

Abstract
Today there exists only one FDA-approved treatment for ischemic stroke; i.e., the serine protease tissue-type plasminogen activator (tPA). In the aftermath of the failed stroke clinical trials with the nitrone spin trap/radical scavenger, NXY-059, a number of articles raised the question: are we doing the right thing? Is the animal research truly translational in identifying new agents for stroke treatment? This review summarizes the current state of affairs with plasminogen activators in thrombolytic therapy. In addition to therapeutic value, potential side effects of tPA also exist that aggravate stroke injury and offset the benefits provided by reperfusion of the occluded artery. Thus, combinational options (ultrasound alone or with microspheres/nanobubbles, mechanical dissociation of clot, activated protein C (APC), plasminogen activator inhibitor-1 (PAI-1), neuroserpin and CDP-choline) that could offset tPA toxic side effects and improve efficacy are also discussed here. Desmoteplase, a plasminogen activator derived from the saliva of Desmodus rotundus vampire bat, antagonizes vascular tPA-induced neurotoxicity by competitively binding to low-density lipoprotein related-receptors (LPR) at the blood-brain barrier (BBB) interface, minimizing the tPA uptake into brain parenchyma. tPA can also activate matrix metalloproteinases (MMPs), a family of endopeptidases comprised of 24 mammalian enzymes that primarily catalyze the turnover and degradation of the extracellular matrix (ECM). MMPs have been implicated in BBB breakdown and neuronal injury in the early times after stroke, but also contribute to vascular remodeling, angiogenesis, neurogenesis and axonal regeneration during the later repair phase after stroke. tPA, directly or by activation of MMP-9, could have beneficial effects on recovery after stroke by promoting neurovascular repair through vascular endothelial growth factor (VEGF). However, any treatment regimen directed at MMPs must consider their pleiotropic nature and the likelihood of either beneficial or detrimental effects that might depend on the timing of the treatment in relation to the stage of brain injury.
Keywords: Atherosclerosis, Clinical trials, Cytokines, CDP-choline, Inflammation, Matrix metalloproteinase, Serine protease, Tissue-type plasminogen activator, Stroke
Introduction
Many studies suggest that proteolytic activity may be involved in a number of central nervous system (CNS) disorders and injuries including stroke. Proteases occur naturally in all organisms and constitute approximately 1-5% of the gene content. These enzymes are involved in a multitude of physiological reactions from simple digestion of food proteins to highly-regulated cascades such as blood-clotting, the complement system, and apoptosis pathways. Proteases present in blood serum (thrombin, plasmin, Hageman factor, etc.) play an important role in blood-clotting as well as clot lysis, and the correct action of the immune system. Proteases are currently classified into six groups according to the nature of their catalytic active site and conditions of action: 1) serine proteases, 2) metalloproteases, 3) cysteine (thiol) proteases, 4) aspartic acid proteases, 5) threonine proteases; and 6) glutamic acid proteases. Threonine and glutamic acid proteases were recently described. This article primarily focuses on the role of tissue plasminogen activator (tPA, a serine protease) and matrix metalloproteinases [MMPs] in stroke [1].
Stroke or “brain attack”: a problem of vast clinical importance
Stroke generally refers to a local interruption of blood flow to the brain and is the leading cause of long-term disability and the third leading cause of death [2]. Approximately 12% of strokes are hemorrhagic (rupture of a cerebral blood vessel), whereas the remaining 88% are ischemic and result from occlusion of a cerebral artery (either thrombolic or embolic). Blockage of a cerebral artery results in interruption of the blood flow and supply of nutrients, glucose and oxygen to the brain. The energy needs of the brain are supplied by metabolism of glucose and oxygen for the phosphorylation of ADP to ATP. Most of the ATP generated in the brain is utilized to maintain intracellular homeostasis and transmembrane ion gradients of sodium, potassium, and calcium. Energy failure results in collapse of ion gradients, and excessive release of neurotransmitters such as dopamine and glutamate [3], ultimately leading to neuronal death and the development of an infarction. Excess glutamate release, triggered by stroke, and consequent stimulation of glutamate receptors, results in activation of phospholipases/sphingomyelinases [3, 4], phospholipid hydrolysis and release of second messengers arachidonic acid and ceramide [3, 5, 6]. Ultimately these processes lead to apoptotic or necrotic cell death.
Focal cerebral ischemia or ‘ischemic stroke’ is caused by a local blockage of a cerebral artery that results in loss of blood flow to a portion of the brain. Stroke is characterized by an ischemic core (infarct) surrounded by a “penumbra” (peri-infarct) region that has partial reduction in blood flow due to presence of collateral arteries. The ischemic core is generally considered unsalvageable, whereas the penumbra may be rescued by timely intervention and is a target for the development of therapeutic treatment. Local arterial blockage can be caused by either a thrombus (a clot that forms at the site of the arterial occlusion) or an embolus (a clot that forms peripherally, dislodges into the arterial circulation and is transported to the brain). Atherosclerosis is the main risk factor for development of these embolisms (Fig. 1). Inflammation poses as one of the important risk factors for ischemic stroke for its role in the initiation, progression and maturation of atherosclerosis. A systemic inflammatory response involving up-regulation of the cytokines tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1) is believed to be instrumental in the formation and destabilization of atherosclerotic plaques [7, 8]. There is considerable clinical data indicating that this systemic inflammation is also associated with unfavorable outcome in stroke patients [9]. However, this inter-relationship of systemic inflammation with stroke pathology has not been well studied.
Fig. 1. An odyssey: Plaque to stroke.
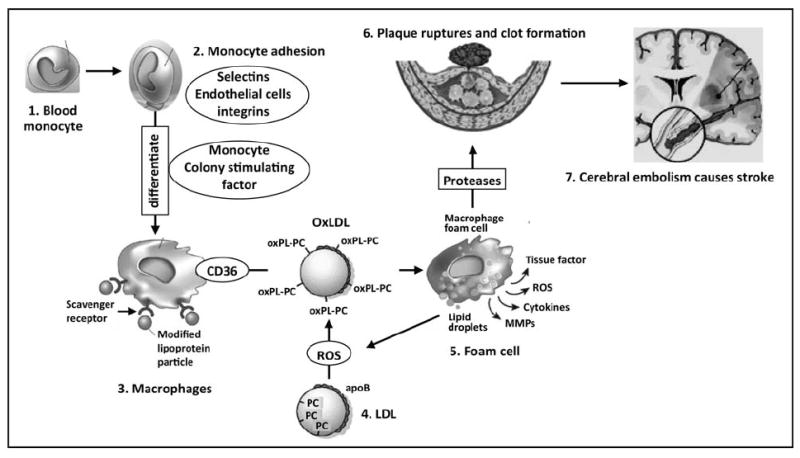
Atherosclerosis is a major risk factor for ischemic stroke [14]. Under inflammatory conditions (OxLDL, homocysteine, cigarette smoke, shear stress and infectious agents such as Chlamydia pneumoniae) endothelia cells of the artery express adhesion molecules that allow monocytes (1) to adhere to endothelia (2). Chemoattractants such as monocyte chemoattractant protein-1 (MCP-1) draw the monocytes through the endothelium into the arterial intima. Once resident in the intima, monocytes differentiate into macrophages (3) in response to locally produced agents such as monocyte colony stimulating factor. LDL (4) under oxidative stress gets oxidized to OxLDL. The macrophages increase expression of scavenging receptors such as CD36, SR-A and SR-B. These scavenger receptors then internalize specifically oxidized LDL (OxLDL, specifically OxPC) particles such that cholesteryl esters accumulate in cytoplasmic droplets, resulting in lipid-loaded macrophages (foam cells, 5). Foam cells produce ROS, which further propagate LDL oxidation, and secrete cytokines and matrix metalloproteinases (MMPs). The MMPs contribute to degradation of the fibrous cap surrounding the plaque, resulting in its rupture and formation of a blood clot (6). If the blood clot dislodges from the plaque, arterial blood flow can carry it to the brain, where it lodges in a cerebral artery (embolism) and causes an ischemic stroke (7).
Atherosclerosis is a risk factor for stroke
Atherosclerosis is believed to be predominantly an inflammatory condition produced as a response to injury [10]. Atherosclerosis is defined by the accumulation in the arterial intima of mainly low-density lipoprotein (LDL)-derived lipids along with apolipoprotein B-100 (apoB100). LDL is the major carrier of cholesterol in the circulation and is composed of one apoB100 together with phosphatidylcholine (PC), sphingomyelin (SM) and unesterified cholesterol (500:200:400 molecules respectively) constituting a surface film surrounding a core of cholesteryl esters and triacylglycerols.
The traditional view of atherosclerosis has been simply the deposition and accumulation of cholesterol, other lipids, and cellular debris within the wall of medium to large arteries, resulting in plaque formation and disturbance of blood flow (Fig. 1). The role of cholesterol in atherosclerosis is well established and has been elegantly reviewed [11]. It is now believed that a complex endothelial injury and dysfunction induced by a variety of factors such as homocysteine, toxins (smoking), mechanical forces (shear stress), infectious agents (such as Chlamydia pneumoniae) and oxidized LDL results in an inflammatory response that is instrumental in the formation and rupture of plaques, one of the greatest risk factors for ischemic stroke [7, 8].
Two critical events involved in atherogenesis involve accumulation and oxidation of LDL in the arterial intima and recruitment of monocytes to the developing lesion. After diffusion through the endothelial cell junctions into the arterial intima, LDL can be retained through interaction of apoB100 and matrix proteoglycans. LDL accumulates in the arterial intima when its rate of influx exceeds the rate of efflux. While the exact mechanisms governing LDL accumulation remain to be elucidated [12], evidence indicates that LDL uptake and retention are increased at plaque sites, which may involve degradation or binding to cellular and matrix components. Once in the arterial intima, LDL can be oxidized to OxLDL through oxidation of polyunsaturated fatty acids of LDL lipids, particularly PC of LDL (note: LDL contains 500 molecules of PC) to form OxPC.
A second critical event in atherosclerosis is an inflammatory response that triggers expression of adhesion molecules (selectins and integrins) in the arterial endothelium, stimulating adhesion of monocytes to the endothelium. Monocytes penetrate into the arterial intima, differentiate into macrophages and eventually become foam cells by binding and endocytosing OxLDL through CD36 scavenging receptors (Fig. 1). Studies showed that oxidized phospholipids bearing the PC headgroup as a ligand on OxLDL mediate uptake by macrophage scavenging receptors such as CD36 [13]. The macrophage foam cells generate reactive oxygen species (ROS), produce TNF-α and IL-1, and MMP-9 that promote atherosclerosis, degrade the fibrous cap, and eventually lead to plaque rupture [14]. Recent studies also showed that carotid atherosclerotic plaques from patients symptomatic of stroke had higher expression of lipoprotein-phospholipase A2 (Lp-PLA2)/PAF acetylhydrolase, lyso-PC, OxLDL, macrophage content and MMP-2 compared to carotid plaques from asymptomatic patients [15].
Increased levels of TNF-α and IL-1 up-regulate expression of adhesion molecules and promote further monocyte recruitment into developing atherosclerotic lesions. Macrophage MMP-9 degrades extracellular matrix (ECM) components including the fibrous cap of atheromatous plaques. Rupture of the fibrous cap exposes the blood to the inner components of the plaque, particularly tissue factor released from apoptotic macrophages. Tissue factor binds to the activated form of coagulation factor VII (proconvertin) and triggers the coagulation cascade (Fig. 2). Most of the enzymes involved in the coagulation cascade are synthesized as zymogens, or inactive enzyme precursors, and are cleaved to release the active forms. The final processes involved in this coagulation cascade include activation of prothrombin to the serine protease thrombin, which converts fibrinogen into fibrin. Thrombin also converts factor XIII is to its active form, factor XIIIa, which then polymerizes fibrin to form a cross linked mesh that creates the clot in conjunction with platelets (Fig. 2). Destabilization of this clot results in release of an embolus into the blood stream, which can be transported to the brain, where it can lodge in a cerebral artery and induce an ischemic stroke (Fig. 1) [7, 8, 10, 16].
Fig. 2. Blood clot formation.

Blood coagulation represents a series of sequential interactive events that lead to the repair of the vascular system following injury, traditionally distinguished in two pathways: the intrinsic and extrinsic pathways. The intrinsic pathway is defined as a cascade that utilizes only factors that are soluble in the plasma, whereas the extrinsic pathway consists of some factors that are insoluble in plasma, e.g. membrane-bound factors like Factor VII. Upon activation, individual glycoproteins serve as enzymes to convert the zymogen (inactive) form of the succeeding glycoprotein to its protease (active) form (identified by “a”). Both pathways produce Factor Xa, which then catalyses the conversion of prothrombin (factor II) to thrombin (factor IIa). Thrombin then converts fibrinogen to soluble fibrin monomers, which spontaneously aggregate. Thrombin also activates Factor XIII, which cross-links the fibrin molecules to form a stable mesh-like structure which traps blood cells, forming a clot.
Oxidized PC (OxPC) is an inflammatory marker
Peroxidation of fatty acids in phospholipids results in an oxidized phospholipid. Scission of the peroxidized fatty acid results in formation of a phospholipid such as OxPC [17] with a fatty acid containing an aldehyde residue, and the aldehyde cleavage fragments malondialdehyde, 4-hydroxynonenal, or acrolein. These reactive phospholipid aldehydes exhibit cytotoxicity by binding to lysine residues of cellular proteins. OxPC itself also changes the membrane properties, resulting in alterations in ion transport and membrane protein function. The presence of OxPC on the apoptotic cell surface has been characterized by EO6 monoclonal antibodies that exclusively bind to OxPC [18]. In addition to OxPC, EO6 antibodies also recognize OxPC bound to lysine residues of proteins. OxPC on apoptotic cells may enhance pro-inflammatory signals and also serve as a marker of inflammation and apoptosis [17, 19, 20]. The presence of OxPC has been demonstrated in multiple sclerosis brain using EO6 monoclonal antibodies [18]. Formation of OxPC species were also shown after permanent focal ischemia in mice [21].
Clinical options for stroke treatment are very limited
Tissue-type plasminogen activator (tPA) is a serine protease that cleaves plasminogen into active plasmin. In plasma, the primary function of plasmin is digestion of fibrin, and thus tPA is used as a thrombolytic agent for treatment of ischemic stroke. Thrombolysis is the breakdown (lysis) of blood clots by pharmacological means, colloquially referred to as clot busting. Clearing the cross-linked fibrin mesh (the backbone of a clot) makes the clot soluble and subject to further proteolysis by other enzymes, thereby restoring blood flow in the occluded blood vessel. More than 12 years after FDA approval, tPA remains the only approved therapy for stroke, but because of its severe inclusion criteria (narrow 3 hr time-to-treatment window and no apparent hemorrhagic complications), <5% of stroke patients get benefit from this clot dissolving agent. A detailed set of criteria for selection of stroke patients for tPA therapy was recently presented [22]. Clinical studies have shown that patients receive little or no benefit if tPA therapy is initiated more than 3 hrs after the onset of stroke, which excludes most stroke patients from tPA treatment due to the time required for transportation to medical facilities and proper diagnosis. Another side of tPA in stroke is that it is more than just a clot bluster (Fig. 3) [1]. In addition to dissolving the blood clot, tPA may damage the basal lamina of the blood vessels, resulting in edema, disruption of the blood-brain barrier (BBB), or hemorrhage (Fig. 3). Thus any patients with evidence of hemorrhage or who have been taking any anti-coagulant medication such as aspirin also are not candidates for tPA therapy.
Fig. 3.

Pleiotropic actions of tPA.
Endogenous functions of tPA in the brain
tPA is expressed widely in the CNS and is involved in mechanisms of synaptic regulation and synaptic plasticity, both during development and in the mature brain. tPA activity is regulated by its endogenous inhibitor neuroserpin, a type of serpin (serine protease inhibitor) [23, 24]. Neuroserpin preferentially reacts with and is present along with tPA in brain regions suggesting that neuroserpin is the selective inhibitor of tPA. tPA facilitates Ca2+ influx via NMDA receptors and dopaminergic transmission via D1 receptors and long-term potentiation (LTP) in the hippocampus [25]. Other effects of tPA on synaptic plasticity are mediated through plasmin, which converts a precursor of brain-derived neurotrophic factor (BDNF) into mature BDNF, critical for LTP in the hippocampus. tPA also promotes degradation of the ECM, which is important for synaptic remodeling and formation of new axonal varicosities [25].
tPA exerts neurotoxic effects in stroke [26]
tPA and plasmin may also target non-fibrin substances in the brain ECM. Experimental studies have shown that tPA knockout mice are protected against kainic acid-induced hippocampal damage and are resistant to focal cerebral ischemic injury [1]. Exogenous tPA exacerbated ischemic injury in both wild-type and tPA-null mice [27]. These studies were conducted in a suture model of stroke where no clot formation is involved [27], thus the effects of tPA on ischemic injury could be evaluated in the absence of its beneficial effects as a clot-dissolving agent. The damaging effects of tPA in stroke may include interaction with the NR1 subunit of the NMDA receptor [28-30], thus amplifying damaging calcium currents during ischemic excitotoxicity. Plasminogen and tPA (either endogenous or therapeutically administered) can enter the brain from the blood or be released by neurons. In the brain, tPA can convert plasminogen to plasmin, which activates protease activated receptor-1 (PAR-1). PAR-1 knockout mice showed a 68% decrease in infarction [31], evidence that the toxic effects of tPA in stroke could also be mediated through activation of PAR-1. tPA amplifies excitotoxic neuronal death by degrading laminin and disruption of pro-survival cell-matrix signaling [1]. tPA also activates MMPs, especially MMP-9 [25], which was shown to be elevated in venous blood from stroke patients that received tPA treatment [32]. In the early times after stroke, MMPs degrade the ECM and vascular basement membrane that leads to BBB breakdown, edema and hemorrhage. It should be noted that tPA, directly or through activation of MMP-9, could have beneficial effects on recovery after stroke by promoting vascular remodeling, angiogenesis, neurogenesis and axonal regeneration [25].
Alternatives and enhancements to tPA therapy
Due to the potential side effects of tPA that aggravate stroke injury and offset the benefits provided by reperfusion of the occluded artery, other thrombolytic agents were sought that would dissolve the clot without neurotoxic side effects and also have a longer half-life in circulation. Some commonly used thrombolytic agents, which all work by plasminogen activation, are summarized in Table 1. The catalytic activity of tPA is rapidly inactivated in the blood stream through binding of protein inhibitor(s), primarily plasminogen activator inhibitor-I (PAI-1). The tPA/PAI-1 complex is cleared from the circulation by the liver via a scavenging receptor, the low-density lipoprotein receptor-related protein-1 (LRP). Because of this, tPA has a half-life in the bloodstream in humans of 5 to 10 min [33]. This short half-life prevents tPA from acting on subsequent and continued vessel occlusions that occur. To date, none of the other thrombolytic agents that have undergone stroke clinical trials has demonstrated efficacy in terms of improved stroke outcome, and some studies were terminated due to increased intracranial hemorrhage. Ancrod, a natural defibrinogenating agent from Malaysian pit viper venom had favorable outcome when given within 3 hr, but not recommended beyond 3 hr period. New trials are still going on (http://www.strokecenter.org/trials/TrialDetail.aspx?tid=701). Desmoteplase, a recombinant form of the plasminogen activator from saliva of the vampire bat Desmodus rotundus, offers promise due to its longer (> 4 hr) half-life in circulation compared to tPA (5-10 min). Desmoteplase has additional advantages to tPA: it is not neurotoxic and is unaffected by ß-amyloid. Desmoteplase also antagonizes the neurotoxicity induced by vascular tPA possibly by competing with tPA for the LRP binding at the BBB and thus preventing tPA access to the brain parenchyma [34](Table 1). However, a phase III clinical trial of desmoteplase was halted since it has failed to demonstrate any beneficial effects in terms of neurological improvements and survival [33, http://www.strokecenter.org/trials/TrialDetail.aspx?tid=515].
Table 1.
Plasminogen activators used for thrombolytic therapy
| Thrombolytic agent | Source | Comments |
|---|---|---|
| t-PA (alteplase) [81, 82] | Recombinant human tPA | Half life in circulation: 5-10 min. FDA approved for stroke treatment in 1996 |
| Reteplase [83] | Recombinant non-glycosylated form of human tPA | Reteplase has a longer half-life (13-16 minutes) than tPA. Tenecteplase has higher fibrin specificity, resistant to inactivation by its endogenous inhibitor PAI-1 |
| Tenecteplase [84] | Genetically modified recombinant tPA | tPA mutants have been evaluated as thrombolytics for myocardial infarction, but their use for ischemic stroke therapy is still under investigation. |
| Anistreplase | Activated complex of streptokinase with human plasminogen | Prepared in inactive anisoylated form, deacylated in circulation to active form with half-life 2 hr, active form has 90 min half-life; used for treatment of myocardial infarction, no information on stroke usage available. |
| Streptokinase [33] | Extracellular metalloenzyme produced by ß-hemolytic streptococcus | Half-life of 20 min. Should not be used again after four days from the first dosing due to ineffectiveness and possible allergic reaction. Clinical trials terminated due to higher mortality and intracranial hemorrhage |
| Urokinase (urokinase-type plasminogen activator, uPA) [85] | Isolated from human urine, also present in blood and the ECM | Not introduced into clinical use due to higher incidence of intracranial hemorrhage |
| Ancrod (Viprinex). Fibrinogenolytic agent. | Purified fraction of Malayasin pit viper venom, cleaves fibrinogen. | Results indicate that Ancrod should not be recommended to stroke patients beyond 3 hr [86]. New trails are ongoing. |
| Desmoteplase (Desmodus salivary plasminogen activator α-1) [30, 33, 34, 87] | A plasminogen activator in the saliva of vampire bats | Half-life > 4 hours, higher fibrin specificity, unaffected by ß-amyloid, no neurotoxicity. Crosses the BBB. IV administration antagonizes the neurotoxicity of vascular tPA by competition for LRP binding and uptake at the BBB; thus limiting the tPA access to brain parenchyma. However, phase III stroke clinical trials were halted since it failed to show improvement in neurological scores and survival. |
Approaches to improve the clinical efficacy of tPA in stroke treatment (Table 2) include intra-arterial catheterization to deliver tPA directly to the clot, intra-arterial injection of tPA together with ultra-sound [35, 36] or with ultra-sound combined with either nano-bubbles or lipid microspheres or phospholipid encapsulated sulfur hexafluoride [36, 37] to aid in the breakdown of the clot. Early clinical trials have indicated that both the frequency of ultrasound and the dose of tPA need to be optimized to achieve efficient lysis without increased intracerebral hemorrhage [35]. tPA has also been combined with mechanical removal of the clot (MERCI™ retriever, a tiny corkscrew threaded through an artery to remove the clot [38], or the Penumbra device, Penumbra, Inc., San Leandro, California, to vacuum the clot bit by bit). These techniques have the drawback that they require arterial catheterization and are therefore more interventional, requiring more specialized personnel and equipment for successful implementation, and may be useful only in large artery occlusions [33]. Additional approaches are to combine tPA thrombolysis with neuroprotective drugs or agents that specifically counteract neurotoxic side effects of tPA (Table 2), although most of these studies are still in the experimental, pre-clinical stages. In one study, activated protein C (APC) was shown to inhibit MMP-9 activation and thereby reduce the hemorrhagic risk and neurotoxicity of tPA in transient brain ischemia and embolic stroke in rodents [39]. The MMP-9 inhibition by APC may have down-side effects during later times where MMP-9 is required for the repair or regeneration phase. Contrarily, recombinant human APC (rhAPC, used for sepsis treatment) alone may have hemorrhagic risk [40, 41].
Table 2.
Approaches to improve tPA efficacy
| tPA delivery directly to the site of the clot (catheterization) |
| tPA + Ultrasound [35, 37] |
| tPA + Ultrasound +gaseous lipid microspheres containing octofluropropane or sulfur hexafluoride (agitate fluid and enhance mechanical grinding of a thrombus) [36] |
| tPA + Mechanical Embolus Removal in Cerebral Ischemia (MERCI™) retriever (a tiny corkscrew threaded through an artery to remove the clot) [38] |
| tPA + clot vacuuming (PENUMBRA Inc., a California based company). |
| tPA + activated protein C (combination showed benefit in experimental studies, clinical trials not yet initiated [39]. Note: APC inhibits MMP-9 that may be needed in the recovery phase after stroke by promoting neurovascular repair through VEGF. APC may also have hemorrhagic risk by itself [40, 41]. |
| tPA + plasminogen activator inhibitor-1 attenuated tPA-mediated signal transduction without compromising its catalytic activity [88]. |
| tPA + neuroserpin (an endogenous tPA inhibitor) [25, 89, 90]. |
| tPA + CDP-choline showed benefit in an animal model of stroke [91]. A high dose of CDP-choline (1000 mg/kg) was better than tPA alone and equally as good as the combination with tPA [92]. Note: One difference between the animal studies vs clinical trials is that animal studies typically use much higher doses of CDP-choline (500-1000 mg/kg i.p. or i.v. immediately after stroke, whereas clinical trials administer 2000 mg/patient within 24 hrs of stroke symptoms [93], for a 70 kg patient receiving 2000 mg citicoline, the dose amounts to 28 mg/kg). Although a dose used in animal studies cannot extrapolate to humans, these represent vastly different dosing. Even 2000 mg/day CDP-choline (citicoline) was safe and had virtually no side effects. On a similar note, CDP-choline encapsulated in liposomes (18 mg/kg i.v.) significantly reduced infarction compared to free CDP-choline (500 mg/kg) in a rat stroke model [5, 74, 75, 94]. |
| tPA + uric acid [42, 43] (may decrease lipid peroxidation and endogenous MMP-9). |
In a recent clinical trial, stroke patients undergoing tPA therapy were shown to have elevated serum levels of endogenous active MMP-9 90 minutes after the end of tPA infusion. Infusion of uric acid immediately after tPA treatment prevented the tPA-induced increase in endogenous active MMP-9 [42, 43].
Matrix metalloproteinases
MMPs are zinc- and calcium-dependent endopeptidases comprised of 24 mammalian enzymes with distinct yet overlapping substrate specificities [44, 45]. MMPs are responsible for the turnover and degradation of the ECM and are involved in wound healing, angiogenesis, and tumor cell metastasis. The ECM is a complex structural entity surrounding and supporting cells that are found within mammalian tissues, often referred to as the connective tissue. The ECM is composed of 3 major classes of molecules: 1) structural proteins, collagen and elastin; 2) specialized proteins, e.g. fibrillin, fibronectin, and laminin; and 3) proteoglycans, composed of a protein core to which is attached long chains of repeating disaccharide units referred as glycosaminoglycans forming extremely complex high molecular weight components of the ECM. Collectively, MMPs are capable of degrading all types of extracellular matrix proteins, but also can process other pro-MMPs and a number of bioactive molecules. They are known to be involved in the cleavage of cell surface receptors, the release of apoptotic ligands (such as the FAS/APO-1/CD95 ligand) and chemokine inactivation. MMPs are also thought to play a major role in cell proliferation, migration (adhesion/dispersion), differentiation, angiogenesis, apoptosis, and host defense. Besides these functions, MMPs act on pro-inflammatory cytokines to regulate various aspects of inflammation.
The regulation of MMP expression is a complex process and tightly regulated [46]. Under normal conditions expression of MMPs are low and induced when remodeling of the extracellular matrix is necessary. Most MMPs are secreted into the extracellular space as inactive zymogens (pro-MMPs). Because activation of pro-MMPs is facilitated by active forms of other MMPs or proteinases, and their activities are controlled by endogenous inhibitors, all of which may be produced in a cell-specific manner, the net functional outcome on MMP activity likely involves interactions among various cell types. In addition to secreted MMPs, six membrane-type MMPs have been identified [47]. MMPs contain a well conserved N-terminal pre-domain signal sequence that has a conserved cysteine which chelates to the catalytic Zn2+, keeping the pro-MMP inactive. The pro-peptide of the zymogen has to be removed by other MMPs or proteases in order to activate them. The classic mechanism of MMP activation involves disruption of the interaction between the zinc molecule on the active site and cysteine in the pro-domain, leading to proteolytic cleavage of the zymogen and production of the mature active form of the enzyme. Among many proteases, plasmin, urokinase-type plasminogen activator (uPA) and tPA are known to serve as important physiological activators of MMPs [46]. In addition to enzymatic activation of MMPs, nitration or oxidation of the inhibitory cysteine can unmask the catalytic site, thereby activating MMPs in the absence of pro-domain cleavage. The activities of MMPs in tissues are further regulated by interaction with tissue inhibitors of MMPs (TIMPs), of which four have been identified. In addition to their inhibitory properties, TIMPs can have a paradoxical role: for example, TIMP-2 binds to pro-MMP-2 and has a key role in its activation. TIMP-1 has a similar role in MMP-9 activation [47].
MMPs in stroke [see for review 46]
Expression of MMPs in the adult brain is generally very low to undetectable, but many MMPs are up-regulated in the brain in response to injury. Neurons, astrocytes, oligodendrocytes, microglia and endothelial cells have all been shown to express MMPs after injury, but the brain regions and cellular sources of expression differ according to the specific MMP and the type of injury [48]. Stroke is associated with a biphasic disruption of the BBB, leading to vasogenic edema and hemorrhage. Experimental studies have indicated that BBB breakdown and hemorrhage result from expression and activation of MMPs. This should be considered within a model of the neurovascular unit [1, 49, 50] (neurons, the microvessels that supply them, endothelium, and supporting cells astrocytes, microglia, and pericytes) in which cell-specific expressions of MMPs interact to determine the final outcome.
Intracerebral injection of MMP-2 resulted in opening of the BBB and hemorrhage, which was inhibited by co-administration of TIMP-2 [48]. After approximately 24 hr, a second phase of severe BBB disruption occurs that is associated with a marked elevation in MMP-9. Mechanistic association of these two events was revealed by MMP-9 deficient mice, which displayed less BBB damage after focal ischemia [48]. While MMP-2 has been implicated in BBB opening, in vitro studies have shown it is not toxic to neurons in hippocampal slice cultures, nor does MMP-2 gene deletion provide neuroprotection in vivo. In contrast, MMP-9 is neurotoxic to hippocampal slices and cultured primary cortical neurons, and MMP-9 gene deletion is neuroprotective in vivo [48]. MMP-3 and MMP-7 may have neuroprotective functions as sheddases by cleaving the transmembrane form of Fas ligand (Fas-L) since the soluble form is less effective in inducing apoptosis compared to the membrane-bound form. MMP-3 may provide additional benefits by increasing the bioavailability of insulin-like growth factor (IGF)-1 through degradation of IGF binding proteins [48], and by cleaving pro-neuronal growth factor to its mature form.
Studies in spontaneously hypertensive rats (SHR) demonstrated significant up-regulation of MMP-9 beginning at 12 hr after permanent middle cerebral artery occlusion (MCAO), which remained elevated for up to 5 days [51]. MMP-2 did not increase during the first 24 hr after MCAO, but was significantly elevated at 5 days. These studies also showed increases in uPA beginning at 12 hr after MCAO, which reached a maximum at 5 days whereas tPA activity decreased at 12 hr and 24 hr and returned to control levels by day 5. There were significant strain differences in MMP-9 activity between SHR and Wistar-Kyoto rats (from which the SHR strain was derived) at 24 hr after permanent MCAO, with SHR showing much higher MMP-9 activity. The two strains showed no difference in uPA activity but there was significantly less tPA in SHR compared to Wistar-Kyoto rats after 24 hr permanent MCAO [51]. In mice, studies using permanent or transient MCAO showed up-regulation of MMP-9 by 1 to 2 hr after ischemia which was rapidly followed by the appearance of active MMP-9. Thus, there are major differences between species and even between strains of rats in the timing and extent of MMP-9 expression and activation.
MMPs are pleiotropic molecules
MMP-9 has been shown to have a dual role in stroke – a pathological role mediating disruption of the BBB, neuronal cell death and hemorrhage after stroke, and a healing role mediating brain regeneration and neurovascular remodeling during the later repair phase. Thus, the therapeutic window for MMP-9 inhibition may be narrow: early (day 1) MMP-9 inhibition reduced infarction at day 14. Benefit was lost when the treatment was delayed until day 3, and stroke pathology was exacerbated when administration was delayed until day 7 [52, 53]. Systemic administration of MMP-9-neutralizing monoclonal antibody significantly reduced infarct size [54]. BB-1101, a broad spectrum MMP inhibitor, attenuated BBB dysfunction during the early phase but did not have any effect on infarct volume at 2 days and had adverse effects on neurologic outcome in rats at 3 and 4 weeks after MCAO [55]. Another broad spectrum MMP inhibitor, BB94, increased apoptosis and hemorrhage size after collagenase-induced intracerebral hemorrhage in mouse [56]. Therefore, it is important to test both the short-term beneficial actions and the possible long-term detrimental actions of MMP inhibitors. These studies all suggest that MMP inhibition may not be a viable therapeutic target since it could have either a beneficial or detrimental effect depending on the timing of the treatment in relation to the stage of brain injury.
TIMPs in stroke [see for review 48]
TIMPs are a family of 20-29 kDa secreted proteins that inhibit the active forms of MMPs through high affinity (10-9 to 10-12 M) non-covalent binding to the MMP catalytic domain in 1:1 ratios. TIMP-1 expression is induced in neurons and astrocytes after transient global ischemia. As a potent MMP-9 inhibitor, TIMP-1 may have a neuroprotective function, at least in the early reperfusion times after injury when MMP-9 is deleterious. This was confirmed using TIMP-1 deficient mice that showed increased MMP-9 activity, BBB injury, and infarct volume 24 hr after 3 hr transient focal cerebral ischemia by suture occlusion [57]. TIMP-1 activity could have negative effects, though, in later times where there is evidence that MMP-9 contributes to the reconstructive phase. TIMP-1 was also shown to protect cultured hippocampal neurons against glutamate-induced excitotoxicity; protection was attributed to a decrease in glutamate-induced Ca2+ influx. While synthetic MMP inhibitors did not replicate this neuroprotection, removal of the MMP inhibitory function of TIMP-1 by point mutation did ablate its neuroprotective effect [58].
TIMP-3 is unique among the TIMP family in that it is bound to the extracellular matrix, suggesting that its action is confined to the cell surface. TIMP-3 is a potent inhibitor of TNF-α converting enzyme (TACE) and of MMP-3 and MMP-7. TIMP-3 expression in adult brain is low to undetectable, but is strongly up-regulated after focal cerebral ischemia, particularly in cortical neurons undergoing apoptotic death [48]. In addition to cleavage of TNF-α, TACE cleaves the TNF-α death-domain containing receptor and releases it from the cell surface. Thus TIMP-3 can mediate apoptosis through inhibition of TACE, MMP-3 and MMP-7, stabilizing the TNF-α receptor and Fas-L. Recent experiments, demonstrating a proapoptotic role for TIMP-3, showed that TIMP-3 null mice had reduced lesion volume following a mild (30 min) transient focal cerebral ischemia, in which caspase-dependent apoptotic death predominates. The protective effect of TIMP-3 deletion was obscured by a more severe (90 min) focal ischemia [59], which is less sensitive to caspase inhibition and involves more of necrotic cell death mechanisms.
Cytokines and Stroke
Cytokines are low molecular weight, soluble proteins that are produced in response to an antigen and were originally described as mediators for regulating the innate and adaptive immune systems. Cytokines thus serve as chemical messengers and include tumor necrosis factors, interleukins, interferons, chemokines, and growth factors. Cytokine biology is particularly complex: one cytokine can act on a number of different cell types (pleiotropic) rather than one type; the same cytokine regulates a number of different functions (multifunctional); and a number of different cytokines can carry out the same function (redundant). The redundancy is due to the utilization of shared key components of intracellular signaling pathways.
There are substantial data from both animal models and clinical studies that TNF-α is up-regulated after stroke. TNF-α is initially synthesized as a 26 kDa membrane bound protein which is cleaved by the Zn-metalloprotease TACE to form 17 kDa mature, soluble TNF-α [60]. TNF-α mediates its effects by binding to two receptors (p55 and p75), both of which are up-regulated in the ischemic brain [61, 62]. The majority of biological effects of TNF-α, including cytotoxicity and inflammatory responses, are mediated through the p55 receptor [61, 62] that contains the death domain. The function of p75 remains obscure, although studies have indicated it may participate in protective signaling following injury [63]. Some of the effects caused by TNF-α in stroke are due to inhibition of glutamate uptake by astrocytes; induction of intracellular adhesion molecule (ICAM-1), increased MMP expression that leads to disruption of the blood-brain-barrier and invasion of inflammatory leukocytes [60]. Although the role of TNF-α in stroke pathology remains controversial, the majority of studies support its deleterious effects in the early phase of stroke injury [64]. The actions of TNF-α can be blocked using specific TNF-α neutralizing antibody or TNF-α binding protein to bind TNF-α and prevent it from interacting with its receptors [65]. Administrations of TNF-α antibody [66] or TNF-α binding protein [66-68] that attenuate the acute phase of TNF-α following cerebral ischemia have consistently demonstrated beneficial effects [61, 65, 68]. Transgenic mice deficient in TNF-α [69] showed dramatic reduction in infarction compared to wild-type mice, and infusion of TNF-α exacerbated infarct volume in focal cerebral ischemia [67].
The over-expression of TNF-α in the early stage after brain injury may be deleterious, whereas later phase expression may facilitate recovery [61, 65]. In support of this proof-of-concept, studies showed that TNF-α null mice had smaller infarcts after MCAO and 1 day reperfusion compared to wild type mice, further evidence that elevated levels of TNF-α during the early reperfusion time are deleterious. TNF-α null mice subjected to controlled cortical impact injury showed fewer neurological deficits compared to wild-type mice at 7 days post-injury. However, wild type mice had better recovery from brain injury by 2-3 weeks post-injury, whereas TNF-α null mice had poor recovery and exhibited greater neurological deficits compared to wild type mice at 4 weeks post-injury [70]. This duality of TNF-α effects (harmful in the early stage after brain injury yet contributing to later reconstruction) parallels the effects of MMPs and could be due to TNF-α regulation of MMPs [68, 71]. This phenomenon has not been investigated in stroke models: studies thus far have not examined the effect of suppressing TNF-α signaling during the later, recovery phase after tMCAO.
CONCLUSIONS
tPA is the only agent that has been shown to improve stroke outcome in clinical trials, despite the many clinical trials that have been conducted. To-date 1,026 drugs have been tested in various animal models, of which 114 underwent clinical evaluation [72]. Preclinical evaluation (animal studies) fostered high expectations for clinical efficacy, however, none of the agents have conclusively demonstrated benefit in stroke clinical trials [73]. CDP-choline, a precursor for synthesis of PC, has been approved for clinical stroke treatment in Europe and Japan. US stroke trials initially showed improvement with CDP-choline treatment, but subsequent trials failed to reproduce this benefit and CDP-choline was deemed safe but ineffective. These results could be due to the long time-to-treatment window (24 hr, average administration time of 13.5 hrs) and oral administration (it is unclear why this route was chosen; no other agent has been administered orally for stroke patients) of CDP-choline in clinical trials [74-76].
The latest failed stroke clinical trial evaluating the nitrone spin trap agent NXY-059 has triggered a major debate concerning the whole approach to development and assessment of potential neuroprotective agents in pre-clinical research. The major recommendations from the National Institutes of Neurological Disorders and Stroke (NINDS) stroke progress review group emphasized a shift from a neurocentric focus to a more composite approach dealing with all the brain cells, with particular attention to the neurovascular unit consisting of neurons, the microvessels that supply them, endothelium, and supporting cells (astrocytes, microglia, and pericytes). Thus at this juncture, while preserving neuronal function after stroke is the ultimate goal, it is time to move away from a concept that focuses only on neuronal injury, a reasonable approach since only <5% brain cells are neurons. Stroke affects non-neuronal cells (astrocytes and glia) that support neurons and axons of neurons that transmit signals. However we should also be realistic in trying to achieve these strategies: for example, can we conceivably develop tissue culture systems that reliably predict responses of the in vivo structure? Even if we achieve this, including the complexity of interactions between all the different cell types [77], how do we also integrate the contribution from blood circulation? This also brings us to self-examine some hard questions recently posed, such as 1) are current rodent stroke models relevant to the clinical situation? (appropriate animal choice such as aged, diabetic, hypertensive, male, female; is the suture model relevant to the clinical situation?) 2) is there any evidence that neuroprotectants reach their target in patients, i.e., the brain [78]? A clinically relevant delivery system for the drug should be used in animal models, i.e., avoid pre-treatment (before induction of stroke), intracisternal or intraventricular injections, which are inappropriate for patient treatment. Experimental data should include evidence that the agent reaches the target brain tissue. These strict selection criteria may largely limit the number of agents that will eventually be brought to stroke clinical trails, but certainly minimize the frustrations and disappointments such as the NXY-059 clinical trials [78-80].
Acknowledgments
This work was supported by grants from NIH/NINDS (NS42008), American Heart Association Greater Midwest Affiliate Grant-in-Aid (0655757Z), UW-School of Medicine and Public Health, UW-Graduate school and UW-Neurological Surgery Department and laboratory resources provided by William S. Middleton VA Hospital (to RMA).
Abbreviations
- ApoB100
apolipoprotein B-100
- BBB
blood-brain barrier
- BDNF
brain-derived neurotrophic factor
- CDP-choline
Cytidine-5’-diphosphocholine (citicoline)
- CNS
central nervous system
- ECM
extracellular matrix
- IGF
insulin-like growth factor
- IL-1
interleukin-1
- LDL
low-density lipoprotein
- Lp-PLA2
lipoprotein-phospholipase A2
- LRP
low-density lipoprotein receptor-related protein-1
- LTP
long-term potentiation
- MCAO
Middle cerebral artery occlusion
- MMPs
matrix metalloproteinases
- OxLDL
oxidized low-density lipoprotein
- OxPC
Oxidized phosphatidylcholine
- PA
Plasminogen activator
- PAI-1
plasminogen activator inhibitor-I
- PAR-1
protease activated receptor-1
- PC
Phosphatidylcholine
- ROS
reactive oxygen species
- Serpin
serine-protease inhibitor
- TACE
TNF-α converting enzyme
- TIMP
tissue inhibitors of MMP
- TNF-α
tumor necrosis factor-α
- tPA
tissue-type plasminogen activator
- uPA
Urokinase-type plasminogen activator
- VEGF
vascular endothelial growth factor
Footnotes
Conflict of interest The authors have no financial conflict of interest
References
- 1.Lo EH, Broderick JP, Moskowitz MA. Stroke. 2004;35:354–356. doi: 10.1161/01.STR.0000115164.80010.8A. [DOI] [PubMed] [Google Scholar]
- 2.Young AR, Ali C, Duretete A, Vivien D. J Neurochem. 2007;103:1302–1309. doi: 10.1111/j.1471-4159.2007.04866.x. [DOI] [PubMed] [Google Scholar]
- 3.Adibhatla RM, Hatcher JF. Free Radic Biol Med. 2006;40:376–387. doi: 10.1016/j.freeradbiomed.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 4.Adibhatla RM, Hatcher JF, Dempsey RJ. AAPS J. 2006;8:E314–E321. doi: 10.1007/BF02854902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adibhatla RM, Hatcher JF, Larsen EC, Chen X, Sun D, Tsao F. J Biol Chem. 2006;281:6718–6725. doi: 10.1074/jbc.M512112200. [DOI] [PubMed] [Google Scholar]
- 6.Mehta SL, Manhas N, Raghubir R. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Emsley HCA, Tyrrell PJ. J Cereb Blood Flow Metab. 2002;22:1399–1419. doi: 10.1097/01.WCB.0000037880.62590.28. [DOI] [PubMed] [Google Scholar]
- 8.Hansson GK, Libby P. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 9.McColl BW, Rothwell NJ, Allan SM. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elkind MS. Neurologist. 2006;12:140–148. doi: 10.1097/01.nrl.0000215789.70804.b0. [DOI] [PubMed] [Google Scholar]
- 11.Maxfield FR, Tabas I. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 12.Nicolo D, Varadhachary AS, Monestier M. Front Biosci. 2007;12:2171–2182. doi: 10.2741/2220. [DOI] [PubMed] [Google Scholar]
- 13.Boullier A, Friedman P, Harkewicz R, Hartvigsen K, Green SR, Almazan F, Dennis EA, Steinberg D, Witztum JL, Quehenberger O. J Lipid Res. 2005;46:969–976. doi: 10.1194/jlr.M400496-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Adibhatla RM, Hatcher JF. In: Lipids in Health and Disease. Quinn PJ, Wang X, editors. Springer-Verlag; New York: [Google Scholar]; Subcell Biochem. 2008;48 in press. [Google Scholar]
- 15.Mannheim D, Herrmann J, Versari D, Gossl M, Meyer FB, McConnell JP, Lerman LO, Lerman A. Stroke. 2008;39:1448–1455. doi: 10.1161/STROKEAHA.107.503193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoll G, Bendszus M. Stroke. 2006;37:1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- 17.Kadl A, Bochkov VN, Huber J, Leitinger N. Antioxid Redox Signal. 2004;6:311–320. doi: 10.1089/152308604322899378. [DOI] [PubMed] [Google Scholar]
- 18.Qin J, Goswami R, Balabanov R, Dawson G. J Neurosci Res. 2007;85:977–984. doi: 10.1002/jnr.21206. [DOI] [PubMed] [Google Scholar]
- 19.Bratton DL, Henson PM. Nat Med. 2005;11:26–27. doi: 10.1038/nm0105-26. [DOI] [PubMed] [Google Scholar]
- 20.Chang M-K, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, Witztum JL. J Exp Med. 2004;200:1359–1370. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao S, Zhang RL, Greenberg ME, Sun M, Chen X, Levison BS, Salomon RG, Hazen SL. J Biol Chem. 2006;281:31298–31308. doi: 10.1074/jbc.M604039200. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein LB. Circulation. 2007;116:1504–1514. doi: 10.1161/CIRCULATIONAHA.106.670885. [DOI] [PubMed] [Google Scholar]
- 23.Miranda E, Lomas DA. Cell Mol Life Sci. 2006;63:709–722. doi: 10.1007/s00018-005-5077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yepes M, Larence DA. Cardiovasc Med. 2004;14:173–180. doi: 10.1016/j.tcm.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Benarroch EE. Neurology. 2007;69:799–802. doi: 10.1212/01.wnl.0000269668.08747.78. [DOI] [PubMed] [Google Scholar]
- 26.Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. J Cereb Blood Flow Metab. 2004;24:945–963. doi: 10.1097/01.WCB.0000137868.50767.E8. [DOI] [PubMed] [Google Scholar]
- 27.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 28.Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 29.Samson AL, Medcalf RL. Neuron. 2006;50:673–678. doi: 10.1016/j.neuron.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Atalaya JP, Roussel BD, Levrat D, Parcq J, Nicole O, Hommet Y, Benchenane K, Castel H, Leprince J, To Van D, Bureau R, Rault S, Vaudry H, Petersen K-U, Santos JS-dO, Ali C, Vivien D. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.1014. [DOI] [PubMed] [Google Scholar]
- 31.Junge CE, Sugawara T, Mannaioni G, Alagarsamy S, Conn PJ, Brat DJ, Chan PH, Traynelis SF. Proc Natl Acad Sci USA. 2003;100:13019–13024. doi: 10.1073/pnas.2235594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ning M, Furie KL, Koroshetz WJ, Lee H, Barron M, Lederer M, Wang X, Zhu M, Sorensen AG, Lo EH, Kelly PJ. Neurology. 2006;66:1550–1555. doi: 10.1212/01.wnl.0000216133.98416.b4. [DOI] [PubMed] [Google Scholar]
- 33.Gravanis I, Tsirka SE. Expert Opin Ther Targets. 2008;12:159–170. doi: 10.1517/14728222.12.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez-Atalaya JP, Roussel BD, Ali C, Maubert E, Petersen K-U, Berezowski V, Cecchelli R, Orset C, Vivien D. Stroke. 2007;38:1036–1043. doi: 10.1161/01.STR.0000258100.04923.84. [DOI] [PubMed] [Google Scholar]
- 35.Shaw GJ, Meunier JM, Lindsell CJ, Holland CK. Stroke. 2008;39:547. [Google Scholar]
- 36.Tsivgoulis G, Alexandrov AV. Neurotherapeutics. 2007;4:420–427. doi: 10.1016/j.nurt.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molina CA, Ribo M, Rubiera M, Montaner J, Santamarina E, Delgado-Mederos R, Arenillas JF, Huertas R, Purroy F, Delgado P, Alvarez-Sabin J. Stroke. 2006;37:425–429. doi: 10.1161/01.STR.0000199064.94588.39. [DOI] [PubMed] [Google Scholar]
- 38.Smith WS for the Multi, M.I. AJNR Am J Neuroradiol. 2006;27:1177–1182. [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernandez JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- 40.Haley M, Cui X, Minneci PC, Deans KJ, Natanson C, Eichacker PQ. Am J Med Sci. 2004;328:215–219. doi: 10.1097/00000441-200410000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Haley M, Cui X, Minneci PC, Deans KJ, Natanson CP, Eichacker PQ. Therapy. 2004;1:123–129. [Google Scholar]
- 42.Amaro S, Gomez-Choco M, Urra X, Cervera A, Obach V. Stroke. 2008;39:594. doi: 10.1111/j.1468-1331.2007.02042.x. [DOI] [PubMed] [Google Scholar]
- 43.Amaro S, Planas AM, Chamorro A. Expert Rev Neurother. 2008;8:259–270. doi: 10.1586/14737175.8.2.259. [DOI] [PubMed] [Google Scholar]
- 44.Parks WC, Wilson CL, Lopez-Boado YS. Nat Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 45.Yong VW. Nature Reviews Neuroscience. 2005;6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- 46.Gasche Y, Soccal PM, Kanemitsu M, Copin JC. Front Biosci. 2006;11:1289–1301. doi: 10.2741/1883. [DOI] [PubMed] [Google Scholar]
- 47.Ethell IM, Ethell DW. J Neurosci Res. 2007;85:2813–2823. doi: 10.1002/jnr.21273. [DOI] [PubMed] [Google Scholar]
- 48.Cunningham LA, Wetzel M, Rosenberg GA. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- 49.Lo EH, Dalkara T, Moskowitz MA. Nat Rev Neurosci. 2003;4:399–414. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 50.del Zoppo GJ. N Engl J Med. 2006;354:553–555. doi: 10.1056/NEJMp058312. [DOI] [PubMed] [Google Scholar]
- 51.Rosenberg GA, Navratil M, Barone F, Feuerstein G. J Cereb Blood Flow Metab. 1996;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 52.Zhao B-Q, Wang SB, Kim H-Y, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- 53.Zlokovic BV. Nat Med. 2006;12:390–391. doi: 10.1038/nm0406-390. [DOI] [PubMed] [Google Scholar]
- 54.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC, Dawson VL. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 55.Sood RR, Taheri S, Candelario-Jalil E, Estrada EY, Rosenberg GA. J Cereb Blood Flow Metab. 2007;28:431–438. doi: 10.1038/sj.jcbfm.9600534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grossetete M, Rosenberg GA. J Cereb Blood Flow Metab. 2008;28:752–763. doi: 10.1038/sj.jcbfm.9600572. [DOI] [PubMed] [Google Scholar]
- 57.Burggraf D, Burk J, Schuhmacher S. Stroke. 2008;39:658. [Google Scholar]
- 58.Tan HK, Heywood D, Ralph GS, Bienemann A, Baker AH, Uney JB. Mol Cell Neurosci. 2003;22:98–106. doi: 10.1016/s1044-7431(02)00024-6. [DOI] [PubMed] [Google Scholar]
- 59.Wetzel M, Li L, Harms KM, Roitbak T, Ventura PB, Rosenberg GA, Khokha R, Cunningham LA. Cell Death Differ. 2008;15:143–151. doi: 10.1038/sj.cdd.4402246. [DOI] [PubMed] [Google Scholar]
- 60.Lovering F, Zhang Y. Curr Drug Targets CNS Neurol Disord. 2005;4:161–168. doi: 10.2174/1568007053544147. [DOI] [PubMed] [Google Scholar]
- 61.Wang CX, Shuaib A. Prog Neurobiol. 2002;67:161–172. doi: 10.1016/s0301-0082(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 62.Botchkina GI, Meistrell ME, Botchkina IL, Tracey KJ. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- 63.Shen Y, Li R, Shiosaki K. J Biol Chem. 1997;272:3550–3553. [PubMed] [Google Scholar]
- 64.Adibhatla RM, Hatcher JF. Front Biosci. 2008;13:1250–1270. doi: 10.2741/2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shohami E, Ginis I, Hallenbeck JM. Cytokine Growth Fact Rev. 1999;10:119–130. doi: 10.1016/s1359-6101(99)00008-8. [DOI] [PubMed] [Google Scholar]
- 66.Lavine SD, Hofman FM, Zlokovic BV. J Cereb Blood Flow Metab. 1998;18:52–58. doi: 10.1097/00004647-199801000-00005. [DOI] [PubMed] [Google Scholar]
- 67.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette R, Lysko PG, Feuerstein GZ. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 68.Hallenbeck JM. Nat Med. 2002;8:1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 69.Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Cell Death Differ. 2001;8:676–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- 70.Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Proc Natl Acad Sci USA. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hosomi N, Ban CR, Naya T, Takahashi T, Guo P, Song X-yR, Kohno M. J Cereb Blood Flow Metab. 2005;25:959–967. doi: 10.1038/sj.jcbfm.9600086. [DOI] [PubMed] [Google Scholar]
- 72.O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 73.Green AR. Br J Pharmacol. 2007;153:S325–338. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adibhatla RM, Hatcher JF. J Neurosci Res. 2002;70:133–139. doi: 10.1002/jnr.10403. [DOI] [PubMed] [Google Scholar]
- 75.Adibhatla RM, Hatcher JF. Neurochem Res. 2005;30:15–23. doi: 10.1007/s11064-004-9681-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adibhatla RM, Hatcher JF. Future Lipidol. 2007;2:403–422. doi: 10.2217/17460875.2.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hallenbeck J, del Zoppo G, Jacobs T, Hakim A, Goldman S, Utz U, Hasan A. Stroke. 2006;37:3035–3042. doi: 10.1161/01.STR.0000248836.82538.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Donnan GA. Stroke. 2008;39:242–248. doi: 10.1161/STROKEAHA.107.493296. [DOI] [PubMed] [Google Scholar]
- 79.Savitz SI. Exp Neurol. 2007;205:20–25. doi: 10.1016/j.expneurol.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 80.Savitz SI, Fisher M. Ann Neurol. 2007;61:396–402. doi: 10.1002/ana.21127. [DOI] [PubMed] [Google Scholar]
- 81.Group, N.S.S. N Eng J Med. 1995;333:1581–1587. [Google Scholar]
- 82.De Keyser J, Gdovinova Z, Uyttenboogaart M, Vroomen PC, Luijckx GJ. Stroke. 2007;38:2612–2618. doi: 10.1161/STROKEAHA.106.480566. [DOI] [PubMed] [Google Scholar]
- 83.Qureshi AI, Ali Z, Suri MF, Kim SH, Shatla AA, Ringer AJ, Lopes DK, Guterman LR, Hopkins LN. Neurosurgery. 2001;49:41–48. doi: 10.1097/00006123-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 84.Haley EC, Jr, Lyden PD, Johnston KC, Hemmen TM. Stroke. 2005;36:607–612. doi: 10.1161/01.STR.0000154872.73240.e9. [DOI] [PubMed] [Google Scholar]
- 85.Furlan A, Higashida R, Wechsler L, Gent M, Rowley H, Kase C, Pessin M, Ahuja A, Callahan F, Clark WM, Silver F, Rivera F for the, P.I. JAMA. 1999;282:2003–2011. doi: 10.1001/jama.282.21.2003. [DOI] [PubMed] [Google Scholar]
- 86.Hennerici MG, Kay R, Bogousslavsky J, Lenzi GL, Verstraete M, Orgogozo JM. Lancet. 2006;368:1871–1878. doi: 10.1016/S0140-6736(06)69776-6. [DOI] [PubMed] [Google Scholar]
- 87.Hacke W, Albers G, Al-Rawi Y, Bogousslavsky J, Davalos A, Eliasziw M, Fischer M, Furlan A, Kaste M, Lees KR, Soehngen M, Warach S for The, D.S.G. Stroke. 2005;36:66–73. doi: 10.1161/01.STR.0000149938.08731.2c. [DOI] [PubMed] [Google Scholar]
- 88.Armstead WM, Nassar T, Akkawi S, Smith DH, Chen X-H, Cines DB, Higazi AA-R. Nat Neurosci. 2006;9:1150–1155. doi: 10.1038/nn1757. [DOI] [PubMed] [Google Scholar]
- 89.Galliciotti G, Sonderegger P. Front Biosci. 2006;11:33–45. doi: 10.2741/1778. [DOI] [PubMed] [Google Scholar]
- 90.Zhang Z, Zhang L, Yepes M, Jiang Q, Li Q, Arniego P, Coleman TA, Lawrence DA, Chopp M. Circulation. 2002;106:740–745. doi: 10.1161/01.cir.0000023942.10849.41. [DOI] [PubMed] [Google Scholar]
- 91.Andersen M, Overgaard K, Meden P, Boysen G. Stroke. 1999;30:1464–1470. doi: 10.1161/01.str.30.7.1464. [DOI] [PubMed] [Google Scholar]
- 92.Gutierrez M, Alonso de Lecinana M, Roda JM, Alvarez-Grech J, Carceller F, Diez-Tejedor E. Stroke. 2008;39:676. [Google Scholar]
- 93.Davalos A. Stroke. 2008;39:e54. [Google Scholar]
- 94.Adibhatla RM, Hatcher JF, Tureyen K. Brain Res. 2005;1058:193–197. doi: 10.1016/j.brainres.2005.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]