

Abstract
Cocaine inhibits survival and growth of rat locus coeruleus (LC) neurons, which may mediate alterations in attention, following in utero exposure to cocaine. These effects are most severe in early gestation during peak neuritogenesis. Prenatal cocaine exposure may specifically decrease LC survival through an apoptotic pathway involving caspases. Dissociated fetal LC neurons or substantia nigra (SN) neurons (control) were exposed in vitro to a pharmacologically active dose of cocaine hydrochloride (500 ng/ml) and assayed for apoptosis using TUNEL and Hoechst methodologies. Cocaine exposure decreased survival and induced apoptosis in LC neurons, with no changes in survival of SN neurons. Activation of apoptotic signal transduction proteins were determined using enzyme assays and immunoblotting at 30 min, 1 h, 4 h and 24 h. In LC neurons, Bax levels were induced at 30 min and 1 h, following cocaine treatment, and Bcl-2 levels remained unchanged at all time points, altering the Bax/Bcl-2 ratio. The ratio was reversed for SN neurons (elevated Bcl-2 levels and transient reduction of Bax levels). Further, cocaine exposure significantly increased caspase-9 and caspase-3 activities at all time points, without changes in caspase-8 activity in LC neurons. In addition, cleavage of caspase-3 target proteins, α-fodrin and PARP were observed following cocaine treatment. In contrast, SN neurons showed either significant reductions, or no significant changes, in caspase-3, 8 or 9 activities or caspase-3 target proteins, α-fodrin and PARP. Thus, cocaine exposure in vitro may preferentially induce apoptosis in fetal LC neurons putatively regulated by Bax, via activation of caspases and its downstream target proteins.
Keywords: noradrenergic, drug abuse, apoptosis, attention, rat, cocaine
Prenatal cocaine exposure has deleterious effects on the developing fetus, (Schenker et al., 1993; Keller and Snyder-Keller, 2000). One prominent behavioral abnormality associated with prenatal cocaine exposure in offspring is attentional dysfunction (Mayes et al., 1998; Singer et al., 2000), however, the cellular mechanisms underlying this deficit has not been identified. A variety of studies implicate the noradrenergic system being affected by prenatal cocaine exposure, specifically locus coeruleus (LC) neurons (Mactutus, 1999; Bayer et al., 2002; Foltz et al., 2004; Dey et al., 2006). Cocaine impairs neurite initiation and elongation in GD 14 and 18 LC neurons (Snow et al., 2001), with pronounced inhibition during early gestation (Snow et al., 2004). Further, prenatal cocaine inhibits LC survival indicated by a decreased number of tyrosine hydroxylase immunoreactive (TH-IR) cells in rat LC when exposed to cocaine in utero, with a 12–13% reduction in cell number, which was associated with decreased LC cell volume (Mactutus et al., 2005). However, an understanding of mechanisms by which maternal cocaine abuse adversely affects the basic cell biology of LC neurons is lacking, and is critical for developing treatments and intervention strategies.
Cocaine exposure may specifically decrease LC survival through activation of apoptotic signal transduction pathways. Although apoptosis or programmed cell death (PCD) is a regulated event in the developing nervous system (Lo et al., 1995; Boonman and Isacson, 1999), dysregulation of this process can lead to abnormal development or disease (Thompson, 1995). Several studies have shown that during development gestational cocaine induces apoptosis in rat heart and murine cerebral wall (Nassogne et al., 1997; Lidow et al., 1999; Xiao et al., 2000; Xiao et al., 2001; Mitchell and Snyder-Keller, 2003; Bae and Zhang, 2005). One of the key regulators of PCD are the members of the bcl-2 family of proteins (Bax, Bak, Bcl-XL, Bcl-2, and others), that regulate mitochondrial integrity and cytochrome c release (Tsujimoto, 1998; Wang, 2001). These proteins function at the mitochondrial level, hence an important determinant of cell death or survival (Gross et al., 1999; Tsujimoto and Shimizu, 2000). Additional regulators of PCD as well as survival are the family of cysteine proteases - the caspases (Campbell and Holt, 2003; Miura et al., 2004), downstream of the Bcl-2 family of proteins (Alnemri et al., 1996; Kaufmann and Hengartner, 2001). During apoptosis, caspases are converted to active proteases, e.g. initiator caspase-8 and/or caspase-9 (Chang and Yang, 2000), which subsequently result in activation and cleavage of effector caspase-3 (Thornberry et al., 1992).
In the present study, we sought to determine whether decreased LC survival following cocaine exposure in vitro was induced by apoptosis and whether this effect was cell specific, to identify a potential underlying mechanism for altered attentional function in offspring exposed to cocaine. Here, we show evidence in vitro to support that cocaine specifically decreases fetal LC neuron survival via an apoptotic pathway that involves activation of Bax and the cysteine protease, caspase-3. These critical proteins could provide targets for future therapeutic strategies.
MATERIALS AND METHODS
Animals
Nulliparous Long Evans timed-pregnant rats were maintained according to NIH guidelines in AAALAC accredited facilities. Food (Pro-Lab Rat, Mouse Hamster Chow No. 3000) and water were available ad libitum. The animal facility was maintained at 21°C + 2 and 50% + 10, relative humidity and had a 12 hr light: 12 hr dark cycle with lights on at 07:00 h (EST). The protocol for the use of rats in this project was approved by the IACUC of the University of Kentucky (00656M2003). The animals were allowed to acclimate for 2 days after shipment.
Tissue culture
Substratum preparation
Tissue culture flasks and chamber slides were coated with poly-L-lysine (100 μg/ml; Sigma, USA.) in sterile calcium-magnesium free phosphate buffered saline (CMF-PBS; pH 7.2) and stored at 37°C overnight in a room air-humidified, HEPA-filtered incubator. The flasks and chamber slides were rinsed twice with sterile double distilled water, then covered with 500 μl of culture media and incubated at 37°C in a 5% CO2-air humidified incubator until required for use.
Tissue dissection
Timed-pregnant rats bearing embryonic day 14 pups were euthanized by CO2 inhalation according to AAALAC regulations. The uterine horns were removed under sterile conditions and placed in a Petri dish containing cold Hank’s Balanced Salt Solution (HBSS; pH 7.3) on ice. The embryos were removed and placed in cold L-15 medium containing 6 mg/ml glucose and 15 μg/ml gentamycin and further dissection was performed in the same medium. The brains were removed and areas containing locus coeruleus (LC -rhombencephalon) (Norbert Konig et al., 1989) and substantia nigra (SN - mesencephalon) (Studer, 1997) neurons were dissected. The tissues were completely dissociated by gentle trituration, using glass Pasteur pipets with increasingly smaller bored fire polished tips. Cells were plated at varying densities on previously prepared tissue culture flasks/slides (see above). The identity of noradrenergic LC neurons and dopaminergic SN neurons was confirmed by immunostaining (Dey et al., 2006).
Media
LC and SN neurons were grown in MEM (Minimum Essential Medium, Earle’s, Invitrogen, USA.), supplemented with 6 mg/ml glucose, 2 mM glutamine, 5% horse serum (Hyclone), 5% fetal bovine serum (Hyclone, USA.), and 15 μg/ml gentamycin. All cultures were grown using the same batch of serum to avoid variability among cultures. Cells were cultured in growth media for four days before it was discarded and culture dishes were either replenished with fresh media (controls: media plus citrate buffer) or media containing 500 ng/ml cocaine (tests).
Cocaine
Cocaine hydrochloride was obtained from the NIDA Drug Supply Program, (RTI International, Research Triangle Park, NC, USA.). Cocaine undergoes rapid degradation and is unstable at neutral or alkaline pH (Baselt, 1983; Isenschmid et al., 1989; Bouis et al., 1990; Vorhees et al., 1995). To increase stability, cocaine was dissolved in sodium citrate buffer (pH 5.0) at a concentration of 10 μg/ml, to prevent degradation into its primary and secondary metabolites, benzoylecgonine (BE) and ecgonine methyl ester (EME) (Vorhees et al., 1995). Further dilutions were made using the growth media such that the final concentration added to the culture dishes was a physiologically-relevant dose of 500 ng/ml (1.5 μM) (Evans et al., 1996; Booze et al., 1997; Mactutus, 1999; Bunney et al., 2000). Control dishes were treated with vehicle (media plus citrate buffer).
Cell survival assay
Cellular damage to LC and SN due to cocaine exposure was determined by measuring the metabolic activity of these cells using a colorimetric MTT assay - [3-(4,5-dimethythiazol-2yl)-2,5 diphenyl tetrazolium bromide, Roche Applied Science, USA.]. Cells were cultured at a density of 1 x 105 in triplicates for each treatment in a 96 well plate. Cells were treated with cocaine at increasing concentrations of 250 ng/ml, 500 ng/ml and 1 μg/ml and incubated for 48 hrs. The resultant purple formazan product indicative of metabolic activity of the cells was solubilized and quantified using a multiwell ELISA spectrophotometer at an absorbance of 570 nm with reference wave length > 650 nm. Mean values were obtained from 3 replicates (n = 4).
Quantitation of Apoptosis
Neuronal apoptosis was analyzed by Terminal Deoxynucleotidyl Transferase (TdT) mediated DNA nick end labeling (TUNEL), using a Fluorescein In Situ Cell Death Detection Kit (Roche Applied Science, USA.). Cells were seeded in chamber slides and cultured for four days and then left untreated or exposed to 500 ng/ml of cocaine. Cell death was measured after 24 and 48 hours of drug treatment. Cell death was confirmed using fluorescent DNA binding Hoechst dye 33258 (1 μg/ml in 1x CMF-PBS; Invitrogen, USA) for 10 min to identify condensed or fragmented nuclei. Neuronal cells were identified by immunocytochemistry (ICC) using the neuronal marker, TUJ1 (anti-βIII tubulin antibody) (Covance, Berkely, CA, USA.). The fixed cells after treatment were triple stained first by performing ICC using the neuronal marker, followed by TUNEL and Hoechst staining, respectively. The stained specimens were visualized using a fluorescent microscope and Axiovision software v. 4.1 (Carl Zeiss Vision GmbH-v 4.1, Germany) and images were merged using multidimentional acquisition. The number of apoptotic cells was counted from 10 randomly selected fields and percentage of apoptotic cells were calculated [number of apoptotic cells / number of total cells x 100]. Mean values were calculated from three replicates (n = 3).
Western Blot Analysis
Embryonic LC and SN neurons were cultured for 4 days and treated with cocaine at 500 ng/ml and harvested at 30 min, 1 h, 4 h and 24 h after treatments, in lysis buffer, described below (caspase activity assay). Cell lysates normalized for protein content were electrophoresed in 12.5% or 5.0% SDS-PAGE gels under reducing conditions in running buffer (25 mM Tris, 192 mM Glycine, 0.1% SDS, pH 8.3; Bio-Rad, USA.) and electrotransferred to PVDF membrane in Towbin-transfer buffer (25 mM Tris, 192 mM glycine, 20% MeOH, pH 8.3; Bio-Rad, USA.) at 100 constant volts for 2 hours. Blots were blocked with 5% milk solution in wash buffer (20 mM Tris, 500 mM NaCl, pH 7.5; Bio-Rad, USA.) for 2 hours and incubated overnight in 5% milk containing anti-Bax monoclonal antibody (Santa Cruz Biotechnology, Inc. USA.), or anti-Bcl-2 polyclonal antibody (Santa Cruz Biotechnology, Inc. USA.), or anti-caspase-3 monoclonal antibody (Cell Signaling Technology, USA.), or anti-alpha fodrin monoclonal antibody (Affiniti Research Products Ltd., Exeter, UK.), or anti-PARP polyclonal antibody (Cell Signaling Technology, USA.). The blots were washed and the bound antigen-antibody was detected by incubation for 1 hour in the appropriate horseradish peroxidase conjugated secondary antibody (Jackson Immunoresearch laboratories, USA.). Mouse anti-actin monoclonal antibody (Chemicon International, USA.) was used as an internal loading control. Immunoreactive proteins were visualized using enhanced chemiluminescence detection system (Amersham Pharmacia Biotechnology, Buckinghamshire, UK.). Results were quantified using the KODAK Imaging system and KODAK 1D Image Analysis Software. The proteins of interest were normalized by actin and expressed as percent control of the protein levels within each group. Mean values were calculated from three replicates (n = 3). Total cell lysates were quantified using the BCA assay kit (Pierce, Rockford, IL, USA.).
Caspase Activity Assay
Caspase 3, 8 and 9 activities were measured using caspase-3 [Ac-DEVD-7- amino-4-methylcoumarin (AMC), Bachem Biosciences, PA, USA.], caspase-8 [Ac-IEPD-AMC, Bachem Biosciences, PA, USA.] and caspase-9 [Ac-LEHD-AMC, Bachem Biosciences, PA, USA.] substrates respectively. Briefly, embryonic LC and SN neurons were cultured for four days and treated with cocaine at 500 ng/ml and harvested at 30 min, 1 h, 4 h and 24 h after treatments, in lysis buffer [25 mM Hepes, pH 7.5 (Sigma, St. Louis, MO, USA.), 5 mM EDTA (Sigma, USA.), 1 mM EGTA (Sigma, USA.), 5 mM MgCl2 (Fisher Scientific, USA.), 10 mM Sucrose (Fisher Scientific, USA.), 5 mM dithiothreitol (DTT; Sigma, USA.), 1 % 3-[-(3-chloramidopropyl) dimethylammonio]-1-propanesulfonic acid (CHAPS, Sigma, USA.), protease inhibitor cocktail (10 μl/ml; Sigma, USA.), and 1 mM PMSF (Sigma, USA.)]. Cell lysates were freeze/thawed three times and centrifuged at 12,000g for 60 min. The resultant cell lysates (supernatants) obtained were incubated in a buffer containing 25 mM HEPES, pH 7.5, 10% sucrose, 0.1% CHAPS, and 1 mM DTT supplemented with 50 μM Ac-DEVD-AMC or Ac- IEPD-AMC or Ac-LEHD-AMC at 37°C in 96-well Costar plates for 1 hour. Caspase 3 activity was confirmed using a caspase-3 specific inhibitor (Ac-DEVD-CHO) (Bachem Biosciences, USA.). The inhibitor was added to the cell lysates 30 min prior to incubation with caspase-3 substrate, Ac-DEVD-AMC. The enzyme catalyzed release of the fluorogenic AMC moieties was quantified in a Wallac Victor III fluorimeter (Perkin Elmer, USA.) using 360 nm excitation and 460 nm emission wavelengths. Caspase activities were expressed as units per microgram of total protein. Induction of Caspase-3, 8 and 9 activities in LC and SN cells were compared with their respective controls. Mean values were obtained from three replicates ( n = 3). Total cell lysates were quantified using the BCA assay kit (Pierce, Rockford, IL, USA.)
Data Analysis
Data were represented as means of + SEM of the mean values of the replicate samples of n = 3-4 experiments. Overall differences between experimental groups were analyzed using Analysis of Variance techniques (BMDP Statistical Software, Release 7, Los Angeles, CA, Dixon, 1993). Specifically, a multifactorial treatment (cocaine vs. control) x region (LC vs. SN) x time (30 min, 1 h, 4 h and 24 h) ANOVA was employed. When a dose response study was conducted, the treatment factor had 4 levels (cocaine dose – 0, 250 ng/ml, 500 ng/ml, 1 μg/ml). Planned comparisons and orthogonal decompositions of main effects were employed and justified given the a priori hypothesized greater vulnerability to cocaine of LC, relative to SN neurons (Mactutus, 1999; Snow et al., 2001; Bayer et al., 2002; Snow et al., 2004; Dey et al., in press) regardless of the presence of significant two- and three-way interaction terms. Correlational analyses and bivariate plots were further employed to examine the degree of relationship between the effects of cocaine on LC vs. SN neurons. An α level of p < 0.05 was considered significant for all statistical tests employed.
Results
Cocaine exposure decreases fetal LC neuron survival
Analysis of neuron survival indicated that cocaine exposure in vitro on E14 LC neurons resulted in a linear dose dependent decrease in survival with increasing concentrations of cocaine [F(3,88) = 8.5, p < 0.005], compared to controls. In contrast, measurement of survival in SN neurons did not reveal any significant decrease in survival with increasing doses of cocaine [F(3,88) = 0.4, p = 0.55] (Fig. 1). Specific planned contrasts indicated a significant different across regions at 500 ng [F(1,88) = 5.6, p < 0.02] and 1 μg [F(1,88) = 5.7, p < 0.02] doses. These data support the hypothesis that the effects of cocaine on neuron survival are cell specific, targeting LC neurons, but not SN neurons.
Figure 1. Cocaine exposure in vitro decreases fetal LC neuron survival.
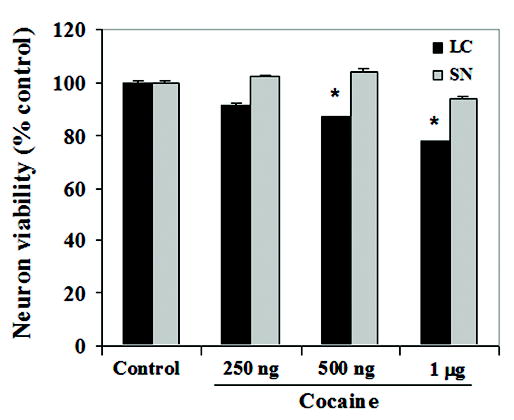
Survival of locus coeruleus (LC) and substantia nigra (SN) neurons in vitro was determined using the MTT assay, a colorimetric measure of metabolic activity. LC neurons demonstrated a linear dose dependent decrease in survival with increasing doses of cocaine [ANOVA: p < 0.005]; [contrasts: 250 ng (p = 0.18); 500 ng (p < 0.02); 1 μg (p < 0.02)]. Survival of fetal SN neurons was not affected by the same dose range of cocaine concentrations, compared to controls [ANOVA: p = 0.55].
Cocaine exposure induces apoptosis in fetal LC neurons
Decreased LC survival in response to cocaine may occur via apoptosis. Consistent with this mechanism, TUNEL analysis indicated positive cell death profiles in cocaine treated LC neurons compared to controls, after 24 h and 48 h, indicated by DNA fragmentation (500 ng/ml) (Fig. 2 A, B, C). Results were confirmed by staining with the DNA binding fluorescent Hoechst dye, which showed neuronal chromatin condensation accompanied by DNA fragmentation in LC neurons (Fig. 2 A, B, C). That the effects were specific to neurons was confirmed by co-staining using immunocytochemistry with the neuronal marker TUJ1(anti-βIII tubulin antibody) (Fig. 2 A, B, C). SN neurons did not reveal abundant TUNEL positive cells in comparison to controls after 24 h and 48 h of exposure to cocaine (Fig. 2 D, E, F). Quantitative analysis showed a significant and dramatic cell death effect of cocaine on LC [F(2,54) = 49.5, p < 0.0001] but no significant increase in cell death was evidenced in the SN [F(2,54) = 0.1, p = 0.92]. (Fig. 2G). Apoptotic cells were expressed as percent cell death [number of apoptotic cells / number of total cells x 100]. Specific planned contrasts indicated a significant different across regions at the 24 h [F(1,54) = 46.5, p < 0.0001] and 48 h time points [F(1,54) = 99.8, p < 0.0001]. The interaction between brain region and time dependent treatment was also noted as statistically significant [F(2,54) = 22.8; p < 0.0001]. These data suggest that cocaine exposure in vitro induces neuronal apoptosis, dramatically increased for LC relative to SN neurons; which could mediate insufficient synaptic connectivity.
Figure 2. Cocaine exposure in vitro induces apoptosis in fetal LC neurons.
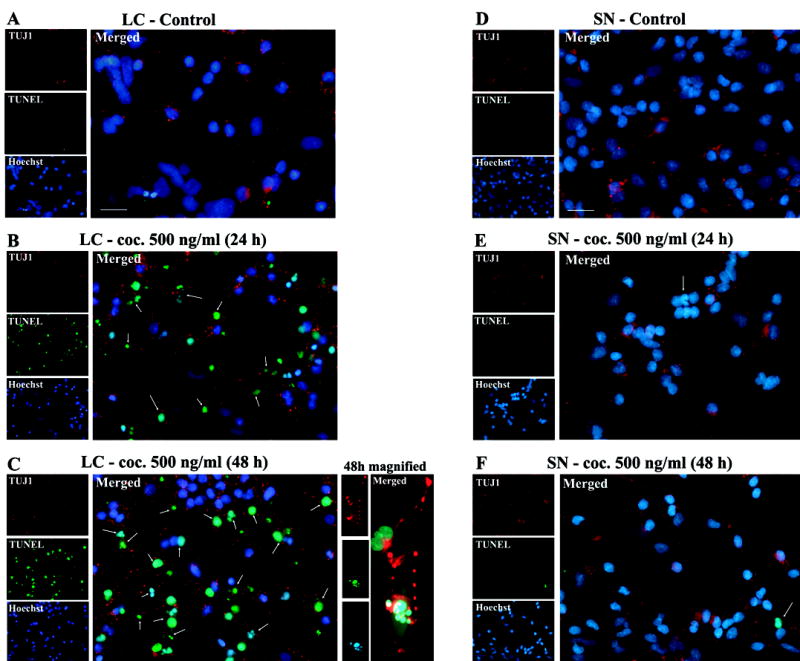
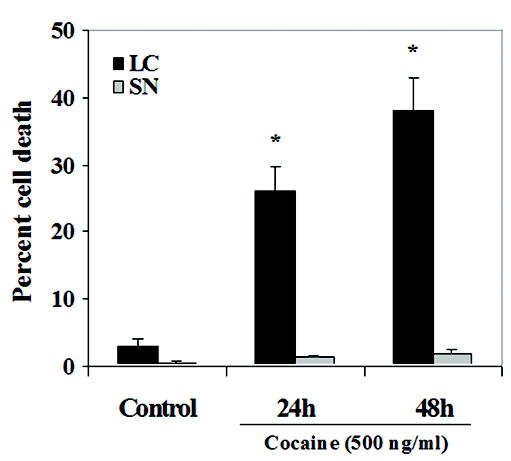
Cells were stained with TUJ1(βIII tubulin antibody) using immunocytochemistry, to confirm effects in neurons, TUNEL ((tdt-mediated dUTP nick end labeling) to identify DNA fragmentation, and DNA binding fluorescent Hoechst dye to identify chromatin condensation, and were visualized by fluorescence microscopy to identify apoptosis in the absence (A, D) and presence of cocaine (500 ng/ml) after 24 h (B, E) and 48 h (C, F) of treatment in LC and SN neurons, respectively. Abundant TUNEL positive cells (green), co-localized with Hoechst (blue) and TUJ1(βIII tubulin) staining (red) were observed, 24 h and 48 h after drug treatment in LC neurons (B, C). TUNEL positive staining was not significant in cocaine treated SN neurons (E, F). Quantitative analysis indicated a significant and dramatic effect for LC neurons [ANOVA: p < 0.0001], and no significant effect on SN neurons [ANOVA: p = 0.92)] (G).
Cocaine induces Bax protein expression in fetal LC neurons
Mitochondrial permeability is believed to be affected by the up-regulation of Bax resulting in the release of cytochrome c into the cytosol, which triggers activation of caspase-9 and subsequently activation of caspase-3 (Golstein, 1997; Tsujimoto, 1998). One of the key regulators of apoptosis is the Bcl-2 family of proteins where Bax is pro-apoptotic and Bcl-2 is anti-apoptotic (Martinou et al., 2000; Dey et al., 2003). Here we determined the effect of cocaine on Bax and Bcl-2 protein levels in E14 LC and SN neurons, in vitro. Cocaine exposure increased Bax protein levels in LC neurons [F(4,20) = 14.5; p < 0.0001 according to a prominent quadratic time course F(1,24) = 37.7; p < 0.0001], with a significant increase at 30 min [F(1,24) = 4.1; p < 0.055], and peak activity at 1 h [F(1,24) = 27.5; p < 0.0001], and a reduction to basal levels by 4 h. No significant changes in Bcl-2 expression were found at any time point analyzed [F(4,20) = 0.03; p = 0.99] (Fig. 3A, C, D). On the other hand, the ratio was reversed for the dopaminergic SN neurons. Significant alterations in Bcl-2 levels [F(4,20) = 4.8; p < 0.007 according to a linear temporal course F(1,24) = 17.0; p < 0.0005] were noted with increases at 1 h [F(1,24) = 5.1; p < 0.03], 4 h [F(1,24) = 10.8; p < 0.003] and 24 h [F(1,24) = 8.8; p < 0.007], compared to controls, accompanied by the lack of any readily discernible temporal course for changes in the proteins levels of Bax, albeit there was some overall evidence for a significant oscillation in levels across time [F(4,20) = 4.0; p < 0.015] which was solely attributable to the significant reduction at 30 min [F(1,24) = 6.7; p < 0.02] (Fig. 3 B, C, D). In addition, the interactions between brain region and time dependent treatment indicated a significant difference for both Bax and Bcl-2 expressions [linear (F(1,24) = 10.0; p < 0.0003) and (quadratic F(1,24) = 15.2; p < 0.0007), respectively]. Further, correlational analyses found no significant relation between the Bax and Bcl-2 expressions of the LC vs. SN (r2 = 0.047; r2 = 0.213, respectively). These data strongly suggest that cocaine exposure selectively affects the pro-apoptotic components of the Bcl-2 family of proteins, which would result in an alteration of the Bax/Bcl-2 ratio in fetal LC neurons.
Figure 3. Cocaine exposure in vitro induces Bax protein expression in fetal LC neurons.
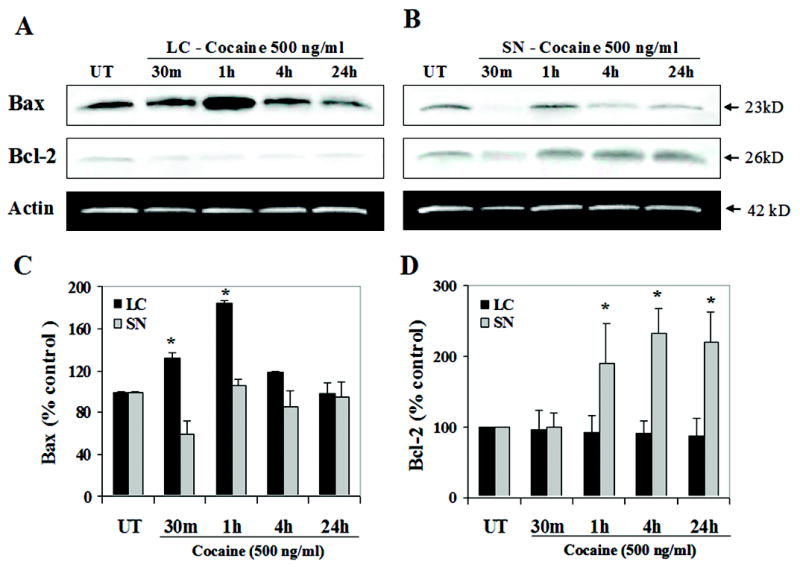
Cell lysates were extracted from control and cocaine treated LC and SN neurons at 30 min, 1 h, 4 h and 24 h after treatment, and 15 μg of cell lysates normalized for protein content was loaded and run on a 12.5 % SDS-polyacrylamide gel. Western blot analyses revealed that cocaine exposure in fetal LC neurons induced Bax protein levels [ANOVA: p < 0.0001] suggesting an induction at 30 min (contrasts: p < 0.055) with peak activity at 1 h (contrasts: p < 0.0001), returning to its basal levels by 4 h, and no significant changes in Bcl-2 levels at any time point analyzed [ANOVA: Fs <1.0] (A, C, D), compared to controls. The ratio was reversed for cocaine treated SN neurons showing significant alterations in the protein levels of Bcl-2 [ANOVA: p < 0.007] and a transient reduction of Bax levels [ANOVA: p < 0.02 ] (B, C, D), compared to controls. Quantitative analysis of Bax (C) and Bcl-2 expression (D) for both LC and SN neurons was performed on blots represented in (A) and (B). Mouse anti-actin monoclonal antibody was used as an internal loading control (A, B).
Cocaine induces caspase-9, and not caspase-8 activity, in fetal LC neurons
Since activation of caspases is important in the induction of apoptosis, we determined if there was a time-dependent cocaine-induced activation of initiator caspases 9 and/or 8, in LC or SN neurons. In LC neurons, cocaine exposure induced caspase-9 activity as indicated by a significant overall effect of time [F(4,10) = 19.3; p < 0.0001] with peak activity at 1 h [F(1,10) = 73.1; p < 0.0001], compared to controls (Fig. 4A). However, no significant changes were observed in caspase-8 activity at the same time-points after cocaine treatment, compared to controls [F(4,10) <1.0] (Fig. 4A). In contrast, E14 SN neurons exposed to cocaine failed to show any significant induction of the initiator caspase-8 or caspase-9 at any time-point tested (Fig. 4B) [F(4,10) = 1.7; p = 0.24; F(4,10) = 2.6; p = 0.10, respectively]. In addition, interactions between brain region and time dependent treatment indicated a significant difference for caspase-9 activity [F(4,20) = 6.0; p < 0.0025], with no significant differences in caspase-8 activity [F(4,20) <1.0]. Further, comparison of caspase-9 and caspase-8 activity between the LC and SN regions did not show any significant correlations (r2 = 0.005; r2 = 0.14, respectively). These data strongly indicate that cocaine exposure preferentially affects the initiation of caspase-9 activity in fetal LC neurons, and may trigger apoptotic cell signaling downstream of caspase-9.
Figure 4. Cocaine exposure in vitro induces caspase-9 activity, and not caspase-8 activity in fetal LC neurons.
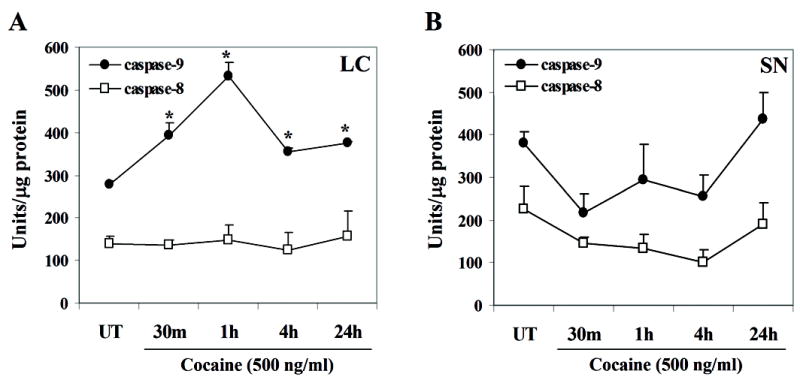
LC and SN neurons were analyzed for initiator caspase-8 and caspase-9 activity, using the caspase-8 (Ac-IEPD-AMC) and caspase-9 (Ac-LEHD-AMC) substrates. Cell lysates obtained from control and cocaine treated LC and SN neurons, normalized for protein content were incubated with the caspase-8 and caspase-9 substrates following the protocol in materials and methods section. In LC neurons caspase-9 activity was significantly induced at all given time points [ANOVA: p < 0.0001] with peak activity at 1 h (contrasts: p < 0.0001), and significant increases at 30 min (p < 0.003), 4 h (p < 0.025), and 24 h (p < 0.008). No change was noted in caspase-8 activity [ANOVA: Fs <1.0] (A). Embryonic SN neurons failed to show any significant alterations in caspase-9 [ANOVA: p=0.24] or caspase-8 activities [ANOVA: p=0.10], compared to controls (B).
Cocaine induces caspase-3 activity in fetal LC neurons
Induction of Bax is believed to induce mitochondrial permeability releasing cyt c, leading to caspase-9 activation and subsequent induction of effector caspase-3 and its target proteins (Tsujimoto, 1998). Since cocaine treated LC neurons showed increased Bax protein levels and subsequent caspase-9 activity, we characterized the downstream events of apoptosis by studying the activated form of caspase-3. Analysis of caspase-3 activity in LC neurons exposed to cocaine showed a significant induction [F(4,28) = 11.5; p < 0.0001] with a temporal course characterized by a prominent quadratic component [F(1,28) = 39.0; p < 0.0001], showing peak activity at 1 h [F(1,28) = 42.9; p < 0.0001], compared to controls (Fig. 5 A). Induction of caspase-3 activity was confirmed by western blot analysis, which revealed significantly reduced forms of inactive pro-caspase-3 (Fig. 5 B, D) again following a prominent quadratic time course [F(1,24) = 8.4; p < 0.008] and showing reduced expression at 30 min [F(1,24) = 7.5; p < 0.015], 1 h [F(1,24) = 10.7; p < 0.003] and 4 h [F(1,24) = 7.2; p < 0.01]. These data complement the results observed in cocaine induced caspase-3 activity (Fig. 5 A). In contrast, cocaine treatment did not induce caspase-3 activity in SN neurons [F(4,28) = 0.8; p = 0.51] (Fig. 5 A). However, western blot analysis of inactive pro-caspase-3 in cocaine treated SN neurons demonstrated a significant induction in the expression of the inactive form, also with a prominent quadratic temporal course [F(1,24) = 17.7; p < 0.0003], compared to controls (Fig. 5 C, D). These observations suggest that cocaine is a positive effector of apoptosis specifically in E14 LC neurons probably via Bax and caspase activation.
Figure 5. Cocaine exposure in vitro induces caspase-3 activity in fetal LC neurons.
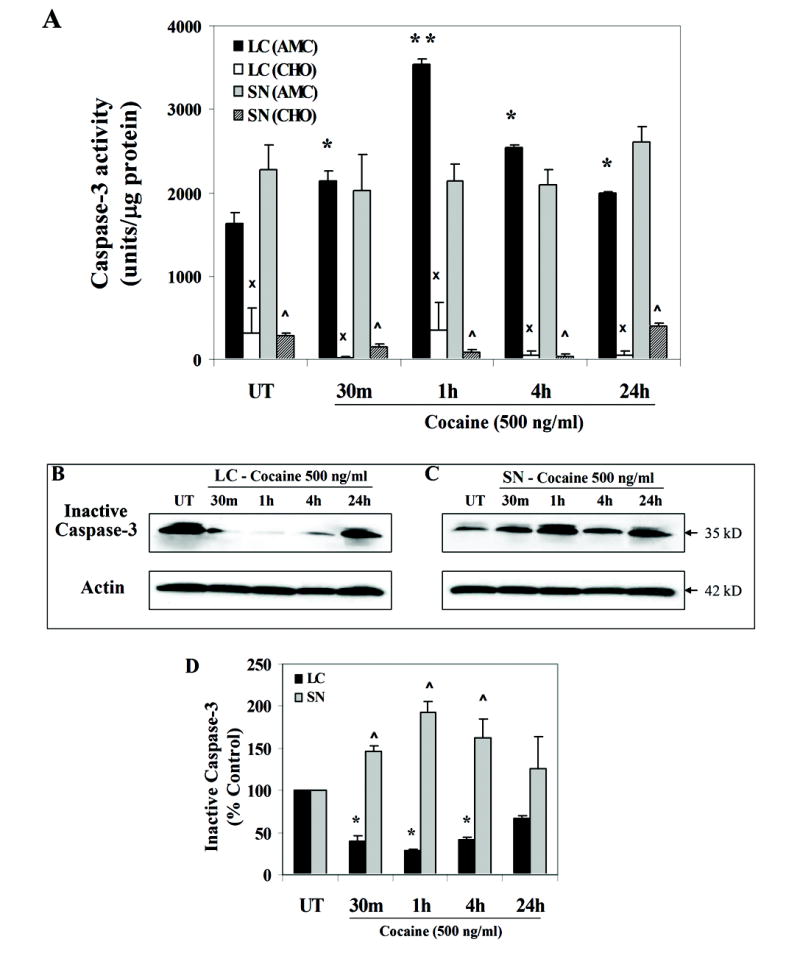
LC and SN neurons were analyzed for the effector caspase-3 activity, using the caspase-3 substrate (Ac-DEVD-AMC). Cell lysates obtained from control and cocaine treated LC and SN neurons, normalized for protein content were incubated with the caspase-3 substrate following the protocol in materials and methods section. Caspase-3 activity was significantly induced at all given time points [ANOVA: p < 0.0001], showing peak activity at 1 h (contrasts: p < 0.0001) compared to controls (A). In contrast, cocaine treatment did not induce caspase-3 activity in SN neurons at any time point studied [ANOVA: Fs <1.0] (A). Caspase-3 activation was significantly reduced by its inhibitor (Ac-DEVD-CHO) in both LC and SN neurons (ANOVA: p < 0.0001) (A). Induction in caspase-3 activity was confirmed by western blot analysis, as evidenced by significantly reduced forms of inactive pro-caspase-3 in a temporal manner [ANOVA: p < 0.008] (B). In SN neurons, analysis of caspase-3 activity by western blotting showed a significant induction of the inactive pro-caspase-3, after cocaine treatment [ANOVA: p < 0.0003] (C). Quantitative analysis of caspase-3 expression for both LC and SN neurons was performed (D). Mouse anti-actin monoclonal antibody was used as an internal loading control (B, C).
Further, caspase-3 activity was inhibited by a highly selective (cell impermeant) caspase-3 inhibitor (Ac-DEVD-CHO), which blocked caspase-3 activity for both LC [F(1,10) = 4424.1; p < 0.0001] and SN neurons [F(1,10) = 253.8; p < 0.0001], when added to lysates 30 min prior to addition of caspase-3 substrate (Ac-DEVD-AMC, Fig. 5 A). In addition, the interaction of brain region and time and lysate treatment was statistically significant [F(4,20) = 7.2; p < 0.0009]. Collectively, these results suggest a protective role of the caspase-3 inhibitor in cocaine-induced apoptosis. Further, comparison on caspase-3 activity between the LC and SN regions did not show any significant correlation (r2 = 0.003). These data show that cocaine exposure targets pro-apoptotic cell signaling components specifically in embryonic LC neurons, and may negatively regulate LC survival.
Cocaine exposure results in cleavage of caspase-3 target proteins, α-fodrin and PARP in fetal LC neurons
Expressions of several nuclear and cytoplasmic proteins are altered due to activation of terminal caspases. We investigated the effect of cocaine exposure on the cortical actin cytoskeletal protein, α-fodrin, cleavage of which induces membrane blebbing and cell shrinkage, and poly(ADP-ribose) polymerase (PARP), a nuclear enzyme that is involved in DNA repair following DNA nicks. Caspase-3 cleaves PARP and inactivates its enzymatic activity (Lazebnik et al., 1994). We performed western blot analysis to identify cleavage products for α-fodrin and PARP in cultured LC and SN neurons exposed to cocaine in vitro.
Cocaine exposure resulted in the activated caspase-3 mediated, α-fodrin cleavage product (120 kDa) in LC neurons in a temporal pattern consistent with time dependent activation of caspase-3. There was an induction of the 120-kDa α-fodrin cleavage product [F(4,20) = 9.6; p < 0.0002] with a temporal course characterized by a prominent linear component [F(1,24) = 10.7; p < 0.003] with significant increases at 1 h and 4 h [ps < 1hr: 0.02, 4 h: 0.009] showing peak activity at 4 h (Fig. 6 A, C) [Data presented as ratio of cleaved product - 120 kDa / (cleaved + uncleaved − 240 kDa)]. In contrast, a significant reduction of cleaved α-fodrin was observed in cocaine treated SN neurons [F(4,20) = 3.9; p < 0.02] (Fig. 6 B, C). In addition, the interaction between brain region and time dependent treatments indicated a significant difference [F(4,20) = 7.6; p < 0.0007].
Figure 6. Cocaine exposure in vitro causes cleavage of caspase-3 target protein, α-fodrin, in fetal LC neurons.
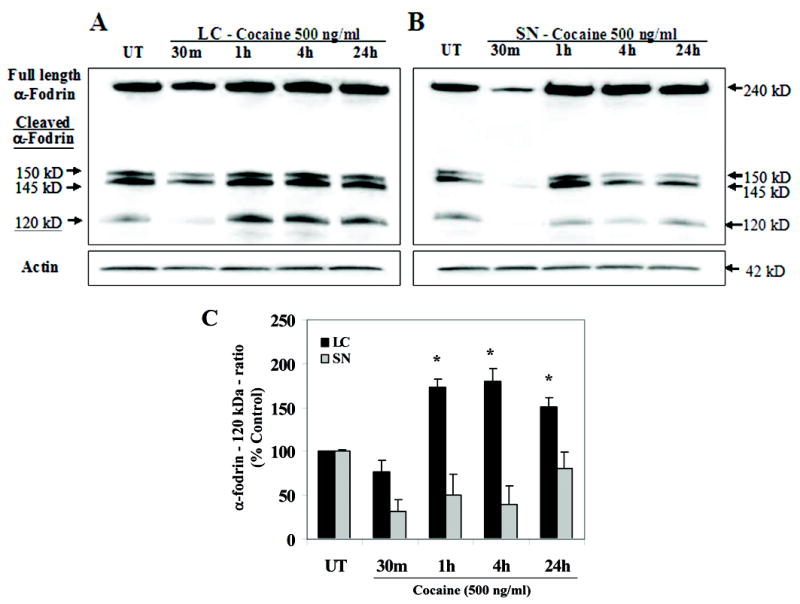
Cell lysates were extracted from control and cocaine treated LC and SN neurons at 30 min (m), 1 h, 4 h and 24 h after treatment and 15 μg of cell lysates were normalized for protein content and run on a 5 % SDS-polyacrylamide gel. Western blot analyses show that cocaine treatment in LC neurons cleaved caspase-3 target protein, α-fodrin into its 120 kDa cleaved product, at 1 h, 4 h and 24 h [ANOVA: p < 0.0002] showing peak increase at 4 h (contrast: p<0.009) (A, C), compared to controls [Data presented as ratio of cleaved product -120 kDa / (cleaved + uncleaved − 240 kDa)]. In contrast, a significant reduction of cleaved α-fodrin was evident in cocaine treated SN neurons [ANOVA: p < 0.02] (B, C), compared to controls. Quantitative analysis of α-fodrin expression for both LC and SN neurons was performed (C). Mouse anti-actin monoclonal antibody was used as an internal loading control (A, B).
Cocaine exposure on fetal LC neurons cleaved the caspase-3 target protein PARP, indicated by the 89 kDa cleaved product [F(4,20) = 38.7; p < 0.0001], with a linear temporal course [F(1,24) = 57.5; p < 0.0001], and significant increases at all time points studied [Fs(1,24) > 6.9; ps < 0.015], showing peak activity at 4 h [F(1,24) = 55.2; p < 0.0001] (Fig. 7 A, C), compared to untreated levels. In contrast, a significant reduction of the cleaved PARP was observed after cocaine treatment in SN neurons [F(4,20) = 25.9; p < 0.0001] with a prominent linear component [F(1,24) = 33.8; p < 0.0001] and notable decreases at all time points studied [Fs(1,24) > 5.7; ps < 0.025] and maximal reduction at 4 h compared to untreated levels [Fs(1,24) = 42.3; p < 0.0001] (Fig. 7 B, C). In addition, the interaction between brain region and time dependent treatment indicated a significant difference [F(4,20) = 62.3; p < 0.0001]. These data suggest that cocaine preferentially induces cell death in fetal LC neurons probably via caspase-3 dependent signaling cascade.
Figure 7. Cocaine exposure in vitro causes cleavage of caspase-3 target protein, PARP, in fetal LC neurons.
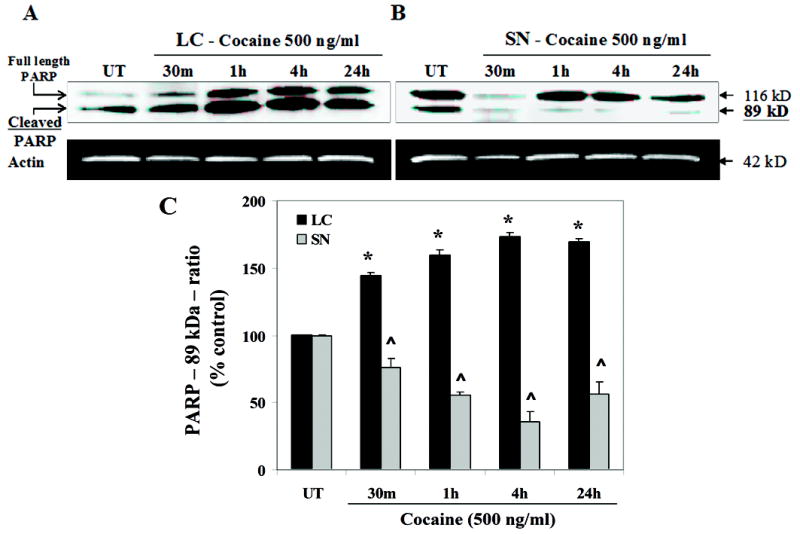
Cell lysates were extracted from control and cocaine treated LC and SN neurons at 30 min (m), 1 h, 4 h and 24 h after treatment and 15 μg of cell lysates were normalized for protein content and run on a 12.5 % SDS-polyacrylamide gel. Western blot analyses show that cocaine treatment in LC neurons cleaved caspase-3 target protein, PARP into its 89 kDa cleaved product [ANOVA: p < 0.0001], showing significant increase at all time points, with peak activity at 4 h (contrast: p < 0.0001) (A, C), compared to controls [Data presented as ratio of cleaved product − 120 kDa / (cleaved + uncleaved − 240 kDa)]. In contrast, a significant reduction of the cleaved PARP was evident in cocaine treated SN neurons [ANOVA: p < 0.0001] (B, C). Quantitative analysis of PARP expression for both LC and SN neurons was performed (C). Mouse anti-actin monoclonal antibody was used as an internal loading control (A, B).
Discussion
One of the most common neurobehavioral anomalies associated with prenatal cocaine exposure is attentional dysfunction, demonstrated both in humans (Delaney-Black et al., 1996; Linares et al., 2005; Savage et al., 2005), and animal models (Bayer et al., 2000; Gendle et al., 2004; Thompson et al., 2005). It is reasonable to conclude that attentional alterations associated with prenatal cocaine (ab)use may be linked to deficits of the basic cell biology of neural systems. Prenatal cocaine exposure results in molecular adaptations or anatomical alterations in brain regions such as the anterior cingulate cortex (ACC) and medial prefrontal cortices, which regulate cognitive and emotional development in animal models (Thompson et al., 2005), alters somatosensory cytoarchitectonics (Ren et al., 2004), and induces deficits in orientation, state control, and motor maturity in non-human primates through cellular mechansims (Lidow, 2003; He and Lidow, 2004). Further, numerous studies have defined a clear role for the noradrenergic system in attentional deficits following prenatal cocaine exposure (Mactutus, 1999; Bayer et al., 2002; Foltz et al., 2004). Consistent with these data, our previous in vivo and in vitro studies show that cocaine exposure specifically impairs the basic cell biology of locus coeruleus (LC), neurons that mediate attention, by inhibition of LC neurite initiation, elongation and branching during early periods of gestation (Snow et al., 2001; Foltz et al., 2004; Snow et al., 2004; Dey et al., 2006).
Important for the present study, cocaine exposure not only targets LC outgrowth, but also LC neuron survival. A decreased number of tyrosine hydroxylase-immunoreactive (TH-IR) cells was noted in the LC, in rats exposed to cocaine in utero (even with dosing restricted to GD11–13), with up to a 12–15% reduction, and under some conditions a reduction in cell volume (Mactutus et al., 2005). In contrast, substantia nigra neurons did not show similar impairments in response to cocaine exposure, demonstrating cellular specificity. The effects are particularly important when considering that prenatal cocaine exposure is most deleterious during early gestation, when LC neurons are undergoing maximal neurogenesis and neuritogenesis.
Thus, in the present study, we chose 1) to determine whether activation of apoptotic pathways may underlie decreased LC survival following cocaine exposure in vitro, and 2) to study this phenomenon at embryonic day 14, [neurogenesis of rat LC neurons = GD 11–13; dopaminergic neurons of the SN = GD 13–16 (Bayer et al., 1993)], when the effects of cocaine in vivo and in vitro are quite detrimental. This is analogous to weeks 5–6 in humans (Lauder and Bloom, 1974; Burgunder and Young, 1990; Bayer et al., 1993), when women abusing cocaine are often unaware of their early pregnancy. In order to continue to address cellular specificity, we compared parameters for LC cocaine exposure to growth and survival of the dopaminergic substantia nigra neurons, given that the dopaminergic system is also clearly affected by prenatal cocaine exposure (Jones et al., 1996; Wang et al., 1996; Jones et al., 2000; Harvey et al., 2001; Stanwood et al., 2001).
Cocaine exposure and apoptotic mechanisms
Our data indicate that cocaine exposure to isolated LC neurons in vitro induce cell death by affecting specific proteins in apoptotic signal transduction pathways. Our results indicate that apoptosis could induce decreased survival in LC neurons at early gestational periods, given that we see clear morphological changes such as chromatin condensation and DNA fragmentation in vitro. Cocaine’s ability to induce apoptosis in isolated fetal LC neurons was further validated by induction of pro-apoptotic Bax protein and no change in anti-apoptotic Bcl-2, thus altering the Bax/Bcl-2 ratio which regulates cellular apoptosis. In addition, study of caspases which are also important regulators of apoptosis revealed that cocaine exposure to fetal LC neurons in vitro activated initiator caspase-9 and downstream effector caspase-3 and its target proteins α-fodrin and PARP. In this study, cocaine showed preferential effects on LC neurons, when compared to isolated fetal SN neurons, since no significant changes were observed in the apoptotic markers studied in cocaine exposed SN neurons, potentially uninfluenced by cocaine exposure. Further, cocaine exposure to SN neurons resulted in increased Bcl-2 levels (Fig. 3 B, D), increased inactive pro-caspase-3 (Fig. 5 C, D) and a decreased ratio of the caspase-3 substrate, PARP (Fig. 7B, C) compared to untreated samples. This suggests a possible protective effect of cocaine over the natural cell death process in SN neurons at the recreational (low) dose used in this study (500 ng/ml), via activation of pro-survival Bcl-2 protein and inhibition of PARP cleavage, perhaps protecting enzymatic activity for DNA repair. This is speculative and will require further studies to determine the role of cocaine on metabolism and survival of SN neurons.
Recent studies in vivo have demonstrated the role of caspase-3 and its substrate in both apoptosis and survival. The role of caspase-3 in survival and differentiation of retinal growth cones has been demonstrated by its requirement for growth cone collapse and chemotropic turning (Campbell and Holt, 2003). Further, caspase-3 deficient mice demonstrated decreased bone mineral density, which was accelerated by administration of caspase-3 inhibitor, in vivo, indicating the functional role of this protein in maintenance of normal bone mass (Miura et al., 2004).
However, cocaine-induced cell death in fetal CNS has been consistently demonstrated by studies both in vitro and in vivo. Cocaine exposure (100 μM – 500 μM) induced apoptosis in cortical neurons of fetal mice (Nassogne et al., 1997; Nassogne et al., 1998). Further, fetal co-cultures exposed to cocaine lead to neurite perturbations followed by neuronal death with no effect on survival of glial cells (Nassogne et al., 1995). In addition, apoptosis indicated by condensed and fragmented nuclei was observed progressively 2–4 days after cocaine exposure in embryonic cerebral neurons, which was blocked by cycloheximide, indicating involvement of proteins (Nassogne et al., 1997; Nassogne et al., 1998). Further studies using micro-array analysis revealed that prenatal cocaine exposure induced several apoptosis related genes in the mouse cerebral wall of E18 fetuses treated with cocaine at 20 mg/kg. Several pro-apoptotic proteins as well as death receptors, members of Bcl-2 family of proteins, caspases and their substrates were elevated indicating that multiple apoptotic pathway mechanisms are affected by cocaine exposure, eventually affecting survival (Novikova et al., 2005).
Cocaine has been implicated in non-nervous system apoptosis as well. Cocaine exposure (10 – 500 μM) on human coronary artery endothelial cells (HCAECs) in vitro induce DNA fragmentation, inhibited by caspase-9, caspase-3 and cyclosporine A inhibitors, indicating the role of cytochrome c, caspase-9 and caspase-3 in cocaine induced apoptosis (He et al., 2000). Similar findings were observed in fetal rat myocardial cells (FRMCs) exposed to 100 μM cocaine in vitro (Xiao et al., 2000). Studies in vivo indicated that FRMCs exposed to cocaine (30 mg/kg and 60 mg/kg) in utero showed an increase in cell death, and increased activities of caspase-3, 8 and 9, and elevated levels of Bax. However, these changes were not observed in maternal heart (Xiao et al., 2001) probably because cocaine crosses the placenta and accumulates in the fetus (DeVane et al., 1989), potentially rendering fetal neurons increasingly susceptible to apoptosis.
In this study a pharmacologically active dose of 500 ng/ml cocaine (1.5 μM) was used to model effects of recreational doses of cocaine on survival of noradrenergic LC neurons, in vitro (Booze et al., 1997). Although cell death in vivo is critical for normal neuronal development, induction of cell death proteins when exposed to cocaine implicates abnormal cell signaling that could play a disruptive role in maintaining cellular integrity and differentiation.
A plausible signal transduction pathway for cocaine mediated LC cell death
Based on our studies, and combined with supportive data from other laboratories, fetal LC neuron cell death in response to cocaine exposure may involve induction of pro-apoptotic Bax and decrease (or no change) in Bcl-2 levels, followed by activation of initiator caspase-9. and effector caspase-3, and cleavage of its target proteins, α-fodrin and PARP (Nicholson et al., 1995; Oliver et al., 1998). The results of this study suggest that these proteins could be targets for cocaine mediated apoptosis in vivo. A putative pathway for the effects of cocaine on LC survival is represented schematically in Fig. 8. It is likely that other proteolytic pathways are also involved in cocaine induced apoptosis, independent of caspase-3 activation, given that the proteins examined in this study represent only a fraction of the apoptotic proteins in the cell and given the vast complexity of intracellular regulation of programmed cell death in general.
Figure 8. Summary of potential apoptotic mechanisms induced by cocaine effects on fetal LC survival.
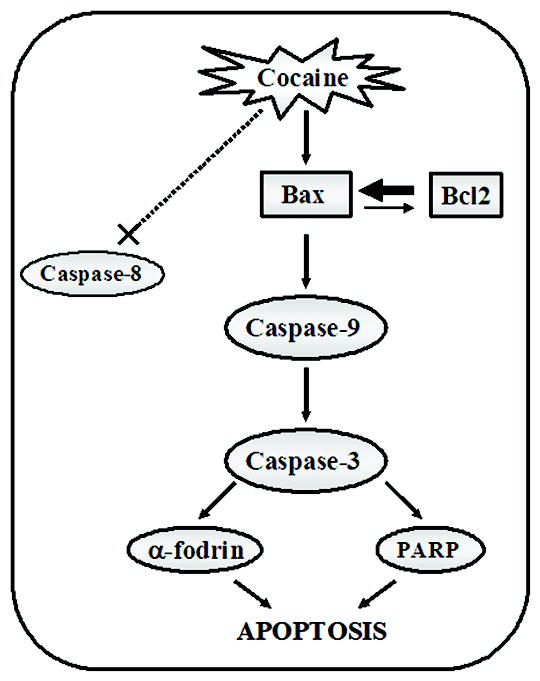
This schematic is limited to the findings of this study. Cocaine exposure on fetal LC neurons potentially altered the Bax/Bcl-2 ratio, elevating Bax protein levels leading to activation of the initiator caspase-9 and effector caspase-3, and no changes in caspase-8. This may lead to cleavage of caspase-3 target proteins - α-fodrin and PARP, suggesting that cocaine exposure on fetal LC neurons is a positive effector of apoptosis at a period of maximal neurogenesis and neuritogenesis via induction of Bax and activation of caspase-9 and caspase-3. However, this schematic is not limited to the possibility of other proteolytic pathways involved in cocaine induced apoptosis in LC neurons, with or without caspase-3 activation.
Conclusion
Apoptosis occurs in normal neuronal development, but enhanced and untimely apoptosis may lead to insufficient growth and synaptic connectivity of LC neurons, which may ultimately underlie the attentional alterations associated with prenatal cocaine exposure in human offspring. This study took a reductionist approach to examine the effects of a single pharmacologically active dose of cocaine administered over a short period of time, to two critical isolated cell types in vitro within the limitations of this type of experimental design. We have identified potential targets for cocaine mediated deficits of LC morphology and outgrowth (Snow et al., 2001; Snow et al., 2004) and survival via apoptotic mechanisms (current studies). These data highlight the importance of identifying critical signal transduction elements in cocaine-mediated cell death to ultimately develop vital treatments and therapeutic drug abuse interventions.
Acknowledgments
This work was supported by NIH grants DA12719 (to DMS), Training Grant NIH T32 NIDCD DC00065 (to SD), DA09160 and HD043680 (to CFM), and DA13137 and DA14401 (to RMB). A preliminary report of these findings was presented at the annual meeting of the Neurobehavioral and Teratology Society, St. Pete, FL, 2005, and the annual meeting of the Society for Neuroscience, Washington D.C, 2005.
Abbreviations
- AAALAC
The Association for Assessment and Accreditation of Laboratory Animal Care
- ACC
anterior cingulate cortex
- Ac-DEVD-AMC
Ac-Asp-Glu-Val-Asp-AMC (AMC = 7-Amino-4methylcoumarin)
- Ac-DEVD-CHO
Ac-Asp-Glu-Val-Asp-CHO (CHO = alhehyde)
- Ac-LEHD-AMC
Ac-Leu-Glu-His-Asp-AMC (AMC = 7-Amino-4methylcoumarin)
- Ac-IEPD-AMC
Ac-Ile-Glu-Pro-Asp-AMC (AMC = 7-Amino-4methylcoumarin)
- α-fodrin
alpha fodrin
- ANOVA
analysis of variance
- BE
benzoyl ecgonine
- BSA
bovine serum albumin
- CHAPS
3-[(3-Cholamidopropyl)Dimethyl-Ammonio]-1-Propanesulfonate
- CMF-PBS
calcium magnesium free-phosphate buffered saline
- Cyt c
cytochrome c
- DTT
dithiothreitol
- EME
ecgonine methyl ester
- E11-13
embryonic day 11-13
- E14
embryonic day 14
- ELISA
enzyme linked immunosorbent assay
- GD 11-13
gestational day 11-13
- GD 13-16
gestational day 13-16
- HBSS
Hank’s balanced salt solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IACUC
Institutional Animal Care and Use Committees
- IV
intravenous
- LC
locus coeruleus
- MEM
minimum essential medium
- MTT
[3-(4,5-dimethythiazol-2yl)-2,5 diphenyl tetrazolium bromide]
- NA
noradrenergic
- NE
norepinephrine
- NIDA
National Institute on Drug Abuse
- PARP
poly (ADP-ribose) polymerase
- PCD
programmed cell death
- PLL
poly-L-lysine
- PMSF
phenylmethylsulfonyl fluoride
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- SEM
standard error measure
- SN
substantia nigra
- TUNEL
terminal deoxynucleotidyl transferase (TdT) mediated DNA nick end labeling
- TH
tyrosine hydroxylase
- TH-IR
tyrosine hydroxylase-immunoreactivity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Bae S, Zhang L. Prenatal cocaine exposure increases apoptosis of neonatal rat heart and heart susceptibility to ischemia-reperfusion injury in 1-month-old rat. Br J Pharmacol. 2005;144:900–907. doi: 10.1038/sj.bjp.0706129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselt RC. Stability of cocaine in biological fluids. J Chromatogr. 1983;268:502–505. doi: 10.1016/s0021-9673(01)95449-4. [DOI] [PubMed] [Google Scholar]
- Bayer LE, Brown A, Mactutus CF, Booze RM, Strupp BJ. Prenatal cocaine exposure increases sensitivity to the attentional effects of the dopamine D1 agonist SKF81297. J Neurosci. 2000;20:8902–8908. doi: 10.1523/JNEUROSCI.20-23-08902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer LE, Kakumanu S, Mactutus CF, Booze RM, Strupp BJ. Prenatal cocaine exposure alters sensitivity to the effects of idazoxan in a distraction task. Behav Brain Res. 2002;133:185–196. doi: 10.1016/s0166-4328(02)00002-5. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Boonman Z, Isacson O. Apoptosis in neuronal development and transplantation: role of caspases and trophic factors. Exp Neurol. 1999;156:1–15. doi: 10.1006/exnr.1999.7056. [DOI] [PubMed] [Google Scholar]
- Booze RM, Lehner AF, Wallace DR, Welch MA, Mactutus CF. Dose-response cocaine pharmacokinetics and metabolite profile following intravenous administration and arterial sampling in unanesthetized, freely moving male rats. Neurotoxicol Teratol. 1997;19:7–15. doi: 10.1016/s0892-0362(96)00180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouis P, Taccard G, Boelsterli UA. Determination of cocaine and norcocaine in plasma and cell cultures using high-performance liquid chromatography. J Chromatogr. 1990;526:447–459. doi: 10.1016/s0378-4347(00)82527-x. [DOI] [PubMed] [Google Scholar]
- Bunney EB, Appel SB, Brodie MS. Cocaine potentiates ethanol-induced excitation of dopaminergic reward neurons in the ventral tegmental area. J Pharmacol Exp Ther. 2000;293:383–389. [PubMed] [Google Scholar]
- Burgunder JM, Young WS., 3rd Ontogeny of tyrosine hydroxylase and cholecystokinin gene expression in the rat mesencephalon. Brain Res Dev Brain Res. 1990;52:85–93. doi: 10.1016/0165-3806(90)90224-m. [DOI] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron. 2003;37:939–952. doi: 10.1016/s0896-6273(03)00158-2. [DOI] [PubMed] [Google Scholar]
- Chang HY, Yang X. Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev. 2000;64:821–846. doi: 10.1128/mmbr.64.4.821-846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney-Black V, Covington C, Ostrea E, Jr, Romero A, Baker D, Tagle MT, Nordstrom-Klee B, Silvestre MA, Angelilli ML, Hack C, Long J. Prenatal cocaine and neonatal outcome: evaluation of dose-response relationship. Pediatrics. 1996;98:735–740. [PubMed] [Google Scholar]
- DeVane CL, Simpkins JW, Miller RL, Braun SB. Tissue distribution of cocaine in the pregnant rat. Life Sci. 1989;45:1271–1276. doi: 10.1016/0024-3205(89)90129-x. [DOI] [PubMed] [Google Scholar]
- Dey S, Mactutus CF, Booze RM, Snow DM. Specificity of prenatal cocaine on inhibition of locus coeruleus neurite outgrowth. Neuroscience. 2006;139:899–907. doi: 10.1016/j.neuroscience.2005.12.053. [DOI] [PubMed] [Google Scholar]
- Dey S, Booze RM, Mactutus CF, Snow DM. Specificity of prenatal cocaine on inhibition of locus coeruleus neurite outgrowth. Neuroscience. doi: 10.1016/j.neuroscience.2005.12.053. in press. [DOI] [PubMed] [Google Scholar]
- Dey S, Spring PM, Arnold S, Valentino J, Chendil D, Regine WF, Mohiuddin M, Ahmed MM. Low-dose fractionated radiation potentiates the effects of Paclitaxel in wild-type and mutant p53 head and neck tumor cell lines. Clin Cancer Res. 2003;9:1557–1565. [PubMed] [Google Scholar]
- Evans SM, Cone EJ, Henningfield JE. Arterial and venous cocaine plasma concentrations in humans: relationship to route of administration, cardiovascular effects and subjective effects. J Pharmacol Exp Ther. 1996;279:1345–1356. [PubMed] [Google Scholar]
- Foltz TL, Snow DM, Strupp BJ, Booze RM, Mactutus CF. Prenatal intravenous cocaine and the heart rate-orienting response: a dose-response study. Int J Dev Neurosci. 2004;22:285–296. doi: 10.1016/j.ijdevneu.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Gendle MH, White TL, Strawderman M, Mactutus CF, Booze RM, Levitsky DA, Strupp BJ. Enduring effects of prenatal cocaine exposure on selective attention and reactivity to errors: evidence from an animal model. Behav Neurosci. 2004;118:290–297. doi: 10.1037/0735-7044.118.2.290. [DOI] [PubMed] [Google Scholar]
- Golstein P. Controlling cell death. Science. 1997;275:1081–1082. doi: 10.1126/science.275.5303.1081. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Harvey JA, Romano AG, Gabriel M, Simansky KJ, Du W, Aloyo VJ, Friedman E. Effects of prenatal exposure to cocaine on the developing brain: anatomical, chemical, physiological and behavioral consequences. Neurotox Res. 2001;3:117–143. doi: 10.1007/BF03033234. [DOI] [PubMed] [Google Scholar]
- He J, Xiao Y, Zhang L. Cocaine induces apoptosis in human coronary artery endothelial cells. J Cardiovasc Pharmacol. 2000;35:572–580. doi: 10.1097/00005344-200004000-00010. [DOI] [PubMed] [Google Scholar]
- He N, Lidow MS. Cerebral cortical abnormalities seen in a non-human primate model of prenatal cocaine exposure are not related to vasoconstriction. Neurotoxicology. 2004;25:419–432. doi: 10.1016/j.neuro.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Isenschmid DS, Levine BS, Caplan YH. A comprehensive study of the stability of cocaine and its metabolites. J Anal Toxicol. 1989;13:250–256. doi: 10.1093/jat/13.5.250. [DOI] [PubMed] [Google Scholar]
- Jones L, Fischer I, Levitt P. Nonuniform alteration of dendritic development in the cerebral cortex following prenatal cocaine exposure. Cereb Cortex. 1996;6:431–445. doi: 10.1093/cercor/6.3.431. [DOI] [PubMed] [Google Scholar]
- Jones LB, Stanwood GD, Reinoso BS, Washington RA, Wang HY, Friedman E, Levitt P. In utero cocaine-induced dysfunction of dopamine D1 receptor signaling and abnormal differentiation of cerebral cortical neurons. J Neurosci. 2000;20:4606–4614. doi: 10.1523/JNEUROSCI.20-12-04606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SH, Hengartner MO. Programmed cell death: alive and well in the new millennium. Trends Cell Biol. 2001;11:526–534. doi: 10.1016/s0962-8924(01)02173-0. [DOI] [PubMed] [Google Scholar]
- Keller RW, Jr, Snyder-Keller A. Prenatal cocaine exposure. Ann N Y Acad Sci. 2000;909:217–232. doi: 10.1111/j.1749-6632.2000.tb06684.x. [DOI] [PubMed] [Google Scholar]
- Lauder JM, Bloom FE. Ontogeny of monoamine neurons in the locus coeruleus, Raphe nuclei and substantia nigra of the rat. I. Cell differentiation. J Comp Neurol. 1974;155:469–481. doi: 10.1002/cne.901550407. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lidow MS. Consequences of prenatal cocaine exposure in nonhuman primates. Brain Res Dev Brain Res. 2003;147:23–36. doi: 10.1016/j.devbrainres.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Lidow MS, Trakht T, Howard RL. Cocaine-induced alterations in the density of monoaminergic receptors in the embryonic guinea pig cerebral wall. Synapse. 1999;32:225–237. doi: 10.1002/(SICI)1098-2396(19990601)32:3<225::AID-SYN8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Linares TJ, Singer LT, Kirchner HL, Short EJ, Min MO, Hussey P, Minnes S. Mental Health Outcomes of Cocaine-Exposed Children at 6 Years of Age. J Pediatr Psychol. 2005 doi: 10.1093/jpepsy/jsj020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo AC, Houenou LJ, Oppenheim RW. Apoptosis in the nervous system: morphological features, methods, pathology, and prevention. Arch Histol Cytol. 1995;58:139–149. doi: 10.1679/aohc.58.139. [DOI] [PubMed] [Google Scholar]
- Mactutus CF. Prenatal intravenous cocaine adversely affects attentional processing in preweanling rats. Neurotoxicol Teratol. 1999;21:539–550. doi: 10.1016/s0892-0362(99)00024-0. [DOI] [PubMed] [Google Scholar]
- Mactutus CF, Hasselrot U, Fitting S, Adams S, Snow DMBJS, Booze RM. Neurobiology and Teratology Society; St Pete, FL: 2005. Prenatal intravenous (IV) cocaine: Cell loss in the locus coeruleus (LC)(Abstract) [Google Scholar]
- Martinou JC, Desagher S, Antonsson B. Cytochrome c release from mitochondria: all or nothing. Nat Cell Biol. 2000;2E:41–43. doi: 10.1038/35004069. [DOI] [PubMed] [Google Scholar]
- Mayes LC, Grillon C, Granger R, Schottenfeld R. Regulation of arousal and attention in preschool children exposed to cocaine prenatally. Ann N Y Acad Sci. 1998;846:126–143. doi: 10.1111/j.1749-6632.1998.tb09731.x. [DOI] [PubMed] [Google Scholar]
- Mitchell ES, Snyder-Keller A. Blockade of D1 dopaminergic transmission alleviates c-fos induction and cleaved caspase-3 expression in the brains of rat pups exposed to prenatal cocaine or perinatal asphyxia. Exp Neurol. 2003;182:64–74. doi: 10.1016/s0014-4886(03)00026-8. [DOI] [PubMed] [Google Scholar]
- Miura M, Chen XD, Allen MR, Bi Y, Gronthos S, Seo BM, Lakhani S, Flavell RA, Feng XH, Robey PG, Young M, Shi S. A crucial role of caspase-3 in osteogenic differentiation of bone marrow stromal stem cells. J Clin Invest. 2004;114:1704–1713. doi: 10.1172/JCI20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassogne MC, Evrard P, Courtoy PJ. Selective neuronal toxicity of cocaine in embryonic mouse brain cocultures. Proc Natl Acad Sci U S A. 1995;92:11029–11033. doi: 10.1073/pnas.92.24.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassogne MC, Evrard P, Courtoy PJ. Selective direct toxicity of cocaine on fetal mouse neurons. Teratogenic implications of neurite and apoptotic neuronal loss. Ann N Y Acad Sci. 1998;846:51–68. [PubMed] [Google Scholar]
- Nassogne MC, Louahed J, Evrard P, Courtoy PJ. Cocaine induces apoptosis in cortical neurons of fetal mice. J Neurochem. 1997;68:2442–2450. doi: 10.1046/j.1471-4159.1997.68062442.x. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Norbert Konig, Mary Beth Wilkie, Lauder J. A Dissection and Tissue Culture Manual of the Nervous System. 1989. Dissection of Monoaminergic Neuronal Groups From Embryonic Rat Brain; pp. 26–29. Chapter 4. [Google Scholar]
- Novikova SI, He F, Bai J, Badan I, Lidow IA, Lidow MS. Cocaine-induced changes in the expression of apoptosis-related genes in the fetal mouse cerebral wall. Neurotoxicol Teratol. 2005;27:3–14. doi: 10.1016/j.ntt.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- Ren J-Q, Malanga CJ, Tabit E, Kosofsky BE. Neuropathological consequences of prenatal cocaine exposure in the mouse. Int J Devl Neurosci. 2004;22:309–320. doi: 10.1016/j.ijdevneu.2004.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage J, Brodsky NL, Malmud E, Giannetta JM, Hurt H. Attentional functioning and impulse control in cocaine-exposed and control children at age ten years. J Dev Behav Pediatr. 2005;26:42–47. [PubMed] [Google Scholar]
- Schenker S, Yang Y, Johnson RF, Downing JW, Schenken RS, Henderson GI, King TS. The transfer of cocaine and its metabolites across the term human placenta. Clin Pharmacol Ther. 1993;53:329–339. doi: 10.1038/clpt.1993.29. [DOI] [PubMed] [Google Scholar]
- Singer LT, Arendt R, Minnes S, Farkas K, Salvator A. Neurobehavioral outcomes of cocaine-exposed infants. Neurotoxicol Teratol. 2000;22:653–666. doi: 10.1016/s0892-0362(00)00092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow DM, Smith JD, Booze RM, Welch MA, Mactutus CF. Cocaine decreases cell survival and inhibits neurite extension of rat locus coeruleus neurons. Neurotoxicol Teratol. 2001;23:225–234. doi: 10.1016/s0892-0362(01)00137-4. [DOI] [PubMed] [Google Scholar]
- Snow DM, Carman HM, Smith JD, Booze RM, Welch M, Mactutus CF. Cocaine-induced inhibition of process outgrowth in locus coeruleus neurons: role of gestational exposure period and offspring sex. Int J Devl Neuroscience. 2004;22:297–308. doi: 10.1016/j.ijdevneu.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Washington RA, Shumsky JS, Levitt P. Prenatal cocaine exposure produces consistent developmental alterations in dopamine-rich regions of the cerebral cortex. Neuroscience. 2001;106:5–14. doi: 10.1016/s0306-4522(01)00256-1. [DOI] [PubMed] [Google Scholar]
- Studer L. Cellular and Developmental Neuroscience. John Wiley & Sons Inc; Los Angeles, CA, USA: 1997. Culture of Substantia Nigra Neurons. [Google Scholar]
- Thompson BL, Levitt P, Stanwood GD. Prenatal cocaine exposure specifically alters spontaneous alternation behavior. Behav Brain Res. 2005;164:107–116. doi: 10.1016/j.bbr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. Bcl-2 family: life-or-death switch. FEBS Lett. 2000;466:6–10. doi: 10.1016/s0014-5793(99)01761-5. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Reed TM, Acuff-Smith KD, Schilling MA, Cappon GD, Fisher JE, Pu C. Long-term learning deficits and changes in unlearned behaviors following in utero exposure to multiple daily doses of cocaine during different exposure periods and maternal plasma cocaine concentrations. Neurotoxicol Teratol. 1995;17:253–264. doi: 10.1016/0892-0362(94)00061-h. [DOI] [PubMed] [Google Scholar]
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- Wang XH, Levitt P, O’Brien Jenkins A, Murphy EH. Normal development of tyrosine hydroxylase and serotonin immunoreactive fibers innervating anterior cingulate cortex and visual cortex in rabbits exposed prenatally to cocaine. Brain Res. 1996;715:221–224. doi: 10.1016/0006-8993(96)00012-1. [DOI] [PubMed] [Google Scholar]
- Xiao Y, He J, Gilbert RD, Zhang L. Cocaine induces apoptosis in fetal myocardial cells through a mitochondria-dependent pathway. J Pharmacol Exp Ther. 2000;292:8–14. [PubMed] [Google Scholar]
- Xiao Y, Xiao D, He J, Zhang L. Maternal cocaine administration during pregnancy induces apoptosis in fetal rat heart. J Cardiovasc Pharmacol. 2001;37:639–648. doi: 10.1097/00005344-200106000-00001. [DOI] [PubMed] [Google Scholar]