

Abstract
The loss of DNA mismatch repair (MMR) is responsible for hereditary nonpolyposis colorectal cancer and a subset of sporadic tumors. Acquired resistance or tolerance to some anti-cancer drugs occurs when MMR function is impaired. 5-Fluorouracil (FU), an anti-cancer drug used in the treatment of advanced colorectal and other cancers, and its metabolites are incorporated into RNA and DNA and inhibit thymidylate synthase resulting in depletion of dTTP and incorporation in DNA of uracil. Although the MMR deficiency has been implicated in tolerance to FU, the mechanism of cell killing remains unclear. Here, we examine the cellular response to fluorodeoxyuridine (FdU) and the role of the MMR system. After brief exposure of cells to low doses of FdU, MMR mediates DNA damage signaling during S-phase and triggers arrest in G2/M in the first cell cycle in a manner requiring MutSα, MutLα, and DNA replication. Cell cycle arrest is mediated by ATR kinase and results in phosphorylation of Chk1 and SMC1. MutSα binds FdU:G mispairs in vitro consistent with its being a DNA damage sensor. Prolonged treatment with FdU results in an irreversible arrest in G2 that is independent of MMR status and leads to the accumulation of DNA lesions that are targeted by the base excision repair (BER) pathway. Thus, MMR can act as a direct sensor of FdU-mediated DNA lesions eliciting cell cycle arrest via the ATR/Chk1 pathway. However, at higher levels of damage, other damage surveillance pathways such as BER also play important roles.
Keywords: MISMATCH REPAIR, 5-FLUOURACIL, DNA DAMAGE, ATR, COLORECTAL CANCER
DNA mismatch repair (MMR) is a highly conserved repair pathway that plays an important role in the detection and correction of DNA mismatches created during replication and recombination. Inactivating mutations in MMR genes cause a greatly increased rate of spontaneous mutation and are the underlying defect in hereditary nonpolyposis colorectal cancer (HNPCC). In addition, MMR defects are associated with a significant proportion of sporadic cancers. Two key MMR proteins, MutS and MutL in prokaryotes and their eukaryotic counterparts, mediate a number of functions in maintaining genome integrity including the correction of DNA biosynthetic errors, suppression of illegitimate recombination, and participation in the cellular response to certain types of DNA damage [Schofield and Hsieh, 2003; Kunkel and Erie, 2005; Iyer et al., 2006]. In its role in correcting mispaired bases arising during replication, MutS targets DNA mismatches and together with MutL, licenses the excision of the newly synthesized strand in the vicinity of the mismatch. DNA synthesis and ligation complete gap repair.
In mammalian cells, the MMR system is also implicated in the cellular response to DNA damage resulting from exposure to SN1 DNA methylating agents, 6-thioguanine, fluoropyrimidines (FPs), cisplatin, reactive oxygen species, ultraviolet light, and some environmental carcinogens [Iyer et al., 2006]. Cell killing mediated by SN1 alkylators such as temozolomide that is commonly used in cancer chemotherapy requires functional MMR proteins. Tolerance or resistance to drug treatment occurs when MMR genes are inactivated. These DNA alkylators produce several covalent DNA modifications, but it is the O6-methylguanine lesion that triggers cell cycle arrest in G2 and apoptosis in an MMR-dependent manner. The checkpoint and apoptotic response is the result of the activation of ATR, a phosphoinositide 3-kinase-related kinase (PIKK), and the subsequent activation of a downstream target of ATR, the checkpoint kinase Chk1. Phosphorylation of p53 in response to O6-methylguanine is also dependent on functional MutSα and MutLα MMR proteins [Duckett et al., 1999].
The FP anti-metabolite 5-fluoro-2′-uridine (FU) is an anti-cancer drug widely used in the treatment of advanced colorectal cancer [Longley et al., 2003]. The mechanism of cell killing by FPs is unclear and is complicated by the fact that FPs are usually administered in the clinical setting in combination with other DNA damaging agents that disrupt replication such as irinotecan, a topoisomerase 1 inhibitor, and oxaliplatin. FP and its metabolites are also incorporated into both RNA and DNA and affect multiple metabolic pathways. For example, FdUMP, a metabolite of FP, inhibits thymidylate synthase (TS), the enzyme that utilizes dUMP as substrate in the only biosynthetic pathway leading to the creation of dTMP. Correspondingly, cells treated with FU exhibit elevated dUTP levels and deoxynucleoside triphosphate precursor pool imbalances. Thus, FP-induced cytotoxicity could be a consequence of DNA fragmentation resulting from extensive excision of uracil in DNA by the base excision repair (BER) pathway. However, mammalian cells lacking Ung, a major uracil-DNA glycosylase, do not show altered sensitivity to FU cell killing [Andersen et al., 2005]. Yet another mechanism for FP cytotoxicity could derive from the consequences of direct incorporation of 5-fluoro-2′-deoxyuridine (FdU), a metabolite of FP, into DNA. FdU upon replication leads to FdU:A and, in principle, occasional FdU:G mispairs (see discussion in Meyers et al. [2004]). Recently, the uracil glycosylase Smug1 was shown to excise FdU from DNA and afford protection from cell killing by FPs in strong support of a pathway for cell killing stemming from direct incorporation of FdU into DNA [An et al., 2007].
The MMR system has been implicated in cell killing by FPs as MMR-deficient cells exhibit increased survival after treatment with FdU [Meyers et al., 2004]. Arrest in G2 is observed within the first cell cycle in MMR-proficient cells, but not in cells missing either MutSα or MutLα [Carethers et al., 1999; Meyers et al., 2003, 2005]. Treatment with a specific inhibitor of TS, Tomudex, elicits a G2 arrest regardless of MMR status suggesting that the contribution of MMR to DNA damage signaling is via incorporation of FPs into DNA as opposed to the inhibition of TS and the resulting imbalance in nucleotide pools. However, the molecular mechanism of MMR-dependent DNA damage signaling triggered by FdU remains unclear. In the clinic, resistance to FU treatment occurs frequently. Since 15% of colorectal tumors exhibit microsatellite instability (MSI) and are presumed to be MMR-deficient, there is concern that this subset of patients might be particularly resistant to FU treatment.
In this study, we examine the cellular response to FdU and assess the role of the MMR system. We ask whether MMR proteins may act as sensors of FdU damage eliciting cell cycle arrest via activation of the ATR/Chk1 pathway.
MATERIALS AND METHODS
CELL LINES, CELL CULTURES AND REAGENTS
HCT116 and HeLaS3 cells were purchased from American Type Culture Collection (Manassas, VA). HCT116 3−6 was kindly provided by Richard Boland (Baylor University Medical Center). Parental HCT116 human colon carcinoma cells have a hemizygous nonsense mutation in the hMLH1 gene located on chromosome 3. HCT116 3−6 was created by microcell transfer of a single normal human chromosome 3 into HCT116 cells [Koi et al., 1994]. HEC59 and HEC59 2−4 cells were kindly provided by Thomas A. Kunkel (NIEHS, National Institutes of Health, Research Triangle Park, NC), in which chromosome 2 (containing wild-type MSH2) was introduced into MSH2-deficient HEC59 human endometrial carcinoma cells to create HEC59 2−4 cells [Umer et al., 1997]. HCT116, HCT116 3−6 and HeLaS3 cells were maintained in DMEM (Mediatech, Herndon, VA) containing 10% FBS (Invitrogen, Carlsbad, CA) in 5% CO2 at 37°C. HEC59 and HEC59 2−4 cells were maintained in DMEM/F12 (1:1, Mediatech) containing 10% FBS, and the HEC59 2−4 cell line was maintained under 400 μg/ml G418 (Sigma, St. Louis, MO) selection. U2OS cell lines conditionally overexpressing ATR-wt (GW33) or kinase-deficient (GK41) protein provided by Paul Nghiem (Massachusetts General Hospital, Boston, MA) were maintained in DMEM supplemented with 10% FBS, 200 μg/ml G418, and 200 μg/ml Hygromycin B (Calbiochem) [Nghiem et al., 2002]. Induction of ATR-wt and ATR-kd was accomplished by supplementing the growth medium with 1 μg/ml doxycycline (Sigma) for 48 h. FdU was obtained from Sigma Chemical Co. DNA synthesis was inhibited by treatment with 5 μM aphidicolin (Sigma).
ANTIBODIES AND WESTERN BLOT ANALYSES
The antibodies used in this study were as follows: anti-Phospho SMC1 (S966) and anti-SMC1 (Bethyl Laboratories, Montgomery, TX), anti-Phospho CHK1 (S317) and anti-CHK1 (AbCam, Cambridge, MA), anti-ATR (AbCam), and anti-MLH1 (BD Pharmingen). Western blotting analyses were carried as previously described with ECLplus chemiluminescent reagents (GE Healthcare) or Odyssey Infrared Imaging System using IRDye 680 or IRDye 800CW secondary antibodies (Odyssey 2.1 LI-COR Biosciences, Lincoln, NE) [Yoshioka et al., 2006].
RECOMBINANT MUTSα
Heterodimeric human MutSα (MSH2-His6-MSH6) was produced by coinfection (MOI = 10) of High Five (Invitrogen) maintained in Express Five (Invitrogen) with 18 mM glutamine (Invitrogen) at 27°C. After 44 h, infected High Five cells were harvested, washed twice with PBS, resuspended in buffer A (25 mM HEPES-KOH, pH 7.5, 500 mM KCl, 20 mM imidazole, and 10% glycerol) containing 0.5% NP-40 and Complete EDTA-free protease inhibitor (Roche Molecular Biochemicals), disrupted by passage through a 25-gauge needle, and centrifuged at 27,000g for 30 min. The supernatants were loaded on a Nickel NTA Superflow column (Qiagen), washed with buffer A (300 mM KCl) and eluted with a linear gradient of 20−200 mM imidazole in Buffer A. Pooled MutSα-containing fractions were loaded onto a heparin-Sepharose column (GE Healthcare) equilibrated in 150 mM KCl buffer B (25 mM HEPES-KOH, pH 7.5, and 10% glycerol). The column was washed extensively in 150 mM KCl Buffer B, and MutSα was eluted in a linear gradient to 1.0M KCl in Buffer B. MutSα-containing fractions were loaded on a HiTrap Q HP 5ml (GE Healthcare) equilibrated in 150 mM KCl Buffer B eluted with a linear gradient to 1.0M KCl in Buffer B. Proteins eluted at 250−300 mM KCl (MutSα). Purified MutSα was judged to be 95% pure or greater by Coomassie staining after SDS-gel electrophoresis, and stored at 4°C. MutSα was used within 1 week of purification. Concentration of MutSα was determined using the theoretical molar extinction coefficient (ε280 = 195,180 M−1 cm−1 for heterodimeric MutSα).
DNA BINDING ASSAYS
MutSα was incubated with 0.5 nM 5’-32P-labeled DNA in binding buffer (25 mM HEPES, pH 7.5, 150 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol, 0.1% NP-40 and 5% glycerol) in 20 μl total volume. After 15 min at 25°C samples were loaded onto 4% native polyacrylamide gels in 0.5 × TBE. After electrophoresis, gels were dried and scanned on a Fuji BAS-2500 PhosphorImager for quantitation. Sequences of oligonucleotides (Midland-PAGE purified) used for duplex DNA substrates were as follows: (X denotes the guanine in G:C, G:FdU and G:T, or adenine in A:FdU) 5′-GTG GAT CCC CCG GGC TGC XGG AAT TCG ATA TCA AG-3′; (Y denotes C, T or FdU) 5′-CTT GAT ATC GAA TTC CYG CAG CCC GGG GGA TCC AC-3′. Annealed duplexes were purified by native PAGE.
CELL CYCLE ANALYSES
Cell cycle distribution was determined by propidium iodide staining and flow cytometry (FCM). Briefly, HCT116 and HCT116 3−6 cells were synchronized in DMEM containing 2 mM hydroxyurea (HU; Sigma) for 14 h or synchronized by serum starvation as described [Meyers et al., 2005]. The cells were then incubated in fresh medium with or without 0.25 μM FdU. Cells were harvested and analyzed by FCM (Becton Dickinson, Franklin Lakes, NJ). Cell cycle populations were quantified using ModFit LT version 3.0 software (Verity Software House, Topsham, ME).
CLONOGENIC SURVIVAL ASSAYS
Clonogenic survival assays after treatment with FdU were performed as follows. Cells were serially diluted (range, 2 × 102−2 × 104 cells/6−12 mm plate), plated in complete medium, and allowed to adhere for ∼15 h. FdU (0−7.5 μM) was then added to the medium for 10 days. For pulse exposure, cells were synchronized by serum starvation, then dissociated with trypsin and replaced at serially diluted densities in fresh, complete medium (range, 2 × 102−2 × 104 cells/6−12 mm plate). Sixteen hours later, just prior to entry into S phase, FdU was added. Two hours later, FdU was removed and cells were grown for 10 days in fresh medium without FdU. Colonies were stained with 0.5% crystal violet, and those colonies of 50 or more cells were scored (as a percentage of the untreated control and after adjustment for plating efficiencies). Experiments were performed twice, each in triplicate.
SINGLE CELL GEL ELECTROPHORESIS (COMET ASSAY)
The induction of DNA single strand break (SSB) in HeLaS3 cell populations by FdU alone (0.25 μM for 24 h) or in combination with 6 mM MX for 24 h was determined by Comet assay. The assay was performed according to the manufacturer's protocol (Trevigen, Gaithersburg, MD). Comet tail moment was analyzed as described [Liu et al., 2005].
STATISTICAL ANALYSIS
Comparisons were made using Student's t-test (unpaired): P-value less than 0.05 taken as statistically significant.
RESULTS
MMR-MEDIATED CELL CYCLE ARREST IN RESPONSE TO FDU
We monitored cell cycle progression of MMR-proficient HCT116 3−6 and MMR-deficient HCT116 cells after treatment with a brief pulse of FdU. Cell lysates were subjected to immunoblotting to confirm the presence and absence of MLH1 protein in HC116 3−6 and HCT 116 cells, respectively (Supplementary Fig. 1A). Cells were synchronized by serum starvation, returned to medium containing 10% FBS for 16 h, and then exposed to a pulse of 0.25 μM FdU for 2 h. After treatment, FdU was removed by washing with PBS, fresh complete medium was added, and cells were allowed to progress through the cell cycle that was monitored by fluorescence activated cell sorting (FACS). FdU caused a significant delay in progression through S-phase and a concomitant increase in cells in G2 for both HCT116 and HCT116 3−6 cells at 20 h, 49% and 59%, respectively (Fig. 1A). However, whereas MMR-deficient HCT116 cells recovered and resumed growth at 32 h (32% in G2), two-thirds of the MMR-proficient HCT116 3−6 cells remained arrested in G2. Eventually, at 72 h post-FdU treatment, the cells recovered, and the G2/M arrest was largely reversed (data not shown). These findings implicated the MLH1 MMR protein in mediating a G2/M arrest in the first cell cycle in response to a low dose of FdU.
Fig. 1.
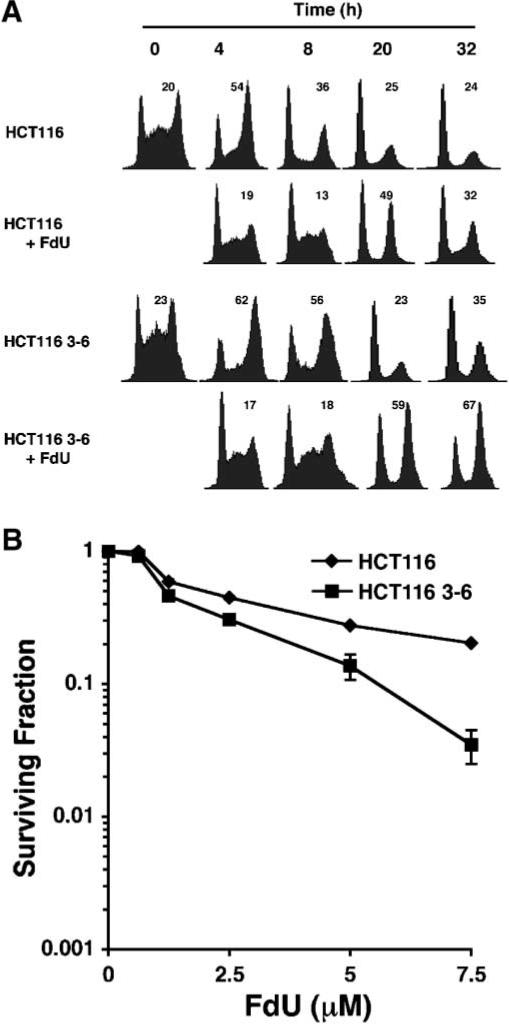
Cell cycle arrest response and cytotoxicity of MMR-deficient and -proficient cells after FdU. A: Kinetics of the G2/M cell cycle arrest in cells after a pulse of FdU. Sixteen hours after release from serum starvation, HCT116 and HCT116 3−6 cells were exposed to 0.25 μM FdU, denoted as 0 h. Two hours later, FdU was removed, and the cells were then incubated in drug-free medium for varying times and analyzed by FCM. The percentage of cells in G2 is indicated in the upper right corner of each histogram. B: Cytotoxicity induced by a 2 h pulse of FdU in MMR-deficient and -proficient cells. Sixteen hours after release from serum starvation, synchronized HCT116 (υ) and HCT116 3−6 (■) cells were treated with various concentration of FdU for 2 h, and scored after 10 days for surviving colonies (as a percentage of untreated control). The experiments were performed twice, each in triplicate. The data represent the mean ± SD. In some case, the symbols hide the error bars.
RESISTANCE OF A MMR-DEFICIENT CELL LINE TO FDU KILLING
Clonogenic assays were performed to determine the survival of HCT116 3−6 and HCT116 cells after limited exposure to FdU. After release from serum starvation, S-phase cells were exposed to varying concentrations of FdU for 2 h. Loss of MutLα led to a modest but reproducible increase in survival amounting to a roughly twofold decrease in the sensitivity of HCT116 cells (D30 = 5.0 μM) compared to HCT116 3−6 (D30 = 2.5 μM); D30 is the concentration of FdU that results in 30% survival (Fig. 1B). Low doses of FdU between 0.625 and 5 μM did not elicit significant cell killing of either HCT116 3−6 or HCT116 cells.
MMR-DEPENDENT ACTIVATION OF THE ATR/CHK1 PATHWAY IN RESPONSE TO FDU
DNA damage checkpoint signaling in response to FdU exposure was monitored by assessing the relative levels of phosphorylation of Chk1 and SMC1. As shown in Figure 2A, SMC1 and Chk1 were phosphorylated in MMR-proficient HCT116 3−6 cells within 2−4 h after treatment with 0.25 μM FdU, but little if any phosphorylation of Chk1 was observed in MLH1-deficient HCT116. A similar dependence on MMR proteins of checkpoint signaling was also observed with MSH2-deficient HEC59 and MMR-proficient HEC59 2−4 cells.
Fig. 2.
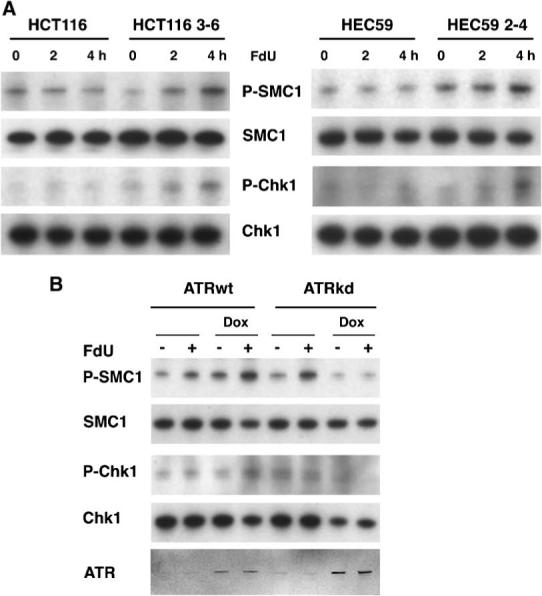
MMR-dependent ATR kinase activation in response to FdU. A: Phosphorylation of Chk1 and SMC1 in MMR-proficient cells. MLH1-deficient HCT116 and MLH1-corrected HCT116 3−6, and MSH2-deficient HEC59 and MSH2-corrected HEC59 2−4 cells were treated with 0.25 μM FdU for 2 and 4 h and harvested. The cell lysates were subjected to Western blotting to monitor phosphoserine 966 of SMC1 and phosphoserine 317 of Chk1. B: ATR-dependent SMC1 and Chk1 phosphorylation in response to FdU. U2OS cells conditionally overexpressing ATR-wt or ATR-kd (kinase deficient) proteins (+doxycycline) were treated with 0.25 μM FdU for 2 h; cell lysates were subjected to Western blotting using indicated antibodies. Induction of ATR-wt and ATR-kd was monitored by Western blotting with anti-ATR antibody.
To determine whether ATR is responsible for this MMR-dependent, FdU-induced checkpoint signaling, we monitored phosphorylation of SMC1 and Chk1 in U2OS cells conditionally overexpressing wild-type ATR or a dominant-negative kinase-deficient variant, ATR-kd, after treatment for 2 h with 0.25 μM FdU (Fig. 2B). The expression of kinase-deficient ATR (+Dox) results in an abrogation of SMC1 and Chk1 phosphorylation in response to FdU indicating that this FdU-induced checkpoint signaling is mediated by ATR. We note that a possible caveat to studies utilizing these cell lines is that MLH1 function in HCT116 and MSH2 function in HEC59 are restored through introduction of complete chromosomes, 3 and 2, respectively. Thus, it is possible that the effects are attributable to some other gene. The fact that both MLH1-deficient and MSH2-deficient cells behave similarly argues that it is the MMR proteins that are involved in checkpoint activation in response to FdU.
FDU CHECKPOINT SIGNALING IN S-PHASE
We asked when in the cell cycle the DNA damage checkpoint is activated after incorporation of FdU into DNA. Synchronized cultures of HCT116 3−6 cells arrested in G1 after treatment with HU were released into complete medium. After varying times, cells were incubated with 0.25 μM FdU for 1 h, and checkpoint signaling was monitored by levels of phosphorylation of SMC1 and Chk1 (Fig. 3A). Cell cycle progression was monitored by FACS. As expected, in the absence of FdU, no FdU-dependent increase in phosphorylation of SMC1 or Chk1 was observed. In cells treated with FdU, the peak of phosphorylated SMC1 and Chk1 occurred at 2 h and coincided with the peak of S-phase. Thereafter, as the cells progressed on to G2/M and subsequently, G1 of the second cell cycle, SMC1 and Chk1 phosphorylation levels did not increase in response to FdU (Fig. 3A). An unavoidable complication is the increase in pSMC1 and pChk1 levels irrespective of FdU treatment that reflects the essential role of ATR in signaling stalled replication brought on by the synchronization of cells in the presence of the replication inhibitor HU. Thus, levels of pSMC1 and pChk1 generally exhibited a gradual increase with time. It is notable that induction of FdU damage signaling in S-phase is only observed in MMR-proficient HCT116 3−6 cells but not in MMR-deficient HCT116 cells (Fig. 3B).
Fig. 3.
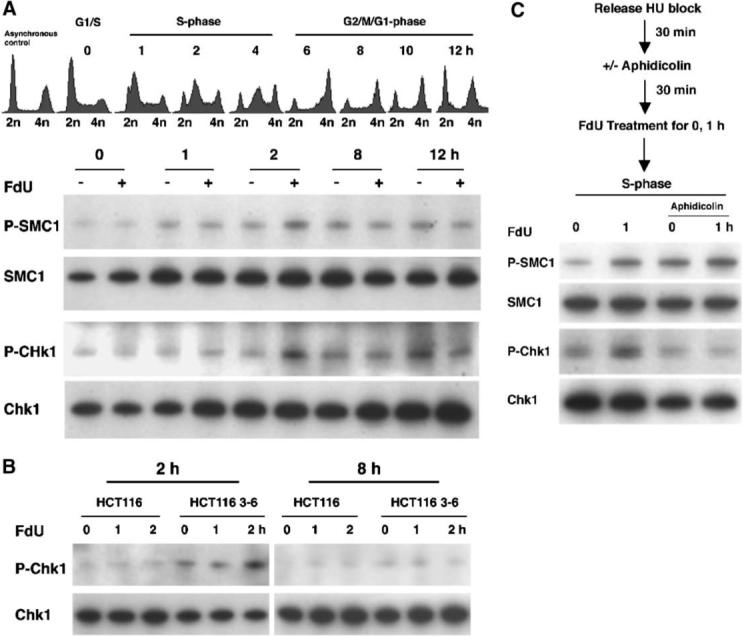
MMR dependent ATR kinase activation coincides with S phase. A: Cell cycle dependence of FdU damage signaling. Synchronized HCT116 3−6 cells were released from an HU block and cultured for varying times in complete medium followed by treatment with or without 0.25 μM FdU for 1 h prior to harvesting. Synchronization was monitored by FCM prior to FdU treatment. Phosphorylated and total SMC1 and Chk1 were detected by Western blotting. B: MMR-dependent ATR kinase activation in S-phase. Both synchronized HCT116 and HCT116 3−6 were released from HU block and cultured in complete medium for 2 or 8 h (left and right panels, respectively), followed by treatment with or without 0.25 μM FdU for 1 or 2 h prior to harvesting. Phosphorylated and total SMC1 and Chk1 were detected by Western blotting. C: DNA damage signaling triggered by FdU depends on DNA replication. After released from HU block, HCT116 3−6 cells were cultured in the presence or absence of aphidicolin follow by treatment with 0.25 μM FdU for 1 h prior to harvesting. The cell lysates were analyzed by Western blotting to monitor phosphorylation of SMC1 and Chk1.
The ability to signal checkpoint machinery upon FdU exposure was examined in cells in which replication was inhibited by aphidicolin. Synchronized cultures of HCT116 3−6 cells were pre-treated with aphidicolin followed by FdU treatment (Fig. 3C). Exposure to FdU in replication-proficient control cultures resulted in SMC1 and Chk1 phosphorylation, as expected. In replication-inhibited cells treated with aphidicolin, modest levels of Chk1 and SMC1 phosphorylation were observed in the absence of FdU (0 h), reflecting a response to arrested DNA synthesis [Guo et al., 2000; Hekmat-Nejad et al., 2000]. Notably, FdU failed to elicit robust levels of SMC1 or Chk1 phosphorylation in the absence of replication. These results are consistent with the notion that the incorporation of FdU into DNA during S-phase is required to trigger the ATR pathway for DNA damage signaling.
MUTSα BINDS PREFERENTIALLY TO FDU DNA ADDUCTS
To determine if the MMR protein MutSα is able to recognize the misincorporated FdU nucleotides, we performed gel shift assays using purified recombinant human MutSα and double-stranded DNA substrates containing a single FdU moiety paired opposite G or A, a G:T mismatch, or a G/C homoduplex control (Fig. 4A,B). We observe that MutSα is able to recognize FdU mispaired opposite guanine, FdU:G, in this particular sequence context, although the affinity for FdU:G is less than that for G:T mispairs. In contrast, MutSα binds FdU/A base pairs with an affinity roughly equivalent to nonspecific binding to a G/C homoduplex control.
Fig. 4.
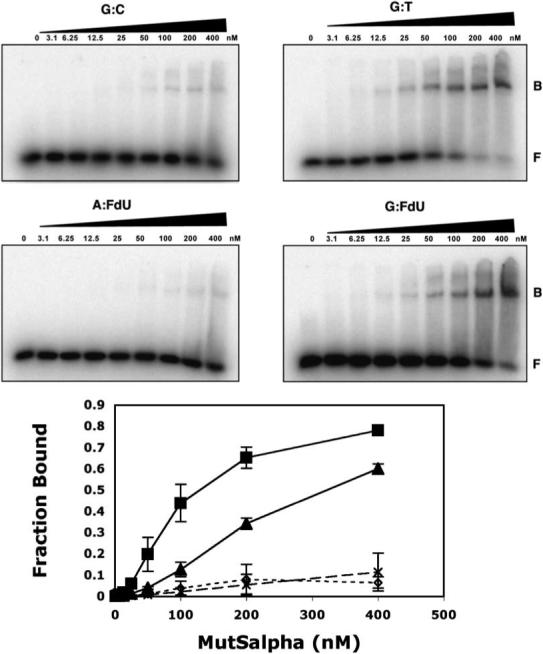
MutSα binds to G:FdU mispairs. Gel mobility shift assays were performed with 35 bp DNA duplexes containing G:C (○), G:T (■), G:FdU (▲) or A:FdU (Δ) base pairs or mispairs, for example, G:T refers to a mismatch in which G is base paired opposite T. 32P-5′-labeled DNA was incubated with the indicated concentration of purified recombinant MutSα and subjected to native gel electrophoresis. Error bars represent ± SD of three independent experiments. Fraction of DNA bound was determined by the ratio of B/(B + F) where “F” denotes free DNA and “B” denotes bound DNA as determined using a Fuji Phosphorimager and ImageGauge v.4.22.
MMR-INDEPENDENT CELL CYCLE ARREST AND ATR ACTIVATION IN RESPONSE TO CONTINUOUS EXPOSURE TO FDU
We monitored cell cycle progression upon continuous exposure to low doses of FdU where the level of incorporation of FdU into DNA would be substantially higher. HCT116 cells (MMR-deficient) and HCT116 3−6 cells (MMR-proficient) were synchronized by serum starvation. After 16 h in medium supplemented with 10% FBS, cells were exposed continuously to 0.25 μM FdU. As shown in Figure 5A, both MMR-proficient and deficient cell lines showed a significant delay in S-phase. The G2/M populations in both HCT116 and HCT116 3−6 cells were increased to similar extents at 24 h after FdU exposure, and the G2/M arrest in the first cell cycle persisted at 32 h in both cell lines. In fact, cells regardless of MMR status were unable to resume cycling at 120 h (data not shown). Similar G2/M cell cycle arrest was found after continuous FdU exposure in cells synchronized with HU (data not shown) as well as in asynchronous cells (Fig. S1B). Consistent with the observed irreversible exit from the cell cycle, extensive cell killing occurred in both HCT116 3−6 and HCT116 cells after continuous treatment with 0.625−7.5 μM FdU (data not shown).
Fig. 5.
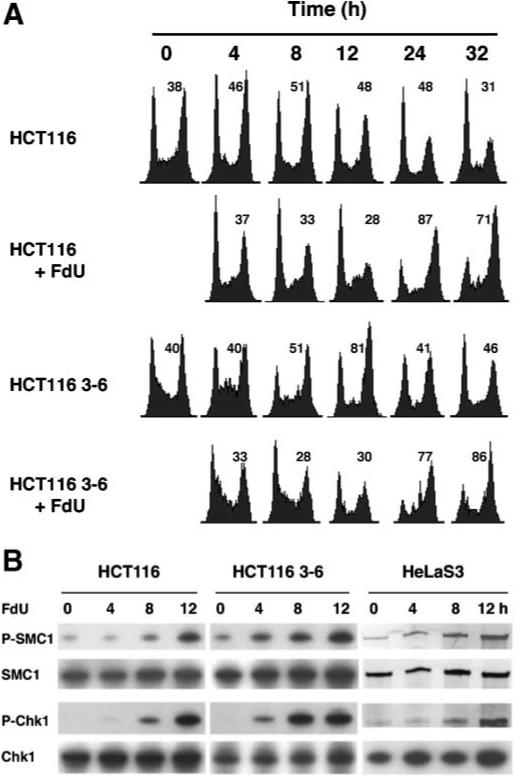
Cell cycle arrest and ATR kinase activation in response to continuous exposure to FdU. A: Kinetics of the G2/M cell cycle arrest in cells after continuous exposure to FdU. Sixteen hours after release from serum starvation, HCT116 and HCT116 3−6 cells were exposed to 0.25 μM FdU, denoted as 0 h. Cells were harvested at indicated times and cell cycle profiles were assayed by FCM. The percentage of cells in G2 is denoted in the upper right of each histogram. B: MMR-independent ATR kinase activation induced by FdU after continuous exposure to FdU. HCT116 and HCT116 3−6 as well as HeLaS3 cells were treated with 0.25 μM FdU continuously for 4, 8, or 12 h and harvested. Cell lysates were subject to Western blotting using indicated antibodies.
We next examined DNA damage checkpoint responses after continuous exposure to FdU. As shown in Figure 5B, after 4 h of exposure to FdU, SMC1 and Chk1 were phosphorylated in MMR-proficient HCT116 3−6 cells, but not in MLH1-deficient HCT116 cells. However, at 8 and 12 h after FdU exposure, SMC1 and Chk1 were phosphorylated in both MMR-proficient HCT116 3−6 and -deficient HCT116 cells to similar extents. These findings imply that other pathways in addition to MMR are involved in FdU-mediated damage signaling and G2/M cell cycle arrest under conditions of increased FdU exposure.
MX INCREASES FDU-INDUCED DNA SSBS AND CYTOTOXICITY
Several BER glycosylases including Smug1, MED1 (MBD4), and TDG mismatch—specific DNA glycosylase, have been implicated in the cellular response to FdU [Hardeland et al., 2003; Turner et al., 2006; An et al., 2007]. Because BER is a major pathway for the generation of SSBs induced by chemical agents, we assessed the role of BER in modulating FdU damage. Methoxyamine (MX), a small molecular inhibitor of BER that blocks the repair of AP sites after glycosylase-mediated removal of a base potentiates the accumulation of damage-induced DNA SSBs and cytotoxicity [Liuzzi and Talpaert-Borle, 1985; Fortini et al., 1990; Taverna et al., 2003]. The ability of MX to sensitize cells to FdU-induced DNA SSBs and cytotoxicity was examined in HeLaS3 cells. First, we confirmed that, as was the case for HCT116 and HCT116 3−6 cells, phosphorylation of SMC1 and Chk1 in HeLaS3 cells occurred after 4, 8, or 12 h of continuous exposure to 0.25 μM FdU (Fig. 5B). DNA SSBs were then monitored by Comet assay. HeLaS3 cells exposed for 24 h to 0.25 μM FdU and 6 mM MX showed significantly increased levels of DNA SSB (detected as a 50% increase in the relative comet tail moment) compared to cells exposed to 0.25 μM FdU in the absence of MX (Fig. 6A,B). Cells treated with MX alone showed no increase in the comet tail compared to mock-treated cells (data not shown). The cytotoxicity resulting from combined MX and FdU treatment was evaluated (Fig. 6C). HeLaS3 cells showed increased sensitivity in the presence of both FdU and MX compared to FdU alone (D30 = 2.7 μM vs. D30 = 3.6 μM, respectively). These data implicate the BER pathway in the production of AP sites in response to FdU.
Fig. 6.
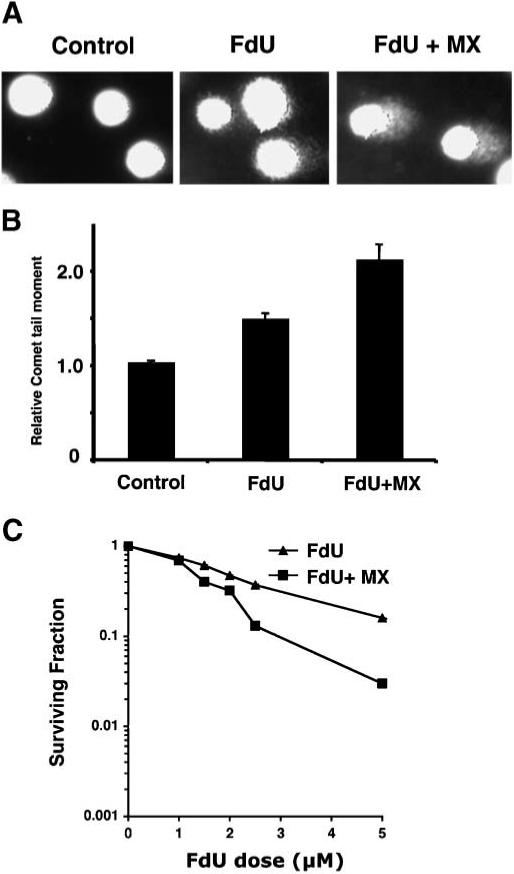
MX potentiates FdU-induced DNA SSBs and cytotoxicity. A: A representative pattern of DNA SSBs (comet tails) produced by the Comet assay. B: Increased comet tail moment in HeLaS3 cells after treatment with 0.25 μM FdU alone or simultaneous treatment with 0.25 μM FdU and 6 mM MX. The experiments were performed in triplicate. Error bars represent ± SD of the mean. C: Effect of MX on cytotoxicity induced by FdU in HeLaS3 cells. Cells were treated for 24 h with 0−5 μM FdU alone (σ) or FdU plus 6 mM MX (■). Surviving colonies were counted 10 days after treatment (as a percentage of the untreated control). The experiments were performed twice, each in triplicate. Error bars represent ± SD of the mean. In some case, the symbols hide the error bars.
DISCUSSION
We examine the role of the MMR system in DNA damage signaling elicited by exposure of mammalian cells to the halogenated uracil analog FdU. We have shown that the MMR proteins MutSα and MutLα are required for a checkpoint response in the first cell cycle after exposure to FdU. Loss of MLH1, a subunit of MutLα, confers a twofold increase in resistance to cell killing by low doses of FdU and abrogates activation of the G2/M checkpoint. Importantly, we demonstrate that DNA damage signaling in response to FdU requires not only MutSα and MutLα, but a functional ATR kinase. Activation of ATR leads to phosphorylation of downstream checkpoint kinases Chk1 and SMC1. Damage signaling is rapid, but reversible, occurs during S-phase, and requires DNA synthesis. Consistent with its role as a DNA damage sensor protein, MutSα specifically recognizes FdU:G in vitro; however FdU:A mismatches are only bound nonspecifically. Prolonged exposure to FdU results in an irreversible G2 cell cycle arrest and cell killing that is largely independent of MMR. In the presence of FdU and MX, an inhibitor of BER, aberrant BER intermediates accumulate resulting in increased cell death. Our data suggest that MMR can act as a damage sensor in response to FdU triggering ATR-dependent cell cycle arrest and cell death. However, other pathways, particularly BER, also play prominent roles in the cellular response.
Two models have been proposed to explain the role of MMR proteins in the DNA damage response. In one scenario, iterative rounds of MMR eventually lead to a lethal double-strand DNA break. In the case of cytotoxic O6-methylguanine residues such “futile cycles” of MMR occur because the lesion resides in the parental strand and is never removed by the MMR machinery that targets only the newly synthesized strand for excision and repair. Multiple rounds of excision at O6-methyl-G:T mismatches is observed in in vitro assays for MMR utilizing a minimal reconstituted system [York and Modrich, 2006]. In a variation on this theme, aberrant processing of O6-methyl-G:T mismatches by MMR may result in a block to replication in the second cell cycle thereby triggering checkpoint activation [Stojic et al., 2004; Mojas et al., 2007]. Nevertheless, in vivo, it appears that an alternate pathway distinct from canonical MMR-mediated excision may exist in which MMR proteins, upon recognizing DNA damage, signal directly to the checkpoint machinery. Separation-of-function alleles in MSH2 and MSH6, encoding the two protein subunits of MutSα, confer a strong mutator phenotype and cancer in mice, but preserve a functional DNA damage signaling pathway [Lin et al., 2004; Yang et al., 2004]. In addition, MutSα, MutLα are required to recruit ATR to sites of O6-methyl-G:T mispairs in vitro and to activate ATR kinase to phosphorylate Chk1 in an in vitro kinase assay [Yoshioka et al., 2006].
The exact molecular mechanism by which MMR activates a cellular DNA damage response to FdU remains to be elucidated. In several respects, the MMR-mediated cellular response to FdU resembles that of SN1 alkylation particularly with regard to signaling through the ATR/Chk1 pathway. However, we note two potentially important differences between the damage response elicited by cytotoxic O6-meG produced by SN1 alkylators and FdU mispairs. First, as mentioned above, O6-methyl-G resides in the parental strand and consequently is never removed by the MMR system whereas FdU is incorporated in the newly synthesized strand in the first round of replication. Second, in the case of O6-methyl-G, the cell cycle arrest is delayed to the second cycle after exposure to SN1 alkylators whereas the observed G2/M arrest triggered by FdU is in the first cell cycle (Fig. 1; see also [Stojic et al., 2004; Meyers et al., 2005; Yoshioka et al., 2006]). Thus, although MMR appears to target a wide variety of lesions including those produced by SN1 DNA methylators, 6-thioguanine, FdU, cisplatin, UV, and some carcinogens, the relative contributions of MMR proteins to excision repair versus damage signaling may differ. In this regard, it is interesting that the contribution of MMR to the cytotoxicity of FdU and two other uracil analogs, 5-iododeoxyuridine (IdU) and 5-bromodeoxyuridine (BrdU), appear to be different. Thus, while MMR-deficient cells are more resistant to killing by FdU than MMR-proficient cells, they are more sensitive than MMR-proficient cells to killing by IdU and BrdU [Berry et al., 2000, 2003]. The role of MMR proteins as damage sensors is supported by the DNA substrate specificity of MutSα. The relatively high affinity for FdU:G mispairs but not FdU:A pairs shown in Figure 4 correlates well with previous results indicating that FdU:G, but not FdU:A, mispairs stimulate the ATPase activity of MutSα [Meyers et al., 2005]. Tajima et al. [2004] observed binding of human MutSα to FdU:A mispairs that was modestly better than a C:T mismatch in surface plasmon resonance biosensor experiments, but FdU:G mispairs were not examined. Would FdU:G mispairs be prevalent enough to trigger MutSα-directed damage signaling? In this regard, it is noteworthy that treatment with FdU has been shown to be both mutageneic and oncogenic (see discussion in Meyers et al. [2004]).
Our data showing that FdU produces lesions that are converted to AP sites (Fig. 6) are consistent with recently published observations implicating the BER pathway in the cellular response to FPs. An et al. [2007] have recently reported that the BER glycosylase Smug1 confers protection from cell killing by FU. Using gene-targeted cell lines deficient in one or both of the mammalian uracil-DNA glycosylases Smug1 and Ung, they demonstrated that the incorporation of FdU into DNA rather than the excision of uracil mediates cell killing. Smug1 by virtue of its ability to remove FdU from DNA confers protection from cell killing; correspondingly, loss of Smug1 results in increased sensitivity to FU. MED1 (MBD4), a BER N-glycosylase that repairs mismatches that occur at CpG methylation sites, also targets halogenated pyrimidines including FdU. Like the loss of MMR, loss of MED1 confers increased survival in the presence of FdU implicating MED1 in the DNA damage response [Cortellino et al., 2003]. Interestingly, MED1 was isolated in a two-hybrid screen using MLH1 as bait [Bellacosa et al., 1999]. Finally, in vitro repair assays suggest that multiple BER enzymes may target FdU/G and FdU/A mispairs [Fischer et al., 2007]. Thus, interactions between components of the MMR and BER pathways and competition between these two pathways for a subset of DNA lesions may be critical in modulating the DNA damage response.
Clinical studies to ascertain the influence of MSI status on overall survival and the efficacy of FU chemotherapy are equivocal. While some studies point to the absence of survival benefit conferred by FU for MMR-deficient microsatellite instability high (MSI-H) colorectal cancer patients suggesting that loss of MMR confers resistance or tolerance to FU [Meyers et al., 2003; Ribic et al., 2003; Benatti et al., 2005; Popat et al., 2005; Jover et al., 2006], other studies find the opposite, that is increased survival among MSI-H patients treated with FU or find no difference ([Lamberti et al., 2007] and references cited within). Conclusions from such studies are potentially confounded by patient sample size, the overall improved survival of patients with MSI-H tumors compared to those with microsatellite-stable tumors observed in some though not all studies, the age of the patient population, and the possibly heterogeneous underpinnings of genomic instability. Our findings suggest another complication in understanding how 5-FU kills cells in which multiple DNA repair pathways may converge on the same lesion. We show that FdU:G mispairs that might arise as rare events during replication in the presence of FdU are targeted by the MutSα and MutLα MMR proteins. These two MMR proteins are also required to mediate a DNA damage response involving the ATR/Chk1 pathway. Our data and the work of other laboratories point to an important role for BER in both removing FdU from DNA and in promoting cell cycle arrest and cell death. Knowledge of the relative contributions of the MMR and BER pathways to both cell killing and DNA repair and temporal and dose thresholds at which each may compete or act synergistically may be very important in predicting the efficacy of 5-FU treatment for different patient populations.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Richard Boland (Baylor University Medical Center) for providing us HCT116 3−6 cells, Paul Nghiem (Massachusetts General Hospital) for providing ATR-wt and ATR-kd U2OS cells, and Tom Kunkel (NIEHS, NIH) for providing HEC59 and HEC59 2−4 cells used in this study. This work was funded by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH.
Grant sponsor: Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases; Grant sponsor: NIH.
Abbreviations used
- MMR
mismatch repair
- HNPCC
hereditary nonpolyposis colorectal cancer
- FP
fluoropyrimidine
- FU
5-fluorouracil
- FdU
5-fluoro-2′-deoxyuridine
- TS
thymidylate synthase
- BER
base excision repair
- SSBs
single strand breaks
- FCM
flow cytometry
- HU
hydroxyurea
- MX
methoxyamine
REFERENCES
- An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007;67:940–945. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- Andersen S, Heine T, Sneve R, Konig I, Krokan HE, Epe B, Nilsen H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis. 2005;26:547–555. doi: 10.1093/carcin/bgh347. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci USA. 1999;96:3969–3974. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatti P, Gafa R, Barana D, Marino M, Scarselli A, Pedroni M, Maestri I, Guerzoni L, Roncucci L, Menigatti M, Roncari B, Maffei S, Rossi G, Ponti G, Santini A, Losi L, Di Gregorio C, Oliani C, Ponz de Leon M, Lanza G. Microsatellite instability and colorectal cancer prognosis. Clin Cancer Res. 2005;11:8332–8340. doi: 10.1158/1078-0432.CCR-05-1030. [DOI] [PubMed] [Google Scholar]
- Berry SE, Davis TW, Schupp JE, Hwang HS, de Wind N, Kinsella TJ. Selective radiosensitization of drug-resistant MutS homologue-2 (MSH2) mismatch repair-deficient cells by halogenated thymidine (dThd) analogues: Msh2 mediates dThd analogue DNA levels and the differential cytotoxicity and cell cycle effects of the dThd analogues and 6-thioguanine. Cancer Res. 2000;60:5773–5780. [PubMed] [Google Scholar]
- Berry SE, Loh T, Yan T, Kinsella TJ. Role of MutSalpha in the recognition of iododeoxyuridine in DNA. Cancer Res. 2003;63:5490–5495. [PubMed] [Google Scholar]
- Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, Bellacosa A. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci USA. 2003;100:15071–15076. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckett DR, Bronstein SM, Taya Y, Modrich P. hMutSa- and hMutLa-dependent phosphorylation of p53 in response to DNA methylator damage. Proc Natl Acad Sci USA. 1999;96:12384–12388. doi: 10.1073/pnas.96.22.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Baerenfaller K, Jiricny J. 5-Fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology. 2007;133:1858–1868. doi: 10.1053/j.gastro.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Fortini P, Bignami M, Dogliotti E. Evidence for AP site formation related to DNA-oxygen alkylation in CHO cells treated with ethylating agents. Mutat Res. 1990;236:129–137. doi: 10.1016/0921-8777(90)90040-c. [DOI] [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardeland U, Bentele M, Jiricny J, Schar P. The versatile thymine DNA-glycosylase: A comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003;31:2261–2271. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol. 2000;10:1565–1573. doi: 10.1016/s0960-9822(00)00855-1. [DOI] [PubMed] [Google Scholar]
- Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: Functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, Pinol V, Xicola RM, Bujanda L, Rene JM, Clofent J, Bessa X, Morillas JD, Nicolas-Perez D, Paya A, Alenda C. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006;55:848–855. doi: 10.1136/gut.2005.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N'-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Lamberti C, Lundin S, Bogdanow M, Pagenstecher C, Friedrichs N, Buttner R, Sauerbruch T. Microsatellite instability did not predict individual survival of unselected patients with colorectal cancer. Int J Colorectal Dis. 2007;22:145–152. doi: 10.1007/s00384-006-0131-8. [DOI] [PubMed] [Google Scholar]
- Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, Umar A, Kucherlapati R, Lipkin M, Kunkel TA, Edelmann W. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- Liu A, Takakuwa T, Fujita S, Ham MF, Luo WJ, Daibata M, Aozasa K. Alterations of DNA damage-response genes ATM and ATR in pyothorax-associated lymphoma. Lab Invest. 2005;85:436–446. doi: 10.1038/labinvest.3700235. [DOI] [PubMed] [Google Scholar]
- Liuzzi M, Talpaert-Borle M. A new approach to the study of the base-excision repair pathway using methoxyamine. J Biol Chem. 1985;260:5252–5258. [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Meyers M, Hwang A, Wagner MW, Boothman DA. Role of DNA mismatch repair in apoptotic responses to therapeutic agents. Environ Mol Mutagen. 2004;44:249–264. doi: 10.1002/em.20056. [DOI] [PubMed] [Google Scholar]
- Meyers M, Hwang A, Wagner MW, Bruening AJ, Veigl ML, Sedwick WD, Boothman DA. A role for DNA mismatch repair in sensing and responding to fluoropyrimidine damage. Oncogene. 2003;22:7376–7388. doi: 10.1038/sj.onc.1206941. [DOI] [PubMed] [Google Scholar]
- Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem. 2005;280:5516–5526. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- Mojas N, Lopes M, Jiricny J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007;21:3342–3355. doi: 10.1101/gad.455407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nghiem P, Park PK, Kim YS, Desai BN, Schreiber SL. ATR is not required for p53 activation but synergizes with p53 in the replication checkpoint. J Biol Chem. 2002;277:4428–4434. doi: 10.1074/jbc.M106113200. [DOI] [PubMed] [Google Scholar]
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield MJ, Hsieh P. DNA mismatch repair: Molecular mechanisms and biological function. Ann Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima A, Hess MT, Cabrera BL, Kolodner RD, Carethers JM. The mismatch repair complex hMutS alpha recognizes 5-fluorouracil-modified DNA: Implications for chemosensitivity and resistance. Gastroenterology. 2004;127:1678–1684. doi: 10.1053/j.gastro.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Taverna P, Hwang HS, Schupp JE, Radivoyevitch T, Session NN, Reddy G, Zarling DA, Kinsella TJ. Inhibition of base excision repair potentiates iododeoxyuridine-induced cytotoxicity and radiosensitization. Cancer Res. 2003;63:838–846. [PubMed] [Google Scholar]
- Turner DP, Cortellino S, Schupp JE, Caretti E, Loh T, Kinsella TJ, Bellacosa A. The DNA N-glycosylase MED1 exhibits preference for halogenated pyrimidines and is involved in the cytotoxicity of 5-iododeoxyuridine. Cancer Res. 2006;66:7686–7693. doi: 10.1158/0008-5472.CAN-05-4488. [DOI] [PubMed] [Google Scholar]
- Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, Edelmann W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- York SJ, Modrich P. Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J Biol Chem. 2006;281:22674–22683. doi: 10.1074/jbc.M603667200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.