. More recently, these lipid mediators have been implicated in physiological and pathophysiological functions in the CNS and PNS neurons (Miettinen et al. 1997; Montine et al. 1999; Liu et al. 2005). PGE2 is a major prostanoid that is involved in a myriad CNS functions (Wolfe and Coceani, 1979; Ho et al. 1999; McCullough et al. 2004). PGE2 is synthesized in the CNS by neurons and glia cells (Bishai and Coceani, 1992; Guay et al. 2004). Cyclooxygenase 2 (COX-2) converts free arachidonic acid to the intermediate prostaglandin H2 (PGH2). PGH2 in turn is converted to PGE2 by the enzyme PGE2 synthase (Thoren et al. 2003). In addition to localized formation in the CNS, PGE2 is synthesized outside the CNS and can reach NTS neurons because this area of the brain lacks a complete blood-brain barrier (Gross et al. 1990).
Functional PGE2 receptors are thought to exist on primary vagal afferents because many distinct types of voltage-gated ionic channels and ligand-gated channels in vagal afferents are modified by exogenous PGE2. These include: calcium-dependent potassium currents (Fowler et al. 1985), leak currents (Undem and Weinreich, 1993), hyperpolarizing-activated cationic currents (Ingram and Williams, 1996), TTX-resistant sodium currents (Gold et al. 2002; Kwong and Lee, 2005), and the ionotropic receptors TRPV1 and 5-HT3 (Kwong and Lee, 2002). In addition, PGE2 binding has been reported in the medial NTS (Matsumura et al. 1992), an area rich in afferent innervation. Thus, it is likely that functional PGE2 receptors are present on vagal afferent nerve terminals in the NTS (Ek et al. 2000). In support of this possibility, Sekiyama et al. (1995) reported that the magnitude of ST evoked excitatory postsynaptic currents (evEPSCs) was facilitated by PGE2 in three of eight NTS neurons examined. The nature of the PGE2 receptors mediating this effect has not yet been clarified.
PGE2 can interact with four subtypes of G-protein-mediated receptors: EP1, EP2, EP3, and EP4 (Narumiya et al. 1999). The NTS has been shown to express all four receptors (Matsumura et al. 1992; Ek et al. 2000; Nakamura et al. 2000). Likewise, EP1–4 receptors are present in the cell bodies of nodose ganglia neurons (Matsumoto et al. 2005), that project their central processes to the NTS. The purpose of this work was to determine the effect of PGE2 on synaptic transmission between ST afferents and second-order NTS neurons.
EXPERIMENTAL PROCEDURES
Slice preparation
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore. Young-Adult Sprague Dawley male rats (n = 42), 14 to 20 days old, were anesthetized with ketamine (30 mg/kg) and decapitated. The brains were rapidly removed, and 150 μm thick brainstem slices were cut at 4°C with a vibratome (VT 1000S, Leica Microsystems Inc. Nusslock, Germany). Coronal slices were kept in a holding chamber that contained artificial cerebrospinal fluid (aCSF) composed of (mM): 124 NaCl, 25 NaHCO3, 1.2 NaH2PO4, 3 KCl, 2 MgSO4, 2 CaCl2, and 15 Glucose. The pH and Osmolarity were adjusted to 7.2 – 7.4, and 305 mOsm, respectively. The aCSF continuously bubbled with 95% O2 - 5% CO2. Following a 30 min recovery period at 30°C, the slices were maintained in the same chamber at room temperature (23 – 25°C). Slices were transferred to the recording chamber to study the effects of PGE2 at 32°C.
Electrical stimulation and recording
Bipolar stimulation electrode (paired 50 μm stainless steel wires, insulated except at the tips) was placed visually in the ST, ipsilateral to the recording site to activate primary sensory afferent fibers (Fig. 1A and 1B). The atlas of Paxinos and Watson (1998) was used for identification and nomenclature of anatomical structures. Constant-voltage pulses were delivered through a stimulus isolation unit, driven by a pulse generator (Grass S88, Astro-Med, West Warwick, RI). The ST was focally stimulated at 0.2 Hz with 10 to 50 V pulses (150 μs duration) for a total of 10 stimuli. To eliminate the potential influence of activation of inhibitory interneurons during ST stimulation, slices were superfused with the aCSF solution containing GABAA and glycine receptor antagonists, bicuculline (10 μM) and strychnine (1 μM), respectively. Since PGE2 can affect also adenosine receptors we add to the aCSF solution the adenosine receptors antagonist, DPCPX (1μM).
Figure 1.
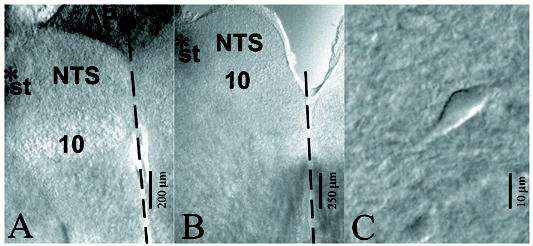
Microscopic views of 150 μm thick brain slices containing the nucleus tractus solitarius (NTS). A, slice from caudal region of the NTS limited by the AP (area postrema), the ST (solitary tract), the midline (dashed line) and the 10 (dorsal motor nucleus of the vagus) (Bregma -13.80 mm). B, Another slice obtained from the intermediate region of the NTS (Bregma -13.30 mm). The asterisk indicates position of stimulating electrode. C, an individual NTS neuron viewed with a microscope equipped with near-infrared differential interference contrast optics.
To test whether ST-NTS responses were monosynaptic, we measured synaptic jitter. We determined the onset latencies for the evEPSCs, the time between the shock artifact and the onset of the evEPSCs. Jitter was calculated as the shock-to-shock variability in the onset latency. An onset latency variability of < 0.5 ms was taken as evidence for a putative monosynaptic response (Chen et al 2002). Paired-pulse ratio is defined as the ratio of the second evEPSC peak amplitude divided by the first evEPSC peak amplitude using a 20 ms interpulse interval.
Recordings were made at 32°C in a submersion type recording chamber perfused with aCSF at 2 ml/min. Patch electrode placement and NTS neuron selection were performed with near-infrared differential interference contrast microscopy, using a 40X (0.9 N.A.) water immersion objective attached to a fixed-stage, upright microscope (Nikon Eclipse, E600 FN, Tokyo, Japan).
Whole-cell recordings
Whole-cell voltage-clamp recordings were obtained from second-order neurons located in the caudal (Fig. 1A) and intermediate part of the NTS (Fig. 1B), with an Axopatch 200A amplifier in conjunction with pClamp 7 software (Axon Instruments, Foster City, CA). Currents were filtered at 2 – 5 KHz and digitized at 5 – 20 KHz with a DigiData 1200 interface (Axon Instruments). The resistances of the patch electrodes ranged from 5 to 8 MΩ. The intracellular recording solution contained (in mM): 144 D-gluconic acid potassium salt, 2 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, 0.2 GTP and 3 ATP, pH 7.2 (310 mOsm). Chemicals for intracellular and extracellular solutions were obtained from Sigma (St. Louis, MO). Neurons included for study had holding currents < 100 pA with membrane potentials ranging between −60 and −65 mV for a 5 min control period, and had input resistances > 200 MΩ. Seal resistances were > 1 GΩ, and series resistance was always < 25 MΩ with 60% compensation.
Analysis of synaptic currents
To determine the action of PGE2 on monosynaptic transmission in the NTS, the following synaptic responses were examined: (1) evEPSC peak amplitude, (2) paired-pulse ratio (PPR), (3) frequency and amplitude of spontaneous EPSCs (sEPSCs), and (4) the TTX-insensitive frequency and amplitude of miniature EPSCs (mEPSCs). For PPR measurements, ST pair stimuli were delivered at an interpulse interval of 20 ms at 0.2 Hz and the peak amplitudes of the first and second paired evEPSCs were recorded for at least 2 min before (control) and for at least 5 min after the onset of PGE2 perfusion. For measurements of mEPSCs, slices were superfused with aCSF containing TTX (1 μM) and the frequency and amplitude of mEPSCs recorded before (2 min) and during a five min perfusion with aCSF containing PGE2. The frequency and the amplitude sEPSCs were analyzed by Mini Analysis Program (Synaptosoft Inc, Fort Lee, New Jersey). The baseline noise was < 8 pA. The threshold for detecting sEPSCs varied between 8 to 15 pA, this variability did not affect the magnitude of decrease in the sEPSC amplitude induced by PGE2. Data are expressed as mean ± S.E.M unless otherwise indicated. Differences were considered to be significant at P < 0.05 for Student paired t-test and P <0.01 for Kolmogorov Smirnov test. The onset latency for each evEPSC was determined from 10 evEPSC delivered at 0.2 Hz. The magnitude of the stimulus artifacts was digitally reduced.
Analysis of passive and active membrane proprieties
For evaluating the neuronal excitability, 750 ms depolarizing current steps (1- and 2-times threshold current, rheobase) were applied in the current-clamp mode and the number of action potential generated were counted following each stimulus. In current clamp, input resistance was calculated by applying 450 ms hyperpolarizing current steps (50 pA) and measuring the peak steady-state amplitude of the resulting voltage transients. Rin was determined at resting membrane potential, −60 to −65 mV. Active membrane proprieties were evaluated by analyzing various action potential waveforms. Action potential (AP) duration was measured at a membrane potential of 0 mV. Overshoot and afterhyperpolarization (AHP) magnitudes of action potential were estimated as the peak amplitude of AP upstroke from 0 mV and down stroke from the resting membrane potential, respectively. AP threshold potential was defined as the membrane potential where the rate of depolarization starts to increase abruptly as determined by differentiation of membrane potential with respect to time (dV/dt). The minimum current needed to evoke an AP was defined as threshold current or rheobase.
Chemicals
Reagents were prepared immediately before use from freshly prepared or frozen stock solutions and dissolved in aCSF. The following chemicals were obtained from Tocris (Ellisville, MO): AP5 (D-2-amino-5-phosphonopentanoic acid), bicuculline, DPCPX (8-Cyclopentyl-1,3-dipropylxanthine), strychnine and CNQX (6-cyano-7-nitroquinoxaline). SC19220 (8-chloro-dibenz(b,f)(1,4)oxazepine-10(11H)-carboxylic acid or 2-acethylhydrazide 10(11)-carboxylic acid), AH 23848 [(4Z)-7-[(rel-1S,2S,5R)-5-((1,1′-Biphenyl-4-yl)methoxy)-2-(4-morpholinyl)-3-oxocyclopentyl)-4-heptenoic acid calcium salt], Sulprostone ((5Z,11α,13E,15R)—11,15-Dihydroxy-9-oxo-16-phenoxy-17,18,19,20-tetranorprosta-5,13-dienoic acid methane sulfonamide), Butaproste ((1R,2R,3R)-3-Hydroxy-2-[(1E,4R)-4-hydroxy-4-(1-propylcyclobutyl)-1-butenyl]-5-oxocyclopentaneheptanoic acid methyl ester), indomethacin, capsaicin, PGE2 and TTX were purchased from Sigma (St Louis, MO).
RESULTS
The activation of cranial visceral afferent axons (and other axons) within the solitary tract (ST) can evoke short-latency, 2.3 ms ± 0.15 excitatory postsynaptic currents (EPSCs) with an onset latency variability (“jitter”) of 0.34 ms ± 0.02, supporting the contention of a monosynaptic connection from the primary sensory afferent terminals originating from the ST to second order NTS neurons (Miles, 1986; Chen et al. 1999; Doyle et al. 2001). Only synapses revealing short latencies and low jitter, as described above, were accepted for study. In 56 neurons studied, the ST-evEPSC averaged 222.4 ± 9.9 pA. In these cells, the whole cell capacitance and membrane input resistance was 20.4 ± 1.3 pF and 335 ± 32.2 MΩ, respectively. Synaptic transmission consistently displayed synaptic depression in response to the paired-pulse stimulation of the ST. The PPR of the evEPSC averaged 0.7 ± 0.03 (n = 56), similar to values reported by Doyle and Andresen (2001), Sekizawa et al. (2003).
PGE2 decreased the evEPSC amplitude
In a series of control experiments, we determined that over a period of five to ten min (in aCSF solution only) the mean evEPSC amplitude was reduced from 0 to 20 % of control (11 ± 8.0 pA; n = 35). Thus, we arbitrarily choose any PGE2 effect greater than 20 % of baseline evEPSC amplitude as a significant PGE2-evoked response. Fifty-six second-order NTS neurons were subjected to perfusion with 100 – 200 nM PGE2-containing aCSF for three to five min. Sixty percent of these neurons showed a PGE2-mediated depression in the amplitude of evEPSCs synaptic transmission averaging of 49 ± 5.0 % (p < 0.01, n = 34; Fig. 2B). The amplitude of evEPSCs synaptic transmission was unaffected in the remaining 22 neurons.
Figure 2.
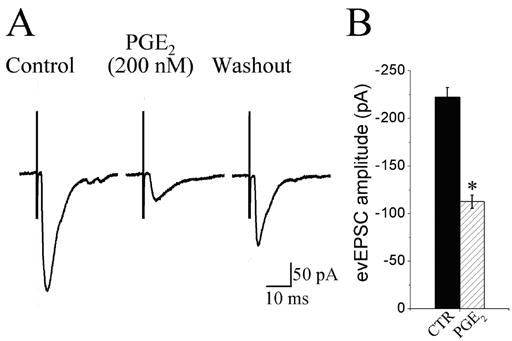
Effect of PGE2 on the amplitude of evoked EPSCs induced by electrical stimulation of the solitary tract. A, Whole-cell voltage clamp recordings of EPSCs sampled from a NTS neuron. Solitary tract (ST) stimulation, 10 V, 150 μs; 0.2 Hz was elicited by placing a stimulating electrode on the ST ipsilateral to the recorded neuron. Each trace is an average of 10 ST-evoked EPSCs (evEPSCs). evEPSCs shown before (control), in the presence of 200 nM PGE2 and 15 min after switching to a drug-free aCSF (washout). B, group data showing that PGE2 significantly reduced the amplitude of the evEPSCs (*P < 0.01; n = 34). Error bars depict ± SEM.
Because a wide range of variability existed in the input resistance (Rin range: 100 to 900 MΩ), we examined whether cells with different Rin values were related to the magnitude of depression in synaptic transmission induced by PGE2. We divided the neurons studied in two groups: a first group with Rin < 300 MΩ and a second group with Rin > 300 MΩ. PGE2-induced decreases in the magnitude of the evEPSCs were of similar value in both groups of neurons (p > 0.05). Thus, PGE2-mediated depression of ST-evEPSCs was unrelated to changes in Rin in the second order NTS neurons.
Effect of PGE2 on passive and active membrane properties
Passive and active membrane proprieties were examined during the peak of the PGE2 effect on synaptic transmission. With the exception of a reduction in resting input resistance (Rin), none of the membrane properties examined were significantly affected by PGE2 treatment (Table 1). The decrease in Rin cannot explain the depression of synaptic transmission because the percentage decrease in evEPSC peak amplitude recorded in 18 neurons did not correlate with the percentage decrease in Rin recorded in the same neurons (r = 0.095, P = 0.73). In addition, changes in evEPSC recorded in voltage- clamp mode should not be influenced by changes Rin recorded in current-clamp mode. The apparent increase in rheobase was not statistically significant due to variability in the data set.
Table 1.
Effect of PGE2 on the passive and active membrane properties. Overshoot measured from 0 mV to peak depolarization of the action potential (AP). Afterhyperpolarization (AHP) measured from the resting membrane potential to the maximal membrane hypepolarization. AP duration measured at one-half peak amplitude. The membrane input resistance shows a significant decrease (* P < 0.05, paired t-test) after PGE2 treatment. The apparent increase in the rheobase after PGE2 treatment was not significantly different (P > 0.05, paired t-test) owing to variability in the data set; 6 of the 10 NTS neurons showed no change. CTR denotes control; PGE2 bath applied at 100 nM. Numbers in parenthesis are number of neurons tested.
| AP Overshoot (mV) | AP AHP (mV) | AP duration (ms) | AP Threshold (mV) | Rheobase (pA) | Number AP at 2X Rheobase | Rin (MΩ) | |
|---|---|---|---|---|---|---|---|
| CTR | 64 ± 3.3 (n = 11) | −6 ± 2.2 (n = 11) | 1.1 ± 0.1 (n = 11) | −25 ± 2 (n = 11) | 44 ± 15 (n = 10) | 13.3 ± 2.2 (n = 12) | 540 ± 46.1 (n = 12) |
| PGE2 | 67 ± 4.4 (n = 11) | −7 ± 2.1 (n = 11) | 1.2 ± 0.1 (n = 11) | −27 ± 2 (n = 11) | 82 ± 26 (n = 10) | 17.9 ± 3.3 (n = 12) | 346 ± 46.0* (n = 12) |
P< 0.05
Effects of PGE2 on capsaicin-sensitive and capsaicin-resistant NTS neurons
Doyle et al. (2002) characterized two classes of medial nucleus tractus solitarius neurons based upon their susceptibility to blockade of afferent synaptic transmission by bath application of 200 nM capsaicin. Capsaicin-sensitive synapses had longer synaptic latencies for evEPSCs relative to capsaicin-resistant synapses suggesting that the former may be associated with slower conducting action potentials mediated by TRPV1-containing C fibers. We tested whether PGE2-induced depression of evEPSCs was associated with capsaicin-sensitive C-fiber synapses. In 15 NTS neurons we added capsaicin (200 nM) to the superfusate at the end of the experiment. In 50 % (8/15) of these neurons 200 nM PGE2 depressed evEPSC by 42.0 ± 7.8 %. Subsequent capsaicin application to the PGE2 sensitive synapses caused a complete block of evEPSC within 10 min (Fig. 3A). By contrast, in the seven NTS neurons where PGE2 did not produce significant depression in the amplitude of the evEPSC, capsaicin application for 10 – 20 min produced no observable change in evEPSCs (Fig. 3B). Thus, it appears that PGE2 exerts its effects on NTS neurons whose synaptic input are likely capsaicin sensitive.
Figure 3.
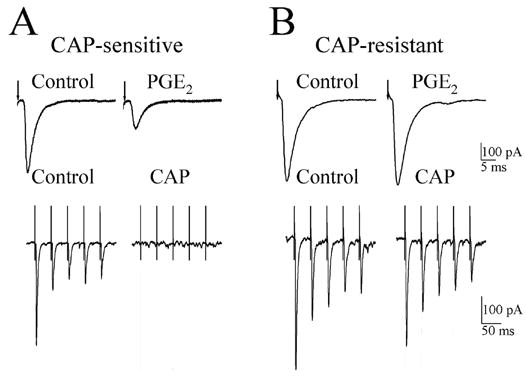
Effect of PGE2 on the amplitude of afferent evoked EPSCs (evEPSCs) recorded in capsaicin-sensitive and capsaicin-resistant second-order NTS neurons. A, top panel, 200 nM PGE2 decreased the evEPSC amplitude recorded in a capsaicin-sensitive neuron. Lower panel, shows five consecutive evEPSCs before (control) and following a 10 min superfusion with 200 nM capsaicin (CAP); note the evEPSCs were completely blocked by CAP treatment. B, top panel, PGE2 (200 nM) did not affect the evEPSC amplitude in CAP-resistant neuron. Lower panel shows a train of five evEPSCs recorded from the same neuron before (control) and 10 min after superfusion with 200 nM CAP. Each trace is an average of 10 responses. The arrows in A and B shows the time of the stimulus artifact.
Nature of the glutamatergic receptors affected by PGE2
Most glutamatergic responses appear to be mediated by AMPA receptors (Andresen and Yang, 1990); However Bonham and Chen (2005) indicated that second order NTS neurons also possess functional NMDA receptors. We were interested in determining which glutamatergic receptors were affected by PGE2. Under the experimental conditions used in which extracellular Mg2+ and the holding membrane potential (−60 mV) suppresses NMDA receptor activity; it is unlikely that NMDA receptors would significantly contributed to the evEPSC. Nonetheless, we examined the effects of bath applying inhibitors of NMDA and AMPA receptors on evEPSCs that were depressed by PGE2. Application of APV (100 μM), a specific antagonist of NMDA receptors, did not measurably affect the evEPSC (n = 4; Fig 4). By contrast, addition of the AMPA/Kainate receptor antagonist CNQX (50 μM) to the APV-containing solution resulted in a complete blockade of the evEPSC (4/4, Fig 4). Thus, we conclude that PGE2 depresses AMPA/Kainate receptor-mediated synaptic transmission in the NTS neurons.
Figure 4.
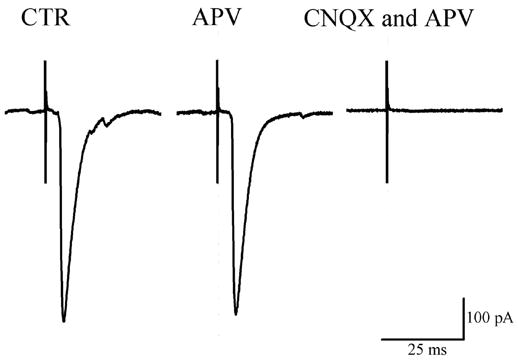
Effect of glutamate receptor antagonists on solitary tract (ST) evoked EPSCs (evEPSCs) recorded in a second order NTS neuron. evEPSC amplitude was not affected by APV (100 μM), but it was completely blocked when CNQX (50μM) was added to the APV solution. Membrane potential held at −60 mV. Each trace is an average of 10 consecutive responses.
Threshold and maximum effect of PGE2
In the current work we did not determine concentration-response relation for PGE2 effect on evEPSCs. However we did estimate threshold and maximum concentrations for the actions of PGE2. In five NTS neurons 3 nM PGE2 did not elicit a measurable change in evEPSCs waveform. Ten nM PGE2 appears to be the minimal concentration necessary to elicit a detectable PGE2 effect on evEPSCs (25.6 ± 12.7 % n = 5). On the other hand, 100 nM PGE2 appears to produce a maximum effect on evEPSCs because a 1000 nM PGE2 elicited no significant (P > 0.05 n = 11) increase in inhibition of evEPSC amplitude greater than that observed with 100 nM (44.5 ± 13.1 % n = 11). Similar results were observed when the same range of concentration of PGE2 were applied to the same neuron (n = 4).
In an additional series of experiments, we investigated whether the steep concentration-response relation might be due to the presence of endogenous PGE2. In, six neurons from control slices, we measured the effect of PGE2 (200 nM) on evEPSCs. Then, six additional slices were incubated at least one hour in an aCSF solution containing indomethacin (3 μM) and the effect of PGE2 (200 nM) on evEPSCs was determined. At this concentration of indomethacin, both cyclooxygenase 1 (COX I) and cyclooxygenase 2 (COX II) enzymatic activity should be abolished (Wolfe and Coceani, 1979). The average decrease in the evoked response in control (47.3 ± 6.1 %) was not significantly different (p > 0.05) from that of indomethacin-treated neurons (52.2 ± 5.5 %), suggesting that endogenous PGE2 does not affect the peak amplitude of the evEPSCs recorded in second order NTS neurons following ST electrical stimulation.
Presynaptic vs. postsynaptic site of PGE2 effect
In order to evaluate whether PGE2-induced depression of synaptic activity was due to a presynaptic effect on ST nerve terminals or at a postsynaptic site on second-order NTS neurons, we measured the PPR of the peak amplitude of the evEPSCs, the frequency and the amplitude of the spontaneous excitatory postsynaptic currents (sEPSC), and action potential-independent synaptic activity, miniature EPSCs (mEPSCs).
PGE2 increases the paired-pulse ratio
We measured PPR of the evEPSCs before and during the peak PGE2-induced depression of the evEPSC. In 70 % of the PGE2-sensitive neurons (24/34) the paired pulse ratio increased by 47.9 ± 9.5 % (paired t-test, p < 0.01, n = 24; Fig 5B). In the other 30 % of these neurons (10/34) five neurons showed a decrease in the PPR and five neurons showed no change in the PPR. As illustrated by the traces in Fig 5A, there was a decrease primarily in the amplitude of the first evEPSC of the paired evEPSCs. The effects of PGE2 on PPR were maximal at approximately 3 – 5 min after the start of perfusion with PGE2 and PPR recovered partially or completely within approximately 20 min after washout of PGE2.
Figure 5.
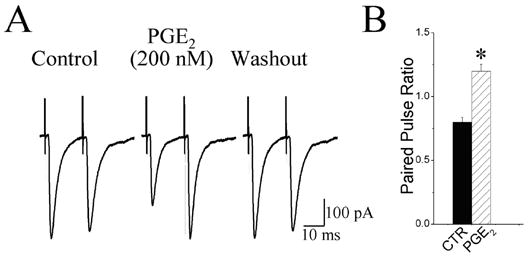
PGE2 increased the paired-pulse ratio. A, Paired pulse ratio for evoked EPSCs was recorded before (control), in the presence of PGE2 (200 nM), and 15 min following superfusion with drug free aCSF (washout). The interval between solitary tract stimulation was 20 ms. B, group data showing that PGE2 significantly (*P < 0.01; n = 24) increased the paired-pulse ratio. Error bars depict ± SEM.
PGE2 depressed the frequency and the amplitude of spontaneous EPSCs
PGE2 (200 nM) decreased the frequency of the sEPSC in 20/34 PGE2 sensitive-neurons (60 %). An example of continuous recordings of sEPSC before, during, and after perfusion with PGE2 is shown in Fig. 6A. The cumulative probability of the sEPSC inter-event intervals was shifted to the right in the presence of PGE2 (Fig. 6B right panel), indicating a decrease in the frequency of events (control, 3.7 ± 1.8 Hz; PGE2 2.2 ± 0.7 Hz P < 0.01 n = 20). In the same group of cells, the cumulative probability of the sEPSC amplitude was shifted to the left (Fig. 6B left panel), indicating a decrease in the amplitude of these events (control, 30 ± 4 pA; PGE2 24 ± 3 pA P < 0.01 n = 20). In the other 40 % of the PGE2 sensitive-neurons (n = 14) PGE2 did not affect the frequency of sEPSCs (control, 1.8 ± 0.5 Hz; PGE2 2.1 ± 0.5 Hz P > 0.05). In the same group of cells, the PGE2 also did not significantly (p > 0.05) affect the amplitude of sEPSCs.
Figure 6.
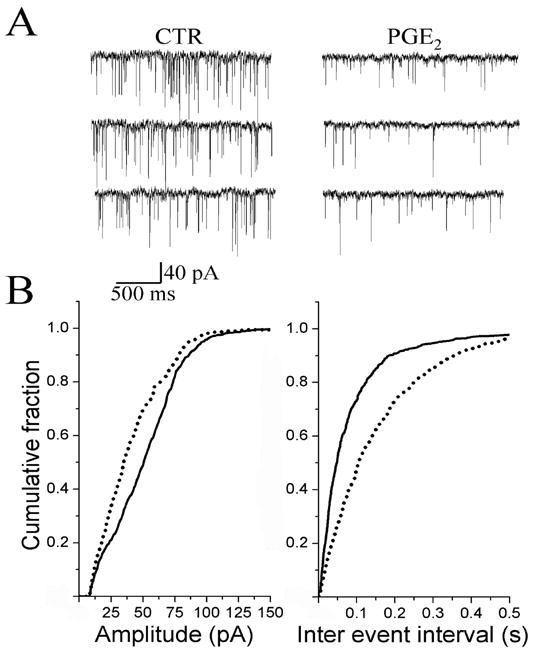
Effect of PGE2 on the frequency and amplitude of spontaneous EPSCs (sEPSCs) recorded in NTS. A, recordings of sEPSCs before (CTR), and during superfusion of PGE2 (200 nM) for 5 min. B, cumulative probability of the amplitude (left panel) and inter-event interval distributions (right panel). Solid lines depict control; dashed lines are data recorded during PGE2 application. PGE2 increased the inter-event interval (p < 0.01) and decreased the amplitude (p < 0.01).
PGE2 depressed the frequency and the amplitude of spontaneous miniature EPSC (mEPSC)
In the presence of 1 μM TTX, PGE2 (200 nM) decreased the frequency and the amplitude of the mEPSC in 50 % of the neurons studied (4/8). An example of continuous recordings of mEPSC before and during treatment with PGE2 is shown in Fig. 7A. The cumulative probability of the mEPSC inter-event intervals was shifted to the right in the presence of PGE2 (Fig. 7B right panel), indicating a decrease in frequency (control, 4.4 ± 1.9 Hz; PGE2, 2.3 ± 1.4 Hz P < 0.05). In the same group of cells, the cumulative probability of the mEPSC amplitude was shifted to the left (Fig. 7B left panel), indicating a decrease in the amplitude (control, 26 ± 7 pA; PGE2, 20 ± 5 pA P < 0.05). Whereas in the other half of neurons studied, mEPSC amplitude and frequency (control, 3.3 ± 1.1 Hz; PGE2, 2.9 ± 1.9 Hz P > 0.05) was not significantly changed after 10 min superfusion with PGE2. These results demonstrate that in the 60 % of the neurons where the peak amplitude of the evEPSC is decreased after PGE2 bath application, the majority of these neurons (60 – 70 %) show: (a) facilitation in the PPR, (b) decrease in the frequency of sEPSC and mEPSC, and (c) decrease in the amplitude of sEPSC and mEPSC. Such results suggest that PGE2 can depress synaptic transmission by both pre- and postsynaptic sites-of-action at second order NTS neurons.
Figure 7.
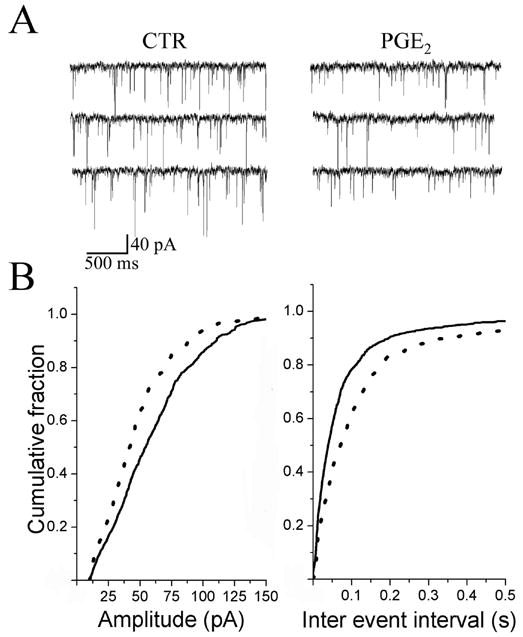
Effect of PGE2 on the frequency and the amplitude of miniature EPSCs (mEPSCs). A, Control (CTR) traces of mEPSCs before and during superfusion of PGE2 (200 nM). TTX (1 μM) was present in control and in the PGE2-containg aCSF. B, cumulative probability of the amplitude (left panel) and inter-event interval distributions (right panel) recorded before (solid line) and during PGE2 superfusion (dashed line). PGE2 increased the inter event interval (p < 0.01) and decreased the amplitude (p < 0.01).
Effects of prostanoid receptors agonist and antagonist on PGE2 modulation of synaptic transmission in the NTS
In order to examine the type of PGE2 receptors underlying the depression of synaptic transmission at NTS synapses, we tested the actions of a panel of PGE2 receptor agonist and antagonist reagents. In four neurons bath application of the EP1 receptor antagonist (SC19220, 20 μM for 5 min) alone did not measurably affect the peak amplitude of the control evEPSCs or control sEPSCs (p > 0.05). This concentration of SC19220 is reported to be five times the EC50 (Rakovska and Milenov, 1984). SC19220 application also did not modify the depressive effects produced by 200 nM PGE2 on synaptic transmission. The peak amplitude of the evEPSC decreased by 45 ± 10% (n = 4). The cumulative probability of the sEPSC inter-event intervals was shifted to the right in the presence of PGE2 plus SC19220 (Fig. 8A right panel), indicating a decrease in frequency (control, 4.6 ± 1.8 Hz; PGE2, 2.4 ± 1.2 Hz P < 0.05). In the same group of cells, the cumulative probability of the sEPSC amplitude was shifted to the left by PGE2 application, indicating a decrease in the amplitude (Fig. 8A left panel). Therefore the EP1 receptor appears not to mediate the PGE2 effects.
Figure 8.
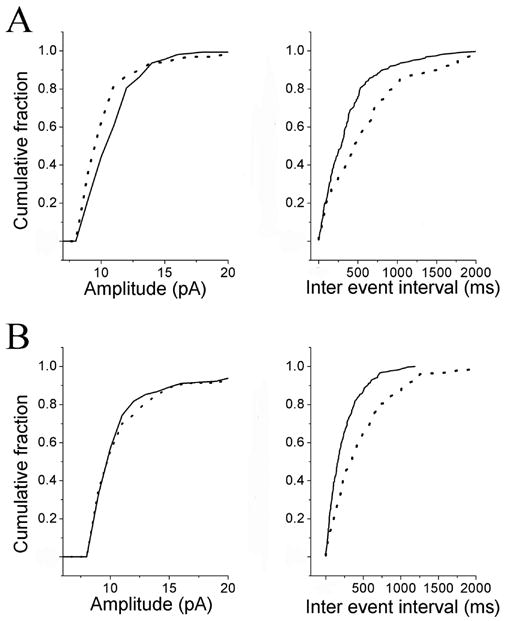
Effects of PGE2 antagonists EP1 and EP2 on the PGE2-induced changes in the frequency and the amplitude of spontaneous EPSCs (sEPSCs). A, cumulative probability of the amplitude (left panel) and inter-event interval distributions (right panel). Solid line shows the effects of an EP1 receptor antagonist (SC19220 30 μM) while the data portrayed by the dotted line shows the effects of PGE2 (200 nM) plus SC19220 30 μM. In the presence of SC19220, PGE2 still increased the inter event interval (p < 0.01) and decreased the amplitude of spontaneous events (p < 0.01). B, cumulative probability of the amplitude (left panel) and inter-event interval distributions (right panel). Solid line shows the effects of EP1 (SC19220 30 μM) plus the EP2 (AH28848 20 μM) receptor antagonists. Dotted line shows the effects of PGE2 (200 nM) in the presence of a mixture of SC19220 30 μM plus AH28848 20 μM. In the presence of this mixture, PGE2 increased the inter event interval (p < 0.01) but did not affect the amplitude of spontaneous events (p > 0.05).
Because SC19220, did not affect any of the synaptic responses measured or modify the PGE2 actions, we simply added an EP4 antagonist to the solution already containing the EP1 antagonist. Thus, in additional four neurons, we tested the effects of a 5 min bath application of a mixture of EP1 (SC19220, 20 μM) and EP4 (AH 23848, 30 μM) receptor antagonists. Bath application of this mixture of antagonists alone did not measurably affect the peak amplitude of the control evEPSC nor the depression in the evEPSCs produced by bath applied 200 nM PGE2. The cumulative probability of the sEPSC inter-event intervals was shifted to the right in the presence of PGE2 (Fig. 8B right panel), indicating that a decrease frequency still occurred (control, 3.7 ± 0.39 Hz; PGE2, 1.8 ± 0.3 Hz P < 0.05). In the same group of cells, the cumulative probability of the sEPSC amplitude was not shifted, indicating no change in the amplitude (Fig. 8B left panel). Thus, EP4 receptors appear to regulate the magnitude of sEPSCs.
In seven neurons bath application of the EP2 receptor agonist (Butaprost, 10 μM for 5 min) alone did not measurably affect the peak amplitude of the control evEPSCs (p > 0.05). In one of these cells the subsequent application of 200 nM PGE2 produced a 51 % decrease in the evEPSC. In three neurons bath application of the EP3 receptor agonist (Sulprostone, 10 μM for 5 min) did not measurably affect the peak amplitude of the control evEPSCs (p > 0.05). In one of these cells, subsequent application of 200 nM PGE2 resulted in a decreased evEPSC by 39 %. In a preliminary survey, EP2 and EP3 agonist produced conflicting results when tested for mimicking PGE2-induced changes in sEPSC. Thus, we did not study the receptor pharmacology of PGE2 actions on these synaptic potentials.
DISCUSSION
Our data shows that PGE2 can depress the synaptic transmission between primary vagal sensory afferent fibers and second-order intermediate and caudal NTS neurons via pre-and postsynaptic sites-of-action. We base this interpretation on several observations: (1) PGE2 altered the paired pulse ratio (PPR) in 85 % of the synapses. Changes between the magnitude of the first and second synaptic responses elicited by two presynaptic stimuli delivered at relatively short intervals (20 ms) can be quantified as the PPR. Changes in the PPR are used to test alterations in the probability of transmitter release, with an increase in the PPR being associated with a decrease in the probability of transmitter release (Thomson, 2000). We observed that, PGE2-elicited increases in the PPR in 70 % of the synapses depressed by PGE2. Thus, it is likely that PGE2 has a presynaptic site-of-action. (2) At the same time that PGE2 increased the PPR, it also inhibited the frequency of spontaneous, EPSC (sEPSCs), and the frequency of action potential-independent (miniature) EPSCs (mEPSCs). Decrease in the frequency of appearance of these currents also suggests PGE2 can act presynaptically. (3) Finally, PGE2 caused a decrease in the amplitudes of the sEPSCs and mEPSCs, an effect traditionally indicative of a postsynaptic site-of-action.
In brain stem preparations, sEPSC are unlikely to arise from spontaneous ST action potential activity. They could emanate from tonically firing interneurons in the brain stem circuits and/or from spontaneous mEPSCs activity. We did not directly examine, in the same cell the relative contribution of AP-dependent (sEPSCs) and AP-independent (mEPSCs) events. However, our population data obtained from measurements of spontaneous events in control solutions and in solutions containing TTX reveal similar frequency of spontaneous events: 3.7 ± 1.8 and 4.4 ± 1.9 Hz for spontaneous events before and in the presence of TTX, respectively. Thus, it appears that the majority of spontaneous activity under the conditions of our experiment appears to be attributable to mEPSCs. These results are in agreement with those reported by Kato and Shigetomi (2001) who observed in NTS neurons that sEPSC and mEPSC frequencies were similar: 4.2 ± 1.0 Hz and 3.9 ± 1.0 Hz in control solution and in the presence of TTX respectively.
ST-mediated evEPSCs are due to release of glutamate (Andresen and Yang, 1990; cf., our Fig. 4) while interneurons within the NTS can release a plethora of neurotransmitters on to second order neurons (Andresen and Kunze, 1994). It is likely that some of PGE2’s presynaptic actions, those affecting sEPSCs, are not unique to glutamatergic synapses and involve some modification of neurosecretion common to most chemical synapses. One potential presynaptic target might be voltage-gated Ca2+ channels in nerve terminals because PGE2 has been reported to decrease voltage-gated Ca2+ currents in both peripheral (Ikeda, 1992; Borgland et al. 2002) and central neurons (Chen and Bazan, 2005).
At the first synapse made by primary vagal sensory neurons in the NTS, PGE2 has been reported to potentiate synaptic transmission in a subpopulation of synapses (Sekiyama et al. 1995). In three of eight synapses tested, PGE2 (1μM) increased the amplitude of the evEPSC. These results would suggest that some central nerve terminals of vagal afferents may be excited by PGE2, like some of the peripheral actions of PGE2. However, our results indicate that the majority of central vagal synapses were depressed by PGE2. In 34/56 NTS neurons tested, 200 nM PGE2 produced a clear depression of the amplitude of the solitary tract-evEPSCs; the 22 neurons that did not show synaptic depression were unaffected by PGE2. We tested whether concentrations of PGE2 larger than 200 nM might cause synaptic potentiation. From threshold concentrations of ~ 10 nM to 1 μM, the largest concentration tested, PGE2 always depressed synaptic transmission at PGE2-sensitive second-order NTS neurons. The difference between our result and those of Sekiyama et al. (1995) could be related to the small number of synapses they tested, and/or to the location of NTS neurons examined in the two studies: we studied second-order intermediate and caudal NTS neurons whereas Sekiyama et al. (1995) did not specify the location of their neurons in the NTS. Different PGE2 receptors subtypes are known to exist (see below) and they may exert opposing effects on synaptic transmission depending upon their distribution within the NTS. In view of our observations that PGE2 produces depression at central synapses in the NTS, we would conclude that central terminals of vagal afferents respond differently to PGE2 than either the peripheral processes or cell body. It is important to note that not all synapses activated via solitary tract stimulation are exclusively vagal in origin; nonetheless, most evEPSCs were depressed by PGE2.
Other primary afferent central terminals respond to PGE2 in various manners. Nakayama et al. (2004) and Malmberg et al. (1995) showed that PGE2 increase neurotransmitter release from the spinal cord nerve terminals of the dorsal root ganglia. By contrast Baba et al. (2001) reported only postsynaptic effects of PGE2 with no modulation of these nerve terminals. PGE2 can increase the calcitonin gene-related peptide release from the central nerve terminals of the trigeminal ganglia neurons (Jenkins et al. 2004). These different studies, together with our data, indicate that PGE2 can have different modulatory actions on primary afferent central nerve terminals. Moreover the effects of PGE2 on the cell bodies of primary afferents neurons do not always reflect the actions of PGE2 at the central nerve terminals of these sensory neurons.
The cellular mechanism underlying the PGE2-mediated depression of the evEPSCs remains to be elucidated. PGE2 can alter many voltage-dependent channels and consequently affect transmitter release. Both INa+ (Kwong and Lee, 2005) and Ih (Ingram and Williams, 1996) are enhanced by PGE2 in vagal afferents, effects that might be expected to lead to an enhancement of transmitter release. PGE2 could depress synaptic currents by inhibiting a specific class of voltage dependent calcium channel (VDCC) in presynaptic nerve terminals. In the cell bodies of the vagal afferents that project their axons into the ST, Huang et al. (2004) reported that 1 μM PGE2 reduced total inward Ca++ current by ~ 30 %. By knowing which class of VDCC underlies this PGE2-induced reduction of Ca++ current, it could be possible to use specific blockers of VDCC and perform occlusion experiments at NTS synapses.
Because PGE2-induced synaptic depression was manifested by both pre- and postsynaptic sites-of-actions we sought evidence whether the same or different subtypes of PGE2 receptors might underlie the effect of PGE2 at each of these sites. Four subtypes of PGE2 receptors, EP1–4, have been cloned and characterized (Narumiya et al. 1999). All four EP receptors are present in cell bodies of the rat vagus nerve (inferior vagal or nodose ganglion neurons; Matsumoto et al. (2005)) and presumably in vagal nerve terminals within the NTS. With the exception of EP2 mRNA, the other EP receptors mRNAs are present in NTS neurons of the rat (Zhang and Rivest, 1999). Application of a panel of PGE2 receptor agonist and antagonist revealed that none of the PGE2 effects on evoked synaptic transmission could be related to the activation of specific PGE2 receptor subtype. However, in the case of sEPSCs, it appears that EP4 receptors regulate sEPSC amplitude. Thus, the presynaptic membrane of the primary sensory neurons and the postsynaptic membrane of second order NTS neurons examined in this study do not appear to possess a functional EP1 – EP4 receptors. However, it should be noted that the strength of these conclusions relies on the specificity of the reagents employed. For example, the EP1 receptor agonist has been reported to have minimum selectivity (Hata and Breyer, 2004). Future experiments with potent and selective PGE2 receptor reagents should help test our conclusions.
We attempted to derive a concentration-effect relation for the actions of PGE2 on the magnitude of the evEPSC. Using population data or recording the effects of many concentrations of PGE2 in the same neuron. Nonetheless, we clearly could delineate a threshold concentration at about 10 nM and a maximum effect at 100 nM. Moreover, there was a significant difference between the effects of 10 nM and 100 nM PGE2. We considered that endogenous presence of PGE2 in the NTS could have obscured the concentration-effects relation. Thus, we tested this possibility by incubating slices with 3 – 10 μM indomethacin. At these concentrations indomethacin acts non-selectively to inhibit both COX I and COX II (O’Banion, 1999). Indomethacin treatment did not affect the amplitude of the evEPSCs nor alter the inhibition produced by PGE2 application. These results indicate that under non-pathological conditions the NTS may possess little endogenous levels of PGE2. This was somewhat of a surprising finding in light of the recent observations that synaptic transmission and membrane excitability in hippocampal CA1 pyramidal neurons of the rat can be dynamically regulated by COX II activity (Chen and Bazan, 2005). Evidently, the expression of COX II may be much higher in the hippocampus than in the NTS, and/or the metabolism of this prostanoid is more efficient in the NTS.
Synaptic transmission from vagal afferents to second-order NTS neurons is characterized as highly efficient, operating with a high probability of release (Doyle and Andresen, 2001). When transmitter release occurs near maximum efficiency synaptic depression following repetitive presynaptic activity is commonly observed: This is clearly evident for solitary tract synaptic transmission in vivo and in vitro (Miles, 1986; Mifflin and Felder, 1990; Doyle and Andresen, 2001; see also references in Bonham and Chen, 2005). These synapses, like many others that have high initial probability of release, can act as low-pass filters (Abbott and Regehr, 2004; Bonham and Chen, 2005). Importantly, neuromodulators acting at presynaptic receptors that reduce the probability of transmitter release can alter the filtering characteristic of these synapses rendering them more reliable. There are many autacoids, cytokines, and neurotransmitters that can presynaptically modulating vagal neurotransmission in the NTS (Andresen and Kunze, 1994; see also references in Rogers et al. 2006). Thus, it is not too surprising that these synapses would also be regulated by lipid mediators such as PGE2. Our results reveal that PGE2, like GABA (Brooks et al. 1992), glutamate (Chen et al. 2002), dopamine (Kline et al. 2002), and Substance P (Sekizawa et al. 2003), also depressed synaptic transmission. By producing synaptic depression PGE2 may convert vagal synapses from a low-pass filter function to a band-pass filter, potentially reducing spike failure in second-order NTS neurons.
Acknowledgments
We are grateful to TD. Gover for helpful criticisms in preparing the manuscript This work was supported by grants from the National Institutes of Health grants (NS-22069 to D.W. and NS-T32-07375 to L.N).
Abbreviations
- aCSF
artificial cerebrospinal fluid
- AHP
afterhyperpolarization
- AMPA
a-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid
- AP
action potential
- CNS
central nervous system
- COX-I
Cyclooxygenase 1
- COX-II
Cyclooxygenase 2
- evEPSCs
evoked excitatory postsynaptic glutamatergic currents
- mEPSCs
miniature excitatory postsynaptic currents
- NMDA
N-Methyl-D-aspartic acid
- NTS
Nucleus Tractus Solitarius
- PNS
peripheral nervous system
- PGE2
Prostaglandin E2
- PPR
paired pulse ratio
- Rin
Input resistance
- sEPSCs
spontaneous excitatory postsynaptic currents
- St
Solitarius Tract
- TTX
tetrodotoxin
Footnotes
Section Editor: Dr. Yoland Smith, Yerkes National Primate Research Center, Emory University, 954 Gatewood Road NE, Atlanta, GA 30329, USA
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott LF, Regehr WG. Synaptic Computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Kunze DL. Nucleus tractus solitarius-gateway to neural circulatory control. Ann Rev Physiol. 1994;56:93–116. doi: 10.1146/annurev.ph.56.030194.000521. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Yang MY. Non-NMDA receptors mediate sensory afferent synaptic transmission in medial nucleus tractus solitarius. Am J Physiol. 1990;259:H1307–H1311. doi: 10.1152/ajpheart.1990.259.4.H1307. [DOI] [PubMed] [Google Scholar]
- Aylwin ML, Horowitz JM, Bonham AC. NMDA receptors contribute to primary visceral afferent transmission in the nucleus of the solitary tract. J Neurophysiol. 1997;77:2539–2548. doi: 10.1152/jn.1997.77.5.2539. [DOI] [PubMed] [Google Scholar]
- Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci. 2001;21:1750–1756. doi: 10.1523/JNEUROSCI.21-05-01750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishai I, Coceani F. Eicosanoid formation in the rat cerebral cortex: Contribution of neurons and glia. Mol Chem Neuropathol. 1992;17:219–238. doi: 10.1007/BF03160012. [DOI] [PubMed] [Google Scholar]
- Bonham AC, Chen CY. Synaptic transmission in the nucleus tractus solitarius (NTS) In: Undem BJ, Weinreich D, editors. Advances in vagal afferent neurobiology, Frontiers in neuroscience. Taylor and Francis Group; Boca Raton: 2005. [Google Scholar]
- Borgland SL, Connor M, Ryan RM, Ball HJ, Christie MJ. Prostaglandin E (2) inhibits calcium current in two sub-populations of acutely isolated mouse trigeminal sensory neurons. J Physiol. 2002;539:433–444. doi: 10.1113/jphysiol.2001.013322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PA, Glaum SR, Miller RJ, Spyer KM. The actions of baclofen on neurons and synaptic transmission in the nucleus tractus solitarii of the rat in vitro. J Physiol. 1992;457:115–129. doi: 10.1113/jphysiol.1992.sp019367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93:929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- Chen CY, Bonham AC. Glutamate suppresses GABA release via presynaptic Metabotropic glutamate receptors neurons in rats. J Physiol. 2005;562:535–551. doi: 10.1113/jphysiol.2004.076885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Horowitz JM, Bonham AC. A presynaptic mechanism contributes to depression of autonomic signal transmission in NTS. Am J Physiol. 1999;277:H1350–H1360. doi: 10.1152/ajpheart.1999.277.4.H1350. [DOI] [PubMed] [Google Scholar]
- Chen CY, Ling EH, Horowitz JM, Bonham AC. Synaptic transmission in nucleus tractus solitarius is depressed by Group II and III but not Group I presynaptic Metabotropic glutamate receptors in rats. J Physiol. 2002;538:773–786. doi: 10.1113/jphysiol.2001.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras RJ, Beckstead RM, Norgren R. The central projections of the trigeminal, facial, glossopharyngeal and vagus nerves: an Autoradiographic study in the rat. J Auton Nerv Syst. 1982;6:303–322. doi: 10.1016/0165-1838(82)90003-0. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Bailey TW, Jin YH, Andresen MC. Vanilloid receptors presynaptically modulate visceral afferent synaptic transmission in nucleus tractus solitarius. J Neurosci. 2002;22:8222–8229. doi: 10.1523/JNEUROSCI.22-18-08222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek M, Arias C, Sawchenko P, Ericsson-Dahlstrand A. Distribution of EP3 prostaglandin E2 receptors subtype in the rat brain: relationship to site of interleukine-1-induced cellular responsiveness. J Comp Neurol. 2000;428:5–20. doi: 10.1002/1096-9861(20001204)428:1<5::aid-cne2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Fowler JC, Wonderlin WF, Weinreich D. Prostaglandins block a Ca2+-dependent slow afterhyperpolarization independent of effects on Ca2+ influx in visceral afferent neurons. Brain Res. 1985;345:345–349. doi: 10.1016/0006-8993(85)91014-5. [DOI] [PubMed] [Google Scholar]
- Gatti PJ, Shirahata M, Johnson TA, Massari VJ. Synaptic interactions of substance P immunoreactive nerve terminals in the baro and chemoreceptor reflexes of the cat. Brain Res. 1995;693:133–147. doi: 10.1016/0006-8993(95)00728-9. [DOI] [PubMed] [Google Scholar]
- Gold MS, Zhang L, Wrigley DL, Traub RJ. Prostagnadin E (2) modulates TTX-RI (Na) in rat colonic sensory neurons. J Neurophysiol. 2002;88:1512–1522. doi: 10.1152/jn.2002.88.3.1512. [DOI] [PubMed] [Google Scholar]
- Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol. 1990;259:R1131–R1138. doi: 10.1152/ajpregu.1990.259.6.R1131. [DOI] [PubMed] [Google Scholar]
- Guay J, Bateman K, Gordon R, Mancini J, Riendeau D. Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J Biol Chem. 2004;279:24866–24872. doi: 10.1074/jbc.M403106200. [DOI] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Pharmacology and Therapeutics. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Ho CY, Gu Q, Hong JL, Lee LY. Prostaglandin E (2) enhances chemical and mechanical sensitivities of pulmonary C fibers in the rat. Am J Respir Crit Care Med. 2000;162:528–533. doi: 10.1164/ajrccm.162.2.9910059. [DOI] [PubMed] [Google Scholar]
- Ho L, Pieroni C, Winger D, Purohit DP, Aisen PS, Pasinetti GM. Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer’s disease. J Neurosci Res. 1999;57:295–303. doi: 10.1002/(SICI)1097-4547(19990801)57:3<295::AID-JNR1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Huang XZ, Won YJ, Park BJ, Cho BP, Lee JW, Jeong SW. Nerve injury alters profile of receptor-mediated Ca2+ channel modulation in vagal afferents neurons of rat nodose ganglia. Neurosci Lett. 2004;364:189–194. doi: 10.1016/j.neulet.2004.04.039. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Williams JT. Modulation of the hyperpolarization-activated current (Ih) by cyclic nucleotides in guinea-pig primary afferent neurons. J Physiol. 1996;492:97–106. doi: 10.1113/jphysiol.1996.sp021292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Prostaglandin Modulation of Ca2+ channels in rat sympathetic neurons is mediated by guanine nucleotide binding proteins. J Physiol. 1992;458:339–359. doi: 10.1113/jphysiol.1992.sp019421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DW, Langmead CJ, Parsons AA, Strijbos PJ. Regulation of calcitonin gene-related pepetide release from rat trigeminal nucleus caudalis slices in vitro. Neurosci Lett. 2004;366:241–244. doi: 10.1016/j.neulet.2004.05.067. [DOI] [PubMed] [Google Scholar]
- Kato F, Shigetomi E. Distinct modulation of evoked and spontaneous EPSCs by purinoceptors in the nucleus tractus solitarii of the rat. J Physiol. 2001;530:469–486. doi: 10.1111/j.1469-7793.2001.0469k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Takacs KN, Ficker E, Kunze DL. Dopamine modulates synaptic transmission in the nucleus of the solitary tract. J Neurophysiol. 2002;88:2736–2744. doi: 10.1152/jn.00224.2002. [DOI] [PubMed] [Google Scholar]
- Kwong K, Lee LY. PGE2 sensitizes cultured pulmonary vagal sensory neurons to chemical and electrical stimuli. J Appl Physiol. 2002;93:1419–1428. doi: 10.1152/japplphysiol.00382.2002. [DOI] [PubMed] [Google Scholar]
- Kwong K, Lee LY. Prostaglandin E2 potentiates a TTX-resistant sodium current in rat capsaicin-sensitive vagal pulmonary sensory neurons. J Physiol. 2005;56:437–450. doi: 10.1113/jphysiol.2004.078725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 receptor in permanent focal cerebral ischemia. Ann Neurol. 2005;57:758–761. doi: 10.1002/ana.20461. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Hamberger A, Hedner T. Effects of Prostaglandin E2 and Capsaicin on behavior and cerebrospinal fluid amino acid concentrations of unanesthetized rats: A microdialysis study. J Neurochem. 1995;65:2185–2193. doi: 10.1046/j.1471-4159.1995.65052185.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Ikeda M, Yoshida S, Tanimoto T, Takeda M, Nasu M. Prostaglandin E2-induced modification of tetrodotoxin-resistant Na+ currents involves activation of both EP2 and EP4 receptors in neonatal rat nodose ganglia neurons. Br J Pharmacol. 2005;145:503–513. doi: 10.1038/sj.bjp.0706212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K, Watanabe Y, Imai-Matsumura K, Connolly M, Koyama Y, Onoe H, Watanabe Y. Mapping of PGE2 binding sites in rat brain using quantitative autoradiography. Brain Res. 1992;581:292–298. doi: 10.1016/0006-8993(92)90720-t. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinene S, Fusco FR, Yrjanheikki J, Keinanen R, Hirvonen T, Roivainen R, Narhi M, Hokfelt T, Koistinado J. Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-D-aspartic acid-receptors and phospholipase A2. Proc Natl Acad Sci USA. 1997;94:6500–6505. doi: 10.1073/pnas.94.12.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifflin SW, Felder RB. Synaptic mechanisms regulating cardiovascular afferent inputs to solitary tract nucleus. Am J Physiol. 1990;259:H653–H661. doi: 10.1152/ajpheart.1990.259.3.H653. [DOI] [PubMed] [Google Scholar]
- Miles R. Frequency dependence of synaptic transmission in nucleus of the solitary tract in vitro. J Neurophysiol. 1986;55:1076–1090. doi: 10.1152/jn.1986.55.5.1076. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ, Morrow JD. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology. 1999;53:1495–1498. doi: 10.1212/wnl.53.7.1495. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Negishi M. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J Com Neurol. 2000;421:543–569. doi: 10.1002/(sici)1096-9861(20000612)421:4<543::aid-cne6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Nakayama Y, Omote K, Kawamata T, Namiki A. Role of Prostaglandin receptor subtype EP1 in Prostaglandin E2-induced nociceptive transmission in the rat spinal dorsal horn. Brain Res. 2004;1010:62–68. doi: 10.1016/j.brainres.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: Structures, Properties, and Functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- O’Banion MK. Cyclooxygenase-2: Molecular biology, pharmacology, and neurobiology. Crit Rev Neurobiol. 1999;13:45–82. doi: 10.1615/critrevneurobiol.v13.i1.30. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Rakovska A, Milenov K. Antagonistic effects of SC-19220 on the response of guinea-pig gastric muscles to prostaglandins E1, E2 and E2α. Arch Int Pharmacody. 1984;268:59–69. [PubMed] [Google Scholar]
- Rogers RC, Nasse JS, Hermann GE. Live-cell imaging methods for the study of vagal afferents within the nucleus of the solitary tract. J Neurosci Methods. 2006;150:47–58. doi: 10.1016/j.jneumeth.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Sekiyama N, Mizuta S, Hori A, Kobayashi S. Prostaglandin E2 facilitates excitatory synaptic transmission in the nucleus tractus solitarii of rats. Neuroscience Lett. 1995;188:101–104. doi: 10.1016/0304-3940(95)11407-n. [DOI] [PubMed] [Google Scholar]
- Sekizawa SI, Joad JP, Bonham AC. Substance P presynaptically depresses the transmission of sensory input to bronchopulmonary neurons in the guinea pig nucleus tractus solitarii. J Physiol. 2003;552:547–559. doi: 10.1113/jphysiol.2003.051326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation, and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Thoren S, Weinander R, Saha S, Jegerschold C, Pettersson PL, Samuelsson B, Hebert H, Hamberg M, Morgenstern R, Jakobsson PJ. Human microsomal prostaglandin E synthase-1. J Biol Chem. 2003;278:22199–22209. doi: 10.1074/jbc.M303227200. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Weinreich D. Electrophysiological properties and chemosensitivity of guinea pig nodose ganglion neurons in vitro. J Auton Nerv Syst. 1993;44:17–33. doi: 10.1016/0165-1838(93)90375-5. [DOI] [PubMed] [Google Scholar]
- Wolfe LS, Coceani F. The role of prostaglandins in the central nervous system. Ann Rev Physiol. 1979;41:669–684. doi: 10.1146/annurev.ph.41.030179.003321. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rivest S. Distribution, regulation and colocalization of the genes encoding the EP2- and EP4-PGE2 receptors in the rat brain and neuronal responses to systemic inflammation. Eur J Neurosci. 1999;11:2652–2668. doi: 10.1046/j.1460-9568.1999.00682.x. [DOI] [PubMed] [Google Scholar]