

Abstract
Through its various metabolites, vitamin A controls essential physiological functions. Both naturally occurring metabolites and novel retinoid analogues have shown effectiveness in many clinical settings that include skin diseases and cancer, and in animal models of human conditions affecting vision. In this review, we analyze several potential retinoid-based therapies from the point of view of drug metabolism and transport to target tissues. We focus on the endogenous factors that affect the absorption, transport, and metabolism of retinoids by taking into account data obtained from the analysis of animal models that lack the enzymes or proteins involved in the storage and absorption of retinoids. We also discuss findings of toxicity associated with retinoids in an effort to improve the outcome of retinoid-based therapies. In this context, we review evidence that esterification of retinol and retinol-based drugs within target tissues provides one of the most efficient means to improve the absorption and to reduce the toxicity associated with pharmacological doses of retinoids. Future retinoid-based therapeutic strategies could involve targeted delivery mechanisms leading to lower toxicity and improved effectiveness of retinoids.
Vitamin A, retinol, plays essential roles in many biological processes including vision, immunity, growth, development, and cellular differentiation. The various functions of vitamin A are carried out by several known physiologically active metabolites including 11-cis-retinal, the visual chromophore (1), and all-trans-retinoic acid (RA1), which controls gene expression through the RA receptor (RAR) (2, 3). Other retinol metabolites that have shown biological activities in vitro are the 9-cis-isomer of RA, which activates both retinoid X receptors (RXRs) and RARs (4), and the retro-retinoids, anhydroretinol (AR), and 14-hydroxy-4,14-retro-retinol (14-HRR), which are involved in the regulation of lymphocyte proliferation (5–7). All-trans-13,14-dihydroxy-retinol was shown to have activity comparable to that of 14-HRR in supporting the proliferation of promyelocytic HL-60 cells and in the activation of T cells (8). The conversion of all-trans-retinol to all-trans-13,14-dihydroretinol by the recently described enzyme all-trans-retinol–all-trans-13,14-dihydroretinol saturase (retinol saturase or RetSat) enzyme leads to the formation of all-trans-13,14-dihydroretinol (9), which can be further oxidized to all-trans-13,14-dihydrore-tinoic acid (10). The metabolism of vitamin A in relation to the formation of known or potential endogenous bioactive retinoids is shown in Figure 1.
Figure 1.
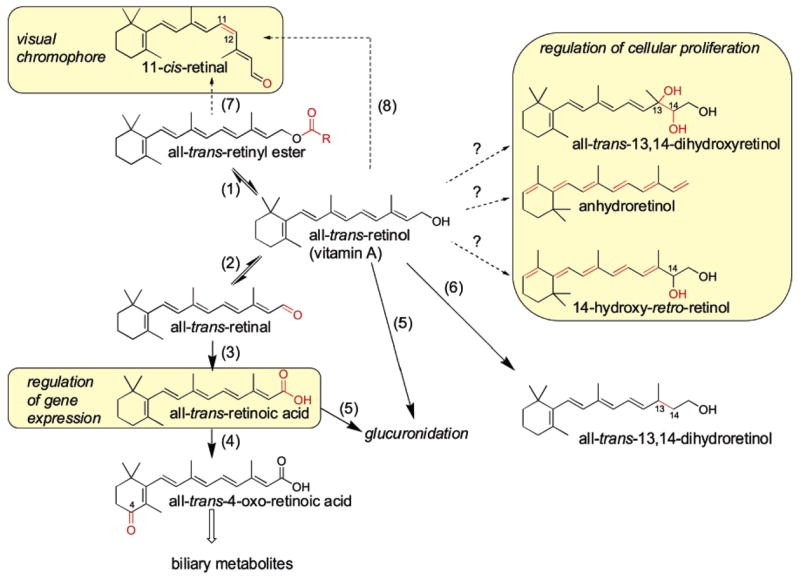
Retinoid metabolism and bioactive retinoids in higher vertebrates. Known endogenous bioactive retinoids are highlighted by yellow boxes and include all-trans-retinoic acid (RA), which regulates gene expression via RAR, 11-cis-retinal, which is the chromophore of the photoreceptor molecule rhodopsin, all-trans-13,14-dihydroxyretinol, and the retro-retinoids, AR and 14-hydroxy-retro-retinol, which control cellular proliferation. All-trans-retinol can be esterified by the LRAT or by the ARAT enzyme to produce retinyl esters that can be hydrolyzed back to all-trans-retinol by retinyl ester hydrolase (REH) (reaction 1). The LRAT enzyme uses phospholipid (PL) as acyl donors and prefers retinol complexed to CRBP as the substrate. The oxidation of all-trans-retinol by short chain dehydrogenase/reductase (SDR) or ADH enzymes leads to the formation of all-trans-retinal (reaction 2), which in turn is oxidized by retinaldehyde dehydrogenases (RALDH) to all-trans-RA (reaction 3). All-trans-RA can be converted to more polar metabolites through oxidation by cytochrome P450 CYP26 enzymes (reaction 4) or through glucuronidation by UDP-glucuronosyltransferase (UGT) enzymes (reaction 5). All-trans-retinol can be saturated by RetSat and leads to the formation of all-trans-13,14-dihydroretinol (reaction 6), which can be oxidized by the same enzymes as those of all-trans-retinol to produce all-trans-13,14-dihydroretinoic acid. The production of 11-cis-retinal occurs in RPE cells and through esterification, isomerization, and oxidation reactions, with isomerization involving the RPE65 enzyme (reaction 7). An alternate pathway for the production of the visual chromophore for cones operates independently of RPE65 (reaction 8). The enzymes involved in the production of all-trans-13,14-dihydroxyretinol, AR, and 14-hydroxy-retro-retinol in vertebrates are not currently known, though a retinol dehydratase from insects was shown to catalyze the formation of AR.
Therapeutic applications for retinoids have made use of both naturally occurring retinoids and synthetic agonists and antagonists of RAR and RXR. Supplementation with all-trans-retinol is used in developing countries to correct vitamin A deficiency (VAD). This treatment is one of the safest and most efficacious therapeutic uses of retinoids and has enormous worldwide impact, saving millions of lives every year at a cost of pennies per treated individual (11). All-trans-RA (tretininoin, Retin-A) and 13-cis-RA (isotretinoin, Accutane) are clinically used in the treatment of severe acne and other dermatological conditions (12). Retinoids like all-trans-RA, 13-cis-RA, N-(4-hydroxyphenyl)retinamide (4-HPR or fenretinide), and non-retinoid synthetic subtype specific RAR and RXR-agonists and modulators are used in the treatment and chemoprevention of various forms of cancer (13–16). With respect to vision, there are several proposed retinoid-based therapies with potential in the treatment of autosomal dominant retinitis pigmentosa (ADRP), Leber congenital amaurosis (LCA), macular degeneration, or Stargardt disease (17). Supplementation of the substitute chromophore 9-cis-retinal can reconstitute the visual pigment in animal models of LCA (18–21). Inhibitors of the visual cycle, such as retinylamine (22) and 13-cis-RA (23), and reduction of serum retinol using 4-HPR (24) have shown effectiveness in animal models of Stargardt disease and macular degeneration by slowing the rate of the visual cycle and preventing the accumulation of all-trans-retinal and the production of phototoxic byproducts such as pyridinium bis-retinoid (A2E) (17). Some of the currently investigated retinoid-based drugs and therapeutic targets are depicted in Figure 2.
Figure 2.
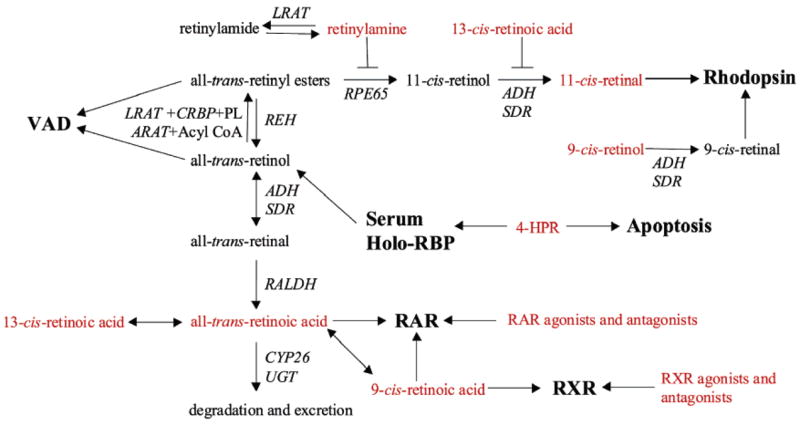
Targets of retinoid treatments. Both all-trans-retinol and all-trans-retinyl esters (palmitate) are effective in the treatment and prevention of vitamin A deficiency (VAD). The reconstitution of rhodopsin with 9-cis-retinal forms isorhodopsin and is an effective means of restoring visual function in several models of Leber congenital amaurosis (LCA). Inhibitors of the visual cycle, retinylamine and 13-cis-RA, can slow the rate of 11-cis-retinal formation in cases where excessive activation of rhodopsin can lead to phototoxicity and accumulation of all-trans-retinal and toxic metabolites as seen in Stargardt disease and age-related macular degeneration. A similar result can also be obtained with N-(4-hydroxyphenyl)retinamide (4-HPR or fenretinide), which binds RBP and leads to its excretion through glomerular filtration. Retinylamine can be amidated by the LRAT enzyme, allowing for the storage of the drug in an inactive form with lower toxicity. Independently of RAR, 4-HPR can lead to the apoptosis of tumor cells. RAR can be modulated by all-trans-RA, 9-cis-RA, and many synthetic agonists and antagonists. The retinoid X receptor can be modulated by 9-cis-RA and several synthetic agonists and antagonists. Both 13-cis- and 9-cis-RA can isomerize to all-trans-RA in vivo. The drug targets are shown in bold font, and the retinoids used in various therapies are shown in red font.
Retinoid therapies have demonstrated clinical efficacy as treatments for many debilitating diseases; however, one must overcome significant obstacles associated with the well-established teratogenic and toxic effects of retinoids. Many of these obstacles could be overcome if the delivery mechanism is more specific. In this review, we focus on factors affecting the delivery of retinol and other retinoids to specific target tissues.
ABSORPTION OF RETINOIDS
Retinoids delivered orally are efficiently absorbed by the intestine. Retinyl esters are hydrolyzed to retinol within the intestinal lumen by the actions of pancreatic triglyceride lipase (25) or at the intestinal brush border by the membrane-bound enzyme phospholipase B (26, 27). The newly generated retinol as well as the retinol obtained from the diet or supplements is taken into the enterocyte as retinol. Because of the lipophilic character of retinol, absorption is enhanced by a fatty meal (28). Within the enterocyte, newly absorbed retinol is re-esterfied through the actions of the enzyme lecithin retinal–acyl transferase (LRAT), which catalyzes the transacylation of a fatty acyl group from lecithin to retinol (29–33). Cellular retinol-binding protein II (CRBP-II), a member of the family of intracellular lipid-binding proteins, which binds retinol with high affinity, helps facilitate retinol uptake into the body providing the retinol as a substrate for LRAT (34, 35). Although LRAT is the predominant enzyme for catalyzing retinyl ester formation within the intestine, a study of Lrat−/− mice indicates that an acyl-CoA-dependent enzyme, referred to as acyl-CoA–retinol acyltransferase (ARAT), may also be involved in retinyl ester formation in the liver, mammary gland, or adipose tissue (32, 36). Ultimately, the retinyl ester newly synthesized by LRAT and/or ARAT is incorporated along with other dietary lipids into nascent chylomicrons that are then secreted from the enterocyte into the lymphatic system (37).
TRANSPORT OF RETINOIDS TO TARGET TISSUES
Role of Chylomicrons and Their Remnants
Nascent chylomicrons in the lymphatic system enter the general circulation through the thoracic duct where they undergo metabolism to form chylomicron remnants. The conversion of chylomicrons to chylomicron remnants involves the actions of lipoprotein lipase (LPL), which hydrolyzes chylomicron triglycerides to free fatty acids, resulting in a smaller particle and facilitating the recruitment to the lipolyzed particle of several apolipoproteins, especially apolipoprotein E (apoE), that are needed for receptor-mediated uptake of the remnants by the liver (38, 39). About 66–75% of retinyl esters contained in chylomicrons are taken up by the liver and resecreted into the circulation as retinol bound to serum retinol-binding protein (RBP) or stored as esters in hepatic stellate cells (also called Ito cells, lipocytes, or fat-storing cells) (40, 41). The remaining 25–33% of the retinyl esters from chylomicrons are taken up by target tissues that include adipose tissue, heart, muscle, lungs, reproductive organs, and bone marrow. The clearance of chylomicron retinoids appears to involve the actions of LPL (42). This reaction most likely involves LPL-catalyzed retinyl ester hydrolysis but possibly also the well-characterized actions of LPL in facilitating whole particle uptake by cells (42, 43). The importance of chylomicron-derived retinoid toward meeting target tissue needs is evidenced by RBP-deficient mice, which, aside from a mild vision phenotype, are phenotypically normal (44–46). As long as RBP-deficient mice are provided sufficient quantities of vitamin A in the diet, they are able to maintain normal retinoid-dependent functions and even to acquire retinyl ester stores in target tissues. When these mutant mice are deprived of dietary retinoid, they are at increased risk of developing symptoms of VAD because they are not able to mobilize hepatic retinyl ester stores through the actions of RBP (44–46). Thus, the delivery of retinoids by chylomicrons and their remnants can be an important route for retinoid delivery to target tissues.
Role of RBP in the Delivery of Retinol
As mentioned earlier, unesterified retinol is transported in the blood as retinol bound to RBP (47). RBP is primarily synthesized and secreted from the liver, where the majority of the body’s retinoid is stored (48, 49). However, RBP also is synthesized and secreted in lesser amounts by extrahepatic tissues, including the eye, adipose tissue, kidney, lungs, and heart (50). RBP forms a one-to-one protein–protein complex with another blood protein transthyretin (TTR), which prevents renal clearance of the retinal–RBP by the glomerulus (51–53). Hepatic secretion of holo-RBP is needed for the efficient export of retinol from liver stores (44–46). There is some controversy regarding how retinol is taken up by cells from the retinal–RBP–TTR complex (54). Retinol rapidly dissociates from RBP and rapidly flip–flops across the plasma membrane (55). Moreover, the rate of dissociation of retinol from RBP is not rate limiting for retinol uptake by cells, which argues that it is not necessary to invoke the actions of a cell surface receptor for RBP to account for retinol uptake into cells (54, 56, 57). Yet, other studies suggest that retinol uptake into cells involves a cell surface receptor for RBP (54, 58–62). One possible receptor is coded by a retinoic acid-responsive gene, STRA6 (stimulated by retinoic acid-6) (63). The STRA6 protein is a multispanning membrane protein that confers RBP binding to transfected cells and mediates the uptake of retinol (62). STRA6 is expressed in several blood–organ barriers, such as Sertoli cells, the yolk sac, chorioallantoic placenta, choroid plexi (64), and RPE (62, 64). Mutations in STRA6 cause severe malformations, such as anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation (65). However, patients affected by deleterious mutations in the RBP gene suffer from night-blindness early in life but are otherwise unaffected (66). A similar phenotype is seen in Rbp−/− mice, manifested as visual impairment early in life (44). These observations argue for additional roles for the STRA6 protein during development that are independent of its role as a receptor for RBP (67).
The retinoid needs for all tissues aside from the eye can be fully met through chylomicron delivery of retinoid or retinoid bound to serum albumin, as is evidenced by the visual phenotype of the Rbp−/− mice (44). Thus, the eye may rely on the STRA6 receptor to ensure sufficient retinoid uptake from RBP to support the visual cycle. However, because the complete absence of RBP does not impair retinoid-dependent functions in other tissues, a cell surface receptor for RBP may not be needed to ensure the normal uptake of retinol by such tissues in times of sufficient dietary vitamin A intake.
RA Delivery by Serum Albumin
RA is far less hydrophobic than retinol or retinyl ester, and it is not absorbed in chylomicrons but rather transported via the portal system bound to albumin because binding to RBP hinders its interaction with TTR (68). Pharmacokinetic studies of many of the synthetic analogues of RA indicate that they are absorbed primarily via the portal system like the natural acidic retinoid forms (69). However, depending on the lipophilic nature of a synthetic retinoid, some of its absorption may involve uptake via nascent chylomicrons, or it may be taken up solely via the portal route. This absorption will depend on the chemical properties of the particular retinoid.
In well-nourished humans and animal models, the circulating levels of all-trans-RA and 13-cis-RA bound to albumin are approximately 0.1 to 0.4% of those of retinal–RBP during fasting (70). Yet, this circulating RA can contribute significantly to tissue pools of RA in well-nourished animals (71). Tracer kinetic turnover studies in rats have shown that, for most tissues, 5–30% of the RA present within the tissue is derived from the circulating RA pool, whereas the brain and liver derive greater than 80% of their RA from the circulating RA pool (71). Thus, despite the low levels of all-trans- and 13-cis-RA in circulation, these acids are rapidly taken up by cells and tissues. Albumin-bound RA can be an important delivery pathway through which cells/tissues normally acquire retinoid. Like retinol, RA undergoes dissociation from albumin prior to traversing the membrane and entering into the cell, and RA is known to flip–flop readily across membrane bilayers. The delivery of synthetic RA analogues via albumin is pharmacologically important, and presumably, this process mimics RA uptake. However, there is little information available in the literature regarding how synthetic retinoids enter cells/tissues from the circulation.
Other Routes of Retinoid Delivery
In humans as well as in animal models, retinyl esters are normally secreted at low levels (1–2% of those of retinal–RBP) from the liver bound to nascent very low-density lipoprotein (VLDL) (72). The physiologic significance of VLDL retinyl ester has not been systematically investigated, but, invoking similarities between VLDL and chylomicron metabolism, it seems likely that the retinyl ester contained within VLDL is destined for uptake by extrahepatic tissues. The circulation also contains low levels of retinol-β-glucuronides and retinoyl-β-glucuronides (levels similar to those of all-trans- and 13-cis-RA) that are fully water soluble (54, 73, 74), hence more easily excreted. Alternatively, these retinoid-β-glucuronides may be used by the body to satisfy tissue retinoid needs because they can be readily hydrolyzed to retinol and RA (54, 73, 74).
ROLE OF LRAT IN THE ABSORPTION AND STORAGE OF RETINOL
As is true for the liver and RPE, LRAT is quantitatively the most important enzyme for catalyzing retinyl ester formation in most tissues (20, 32, 75). Consequently, LRAT is essential for the accumulation of retinoids within the liver and eye. In the liver, retinyl esters are found predominantly in hepatic stellate cells, which represent the main cellular retinoid storage site in the body. RPE cells also represent an important storage of retinyl esters reserved mostly for use in the production of the visual chromophore. The importance of LRAT in allowing tissues to accumulate and store retinol is evidenced by the complete absence of retinyl esters in nearly all of the tissues that have been examined in Lrat−/− mice. The one exception to this general statement is adipose tissue, where retinyl ester levels are 2- to 3-fold elevated in Lrat−/− mice compared to those in age- and gender-matched wild-type mice (20, 32). Although adipocytes have long been known to be important cellular sites of retinyl ester accumulation in the body accounting for the second largest retinoid store after the liver, little is known regarding how retinol is processed and becomes esterified within this cell type (76–80). LRAT is not needed to catalyze retinyl ester formation in adipose tissue but it is not presently clear which enzyme is responsible for retinyl ester formation within this tissue. The diacylglycerol acyltransferase 1 (DGAT1) enzyme, which catalyzes the formation of triglyceride from diacylglycerol and an acyl-CoA, catalyzes acyl-CoA-dependent retinol esterification in vitro, hence exhibiting ARAT activity (32, 81, 82). However, DGAT1, although highly expressed in adipose tissue, does not seem to be the only enzyme involved in catalyzing retinyl ester formation, because adipose tissue retinyl ester levels are the same for Lrat−/− mice and Lrat−/−/Dgat1−/− double knockout mice (Blaner, W. et al., unpublished observations). Thus, it remains unclear which other enzymes can catalyze retinyl ester formation and facilitate the accumulation of retinoid stores within adipose tissue.
In addition to allowing for retinoid accumulation within tissues, the sequestration of retinol as retinyl ester also serves to limit the generation of RA during development (83) and acts as a protective mechanism following the consumption of high doses of dietary vitamin A (84). Although the levels of serum retinol and RA are within the normal range, Lrat−/− mice are not capable of sequestering retinol as retinyl esters, thus exhibiting signs of RA toxicity when maintained on a vitamin A-sufficient chow diet. This observation is also supported by the upregulation in Lrat−/− mice of Cyp26 genes that are known to have central roles in the detoxification (catabolism) of RA (75). In addition, when we employed microarray analysis to identify other differentially expressed genes in the livers of Lrat−/− mice and their wild-type counterparts, we found that among the genes upregulated in the liver of Lrat−/− mice maintained on a standard chow diet were several isoforms of the UDP-glucuronosyltransferase 1 (UGT1) family of enzymes (85) (Table 1). The UGT1 locus is known to be subject to regulation by RA (86). As mentioned above, β-glucuronidation products of all-trans-RA, 13-cis-RA, 4-oxo-RA, and 5,6-epoxy-RA are normally present at low levels in the blood and bile and at greater concentrations following supplementation with large doses of retinol or RA (87–89). Elevated expression levels of UGT1 in the livers of Lrat−/− mice could be taken to suggest that the liver is sensing excessive levels of RA, leading to the induction of detoxification mechanisms.
Table 1.
Differentially Expressed Genes in the Liver of Lrat−/− Mice vs WTa
| gene symbol | gene description | accession no. | fold change Lrat−/− WT |
|---|---|---|---|
| upregulated in Lrat−/− mice vs WT | |||
| Ugt1a9 | UDP-glucuronosyltransferase 1 family A9 | S57479 | 7.9 |
| Apoa4 | apolipoprotein A-IV | M64249 | 6.0 |
| Mt1 | metallothionein 1 | BC027262 | 4.4 |
| Alas1 | aminolevulinate synthase 1 | M63245 | 4.4 |
| Gdpd3 | Glycerophosphodiester phosphodiesterase domain containing 3 | NM_024228 | 3.6 |
| Cxcl14 | chemokine (C-X-C motif) ligand 14 | AF352785 | 3.3 |
| Hspa8 | heat shock protein 8 | X05837 | 3.1 |
| Serpina4-ps1 | serpin A4, pseudogene 1 | BC024071 | 3.0 |
| Hsp110 | heat shock protein 110 | AK075731 | 3.0 |
| Dhtkd1 | dehydrogenase E1 and transketolase domain containing 1 | AK050057 | 2.7 |
| Hspa1b | heat shock protein 1B | M12573 | 2.5 |
| Lcn13 | Lipocalin 13 | NM_153558 | 2.5 |
| Cap1 | adenylate cyclase-associated protein 1 | NM_007598 | 2.4 |
| Tppp | tubulin polymerization-promoting protein | NM_182839 | 2.3 |
| Tsc22d3 | TSC22-related inducible leucine zipper 3 | AF024519 | 2.3 |
| downregulated in Lrat−/− mice vs WT | |||
| Clec2h | C-type lectin domain family 2, member h | NM_053165 | 0.49 |
| MGC60813 | Unknown | BC058613 | 0.45 |
| Tubb2a | tubulin, beta 2 a | NM_009450 | 0.44 |
| Cml5 | camello-like 5 | AK007530 | 0.43 |
| Cg10671 | Cg10671 like | BC048823 | 0.43 |
| Mm.2121 | Unknown | NM_026790 | 0.38 |
| Tiam2 | T-cell lymphoma invasion and metastasis 2 | NM_011878 | 0.38 |
| Cebpe | CCAAT/enhancer binding protein epsilon | NM_207131 | 0.37 |
| Hsd3b5 | Hydroxy-delta-5-steroid dehydrogenase | NM_008295 | 0.32 |
| Camk2b | calcium/calmodulin-dependent protein kinase II, beta | AK083344 | 0.19 |
| Gadd45g | growth arrest and DNA-damage-inducible 45 gamma | AK002237 | 0.15 |
Total RNA from the three livers of 3 month old Lrat−/− (or WT) mice maintained on a standard diet were pooled to generate one sample. We used two different pools of hepatic RNA from Lrat−/− mice to compare them with two different pools of WT mice. Each sample of pooled RNA was detection-labeled and hybridized in duplicate on a mouse genomic microarray using a service provided by NimbleGen System, Inc. (Madison, WI) as described previously (139). The microarray contained 37,364 genes and covered the entire mouse transcriptome as represented by the University of California, Santa Cruz database (build HG 17), using a minimum of 11 probes per gene. The expression of genes was normalized according to the probe signal, and the average signal for each gene was normalized for each sample replicate. Array data for samples across the whole study were normalized by NimbleGen Systems, Inc. (Madison, WI), using the robust multichip analysis feature of the data analysis package found at http://www.bioconductor.org. Project-wide spreadsheets of robust multichip analysis results were exported to Microsoft Excel. Expression level ratios for all of the possible pairwise comparisons, comprising a four-way comparison of two Lrat−/− and two WT samples, were calculated. These pairwise ratios were imported to Microsoft Access and mined for credible fold changes. Changes in gene expression greater than or equal to 2-fold in at least three of the four comparisons were considered significant.
Esterification by LRAT leads to active recruitment of the retinoid from circulation as well as the establishment of a long-lived storage pool. This property of LRAT can be manipulated for therapeutic advantage for the delivery of retinol and other retinoids with an alcohol functional group. Another potential retinoid-based therapeutic retinylamine has a reactive amine group instead of alcohol, yet it can also be recruited and stored via amidation by LRAT (90) (Figure 2). The conversion of retinylamine to pharmacologically inactive retinylamides occurs in vitro and in vivo in the liver and RPE and leads to the safe storage of this potent inhibitor, which can be released after hydrolysis back to free retinylamine. This transformation leads to a prolonged therapeutic effect and lower toxicity of this compound in comparison with those of other proposed visual cycle inhibitors such as 13-cis-RA (90). In summary, the presence of LRAT in the target tissue can ensure the efficient and active uptake of retinol and other retinoids with a functional hydroxyl group as well as retinoids that can be amidated by LRAT.
ROLE OF CRBPS IN THE UPTAKE OF RETINOID BY SPECIFIC TISSUES
Three cellular-retinol-binding proteins, CRBP-I, II, and III, are known to facilitate the esterification of retinoids by LRAT. The physiologic roles of LRAT and the CRBPs in the absorption and storage of retinol have been best studied in knockout animal models. CRBP-I is expressed in the liver, kidney, and eye, and Crbp-I−/− mice display lower levels of hepatic retinyl esters and reduced numbers of lipid droplets in hepatic stellate cells than wild-type mice. Crbp-I−/− mice succumb more easily to symptoms of VAD than wild-type mice if they are maintained on a vitamin A-restricted diet (91). CRBP-II is expressed primarily in the intestine, and its lack in Crbp-II−/− mice results in decreased levels of stored retinoids and increased neonatal mortality, especially during dietary vitamin A restriction (92). CRBP-III is expressed in the heart, muscle, adipose, and mammary tissues (93). Crbp-III−/− mice have difficulty incorporating retinol into milk. In some cases, the loss of one form of CRBP can be partly compensated for by the upregulation of other CRBPs, as is the case of the upregulation of CRBP-III in the absence of CRBP-I and vice versa (94). Although the studies of Crbp−/− mice demonstrate that the absence of these proteins results in a much less severe metabolic phenotype than that observed with Lrat−/− mice, these studies do show that like LRAT, the CRBPs have an important role in facilitating retinol uptake, esterification, and storage by cells (20, 32, 91–93).
The critical role of LRAT in catalyzing the esterification and thus the uptake and storage of retinol suggests that this enzyme may be a useful target for assuring the specific uptake of a retinoid therapeutic agent by a target tissue. Coupled with the hydrolysis of the esters, this potentially provides a way to localize release of the drug within a specific cell type or tissue. The release of stored retinoid drugs from their ester pools depends on the activity of retinyl ester hydrolases in the respective target tissues. Manipulating this reaction may provide a means for a controlled delivery of therapeutic retinoids, which, in principle, would allow for the usage of lower administered doses and hence lower toxicity. Moreover, the expression of LRAT is subject to regulation by RA (95, 96). STRA6 was described as an RA-responsive gene (63), and modulation of the levels of STRA6 in WiDr colon adenocarcinoma cells by RA leads to the increased uptake of retinol in these cells (62). As a result, induction of the expression of LRAT or STRA6 using RA could increase the delivery of retinol to target tissues. Indeed, co-administration of RA effectively increases the delivery of retinol to lungs (97, 98). In addition, the expression of CRBP-I is also upregulated via RAR action (99), whereas CRBP-II expression is controlled via RXR (100). In fact, agonists of RXR have been shown to affect the esterification of retinol in certain cell types (101). Possibly, ectopic expression of STRA6, LRAT, and/or CRBP via gene therapy or transcriptional regulation could help ensure the delivery of retinoids to specific target tissues or cell types. Both LRAT and CRBP are downregulated in many tumor cell types (102–117); therefore, less retinol is available for oxidation to produce RA in cancer cells. Upregulation of the retinoid pool in tumor cells might lead to higher endogenous levels of RA at the tumor site enabling the anticarcinogenic activities of this agent. Future studies could examine the feasibility and therapeutic effectiveness of increasing the intracellular retinoid pool through the upregulation of the enzymes and accessory proteins involved in the generation of retinyl esters.
RETINOL TOXICITY AND THE SIDE EFFECTS ASSOCIATED WITH RETINOID TREATMENT
Excess levels of vitamin A lead to a wide range of deleterious effects on health. Retinoids present a serious teratogenic risk and can lead to craniofacial, cardiac, thymic, and central nervous system malformations (118), which are the most severe manifestations of toxicity associated with retinoid therapy. Teratogenic effects are seen for both natural and synthetic retinoids used in clinical trials (119, 120), but these are less pronounced for preformed retinol supplements (121). The administration of retinyl palmitate to rats, mice, and hamsters led to exencephaly, with frequent occurrences of cleft palate, spina bifida, eye defects, hydrocephaly, and shortening of the mandible and maxilla (122–125). The teratogenic effects are believed to stem mainly from the transcriptional activities of retinoids, mediated via the activation of RAR or RXR. The teratogenic consequences of retinoid supplementation during pregnancy depend on the stage of development, the nature of the retinoid metabolite, and the genetic and environmental factors (120). Caution must be exercised in prescribing retinoids to ensure that pregnancy is avoided during treatment (126). Additionally, the administration of retinoids to young animals and children results in delayed growth as a result of premature epiphyseal plate closure (127, 128).
Less severe consequences of retinoid supplementation result in acute and chronic forms of retinoid toxicity. Acute intoxication with vitamin A occurs following the ingestion of large amounts of vitamin A over a short period of time, but cases of acute toxicity are relatively rare. The clinical effective dose for vitamin A administration in the case of degenerative eye diseases such as retinitis pigmentosa (15,-000 IU/day) is below the maximal recommended dose (25,-000 IU/day) and was shown to be well tolerated after 12 years of treatment (129, 130). In the few documented cases, recovery from acute retinoid toxicity is rapid once intake is reduced (131, 132). Chronic effects are manifested as drying, ulceration and desquamation of the skin, anorexia, anemia, and bone loss. Liver damage leading to fibrosis and hepatic stellate cell activation are both seen in patients affected by hypervitaminosis A (133). Upon activation, hepatic stellate cells lose their capacity to store retinyl esters and secrete collagen, leading to cirrhosis (132, 134). Oxidation of retinol generates RA, which stimulates osteoclast formation and activity leading to bone resorption and hypercalcemia (135, 136). Bone loss could represent a health issue in the elderly, leading to osteopenia, osteoporosis, and increased risk of hip fracture (137, 138).
CONCLUSIONS
Vitamin A metabolites, including RAs and their synthetic derivatives, collectively known as retinoids, modulate the rates of transcription of the genes involved in the regulation of cell proliferation and differentiation and thus exhibit chemotherapeutic and chemopreventive activities in a number of human cancers. Another vitamin A metabolite, 11-cis-retinal, serves as the visual chromophore, and several proposed retinoid-based therapies show promise in the treatment of eye diseases such as ADRP, LCA, and macular degeneration. Retinoid-based therapy is, however, often confounded by the toxicity of these compounds at pharmacological doses. Recent observations suggest that factors that regulate the uptake and storage of retinoids in cells may serve as targets for novel retinoid-based therapeutic strategies. Such factors include LRAT, the enzyme that catalyzes retinoid esterification and storage, and CRBPs, proteins that cooperate with LRAT in regulating retinoid uptake and metabolism. Manipulating the levels or activities of these proteins may allow for a better control of the levels of active retinoids in specific tissues, thereby reducing the amounts needed for efficacious therapy and targeting the agents to specific tissues.
Footnotes
This research was supported in part by U.S. Public Health Service Grants EY01730, EY015399, and EY08061 from the National Eye Institute, National Institutes of Health, Bethesda, MD (to K.P.) and Grant DK068437 from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD (to W.S.B.).
Abbreviations: A2E, pyridinium bis-retinoid; ADH, medium-chain alcohol dehydrogenases; ADRP, autosomal dominant retinitis pigmentosa; apoE, apolipoprotein E; AR, anhydroretinol; ARAT, acyl-CoA–retinol acyltransferase; CRBP, cellular retinol-binding protein; DGAT1, diacylglycerol acyltransferase 1; 4-HPR, N-(4-hydroxyphenyl)retinamide (or fenretinide); 14-HRR, 14-hydroxy-4,14-retro-retinol; LCA, Leber congenital amaurosis; LPL, lipoprotein lipase; LRAT, lecithin–retinol acyltransferase; RA, retinoic acid; 13-cis-RA, 13-cis-retinoic acid (or isotretinoin, Accutane); RALDH, retinaldehyde dehydrogenase; RAR, retinoic acid receptor; RBP, retinol-binding protein; RetSat, all-trans-retinol–all-trans-13,14-dihydroretinol saturase (or retinol saturase); RPE65, retinal pigmented epithelial-specific 65 kDa protein; RXR, retinoid-X receptor; SDR, short-chain dehydrogenase/reductase; STRA6, stimulated by retinoic acid 6; TTR, transthyretin; UGT1, UDP-glucuronosyltransferase 1; vitamin A, all-trans-retinol; VAD, vitamin A deficiency; VLDL, very low-density lipoprotein.
References
- 1.Wald G. Molecular basis of visual excitation. Science. 1968;162:230–239. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- 2.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 3.Mark M, Ghyselinck NB, Chambon P. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol. 2006;46:451–480. doi: 10.1146/annurev.pharmtox.46.120604.141156. [DOI] [PubMed] [Google Scholar]
- 4.Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, Thaller C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 5.Buck J, Derguini F, Levi E, Nakanishi K, Hammerling U. Intracellular signaling by 14-hydroxy-4,14-retro-retinol. Science. 1991;254:1654–1656. doi: 10.1126/science.1749937. [DOI] [PubMed] [Google Scholar]
- 6.Buck J, Grun F, Derguini F, Chen Y, Kimura S, Noy N, Hammerling U. Anhydroretinol: a naturally occurring inhibitor of lymphocyte physiology. J Exp Med. 1993;178:675–680. doi: 10.1084/jem.178.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derguini F, Nakanishi K, Hammerling U, Buck J. Intracellular signaling activity of synthetic (14R)-, (14S)-, and (14RS)-14-hydroxy-4,14-retro-retinol. Biochemistry. 1994;33:623–628. doi: 10.1021/bi00169a001. [DOI] [PubMed] [Google Scholar]
- 8.Derguini F, Nakanishi K, Hammerling U, Chua R, Eppinger T, Levi E, Buck J. 13,14-Dihydroxy-retinol, a new bioactive retinol metabolite. J Biol Chem. 1995;270:18875–18880. doi: 10.1074/jbc.270.32.18875. [DOI] [PubMed] [Google Scholar]
- 9.Moise AR, Kuksa V, Imanishi Y, Palczewski K. Identification of all-trans-retinol:all-trans-13,14-dihydroretinol saturase. J Biol Chem. 2004;279:50230–50242. doi: 10.1074/jbc.M409130200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moise AR, Kuksa V, Blaner WS, Baehr W, Palczewski K. Metabolism and transactivation activity of 13,14-dihydroretinoic acid. J Biol Chem. 2005;280:27815–27825. doi: 10.1074/jbc.M503520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humphrey JH, West KP, Jr, Sommer A. Vitamin A deficiency and attributable mortality among under-5-year-olds. Bull W H O. 1992;70:225–232. [PMC free article] [PubMed] [Google Scholar]
- 12.Chivot M. Retinoid therapy for acne. A comparative review. Am J Clin Dermatol. 2005;6:13–19. doi: 10.2165/00128071-200506010-00002. [DOI] [PubMed] [Google Scholar]
- 13.Chomienne C, Ballerini P, Balitrand N, Amar M, Bernard JF, Boivin P, Daniel MT, Berger R, Castaigne S, Degos L. Retinoic acid therapy for promyelocytic leukaemia. Lancet. 1989;2:746–747. doi: 10.1016/s0140-6736(89)90812-x. [DOI] [PubMed] [Google Scholar]
- 14.Hong WK, Sporn MB. Recent advances in chemoprevention of cancer. Science. 1997;278:1073–1077. doi: 10.1126/science.278.5340.1073. [DOI] [PubMed] [Google Scholar]
- 15.Simeone AM, Tari AM. How retinoids regulate breast cancer cell proliferation and apoptosis. Cell Mol Life Sci. 2004;61:1475–1484. doi: 10.1007/s00018-004-4002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hail N, Jr, Kim HJ, Lotan R. Mechanisms of fenretinide-induced apoptosis. Apoptosis. 2006;11:1677–1694. doi: 10.1007/s10495-006-9289-3. [DOI] [PubMed] [Google Scholar]
- 17.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, Pittler SJ, Stone EM, Jacobson SG, Palczewski K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA. 2000;97:8623–8628. doi: 10.1073/pnas.150236297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Hooser JP, Liang Y, Maeda T, Kuksa V, Jang GF, He YG, Rieke F, Fong HK, Detwiler PB, Palczewski K. Recovery of visual functions in a mouse model of Leber congenital amaurosis. J Biol Chem. 2002;277:19173–19182. doi: 10.1074/jbc.M112384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batten ML, Imanishi Y, Tu DC, Doan T, Zhu L, Pang J, Glushakova L, Moise AR, Baehr W, Van Gelder RN, Hauswirth WW, Rieke F, Palczewski K. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med. 2005;2:e333. doi: 10.1371/journal.pmed.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeda A, Maeda T, Palczewski K. Improvement in rod and cone function in mouse model of Fundus albipunctatus after pharmacologic treatment with 9-cis-retinal. Invest Ophthalmol Visual Sci. 2006;47:4540–4546. doi: 10.1167/iovs.06-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci USA. 2005;102:8162–8167. doi: 10.1073/pnas.0503318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radu RA, Mata NL, Nusinowitz S, Liu X, Travis GH. Isotretinoin treatment inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Novartis Found Symp. 2004;255:51–63. discussion 63–67, 177–178. [PubMed] [Google Scholar]
- 24.Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, Widder K, Travis GH, Mata NL. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Visual Sci. 2005;46:4393–4401. doi: 10.1167/iovs.05-0820. [DOI] [PubMed] [Google Scholar]
- 25.van Bennekum AM, Fisher EA, Blaner WS, Harrison EH. Hydrolysis of retinyl esters by pancreatic triglyceride lipase. Biochemistry. 2000;39:4900–4906. doi: 10.1021/bi9927235. [DOI] [PubMed] [Google Scholar]
- 26.Rigtrup KM, Kakkad B, Ong DE. Purification and partial characterization of a retinyl ester hydrolase from the brush border of rat small intestine mucosa: probable identity with brush border phospholipase B. Biochemistry. 1994;33:2661–2666. doi: 10.1021/bi00175a039. [DOI] [PubMed] [Google Scholar]
- 27.Harrison EH. Mechanisms of digestion and absorption of dietary vitamin A. Annu Rev Nutr. 2005;25:87–103. doi: 10.1146/annurev.nutr.25.050304.092614. [DOI] [PubMed] [Google Scholar]
- 28.Tso P, Lee T, DeMichele SJ. Randomized structured triglycerides increase lymphatic absorption of tocopherol and retinol compared with the equivalent physical mixture in a rat model of fat malabsorption. J Nutr. 2001;131:2157–2163. doi: 10.1093/jn/131.8.2157. [DOI] [PubMed] [Google Scholar]
- 29.MacDonald PN, Ong DE. A lecithin:retinol acyltransferase activity in human and rat liver. Biochem Biophys Res Commun. 1988;156:157–163. doi: 10.1016/s0006-291x(88)80818-0. [DOI] [PubMed] [Google Scholar]
- 30.MacDonald PN, Ong DE. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J Biol Chem. 1988;263:12478–12482. [PubMed] [Google Scholar]
- 31.Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, Bok D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J Biol Chem. 1999;274:3834–3841. doi: 10.1074/jbc.274.6.3834. [DOI] [PubMed] [Google Scholar]
- 32.O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K, Blaner WS. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT) J Biol Chem. 2005;280:35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, Palczewski K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herr FM, Ong DE. Differential interaction of lecithin-retinol acyltransferase with cellular retinol binding proteins. Biochemistry. 1992;31:6748–6755. doi: 10.1021/bi00144a014. [DOI] [PubMed] [Google Scholar]
- 35.Ong DE. Cellular transport and metabolism of vitamin A: roles of the cellular retinoid-binding proteins. Nutr Rev. 1994;52:S24–S31. doi: 10.1111/j.1753-4887.1994.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 36.Randolph RK, Winkler KE, Ross AC. Fatty acyl CoA-dependent and -independent retinol esterification by rat liver and lactating mammary gland microsomes. Arch Biochem Biophys. 1991;288:500–508. doi: 10.1016/0003-9861(91)90227-a. [DOI] [PubMed] [Google Scholar]
- 37.Blomhoff R, Green MH, Berg T, Norum KR. Transport and storage of vitamin A. Science. 1990;250:399–404. doi: 10.1126/science.2218545. [DOI] [PubMed] [Google Scholar]
- 38.Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res. 1997;38:2173–2192. [PubMed] [Google Scholar]
- 39.Redgrave TG. Chylomicron metabolism. Biochem Soc Trans. 2004;32:79–82. doi: 10.1042/bst0320079. [DOI] [PubMed] [Google Scholar]
- 40.Goodman DW, Huang HS, Shiratori T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. J Lipid Res. 1965;6:390–396. [PubMed] [Google Scholar]
- 41.Packer L. Carotenoids and Retinoids: Molecular Aspects and Health Issues. AOCS Press; Champaign, IL: 2005. [Google Scholar]
- 42.van Bennekum AM, Kako Y, Weinstock PH, Harrison EH, Deckelbaum RJ, Goldberg IJ, Blaner WS. Lipoprotein lipase expression level influences tissue clearance of chylomicron retinyl ester. J Lipid Res. 1999;40:565–574. [PubMed] [Google Scholar]
- 43.Otarod JK, Goldberg IJ. Lipoprotein lipase and its role in regulation of plasma lipoproteins and cardiac risk. Curr Atheroscler Rep. 2004;6:335–342. doi: 10.1007/s11883-004-0043-4. [DOI] [PubMed] [Google Scholar]
- 44.Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, Freeman S, Cosma MP, Colantuoni V, Gottesman ME. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999;18:4633–4644. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogel S, Piantedosi R, O’Byrne SM, Kako Y, Quadro L, Gottesman ME, Goldberg IJ, Blaner WS. Retinol-binding protein-deficient mice: biochemical basis for impaired vision. Biochemistry. 2002;41:15360–15368. doi: 10.1021/bi0268551. [DOI] [PubMed] [Google Scholar]
- 46.Quadro L, Hamberger L, Gottesman ME, Wang F, Colantuoni V, Blaner WS, Mendelsohn CL. Pathways of vitamin A delivery to the embryo: insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology. 2005;146:4479–4490. doi: 10.1210/en.2005-0158. [DOI] [PubMed] [Google Scholar]
- 47.Kanai M, Raz A, Goodman DS. Retinol-binding protein: the transport protein for vitamin A in human plasma. J Clin Invest. 1968;47:2025–2044. doi: 10.1172/JCI105889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blomhoff R, Rasmussen M, Nilsson A, Norum KR, Berg T, Blaner WS, Kato M, Mertz JR, Goodman DS, Eriksson U, et al. Hepatic retinol metabolism. Distribution of retinoids, enzymes, and binding proteins in isolated rat liver cells. J Biol Chem. 1985;260:13560–13565. [PubMed] [Google Scholar]
- 49.Blaner WS, Hendriks HF, Brouwer A, de Leeuw AM, Knook DL, Goodman DS. Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells. J Lipid Res. 1985;26:1241–1251. [PubMed] [Google Scholar]
- 50.Soprano DR, Blaner WS. The Retinoids: Biology, Chemistry, and Medicine. 2. Raven Press; New York: 1994. Plasma Retinol Binding Protein. [Google Scholar]
- 51.Peterson PA. Studies on the interaction between prealbumin, retinol-binding protein, and vitamin A. J Biol Chem. 1971;246:44–49. [PubMed] [Google Scholar]
- 52.Melhus H, Nilsson T, Peterson PA, Rask L. Retinol-binding protein and transthyretin expressed in HeLa cells form a complex in the endoplasmic reticulum in both the absence and the presence of retinol. Exp Cell Res. 1991;197:119–124. doi: 10.1016/0014-4827(91)90488-g. [DOI] [PubMed] [Google Scholar]
- 53.van Bennekum AM, Wei S, Gamble MV, Vogel S, Piantedosi R, Gottesman M, Episkopou V, Blaner WS. Biochemical basis for depressed serum retinol levels in transthyretin-deficient mice. J Biol Chem. 2001;276:1107–1113. doi: 10.1074/jbc.M008091200. [DOI] [PubMed] [Google Scholar]
- 54.Vogel S, Gamble MV, Blaner WS. The Handbook of Experimental Pharmacology: Retinoids: The Biochemical and Molecular Basis of Vitamin A and Retinoid Action. Vol. 139. Springer; New York: 1999. Retinoid Uptake, Metabolism and Transport. [Google Scholar]
- 55.Noy N, Xu ZJ. Kinetic parameters of the interactions of retinol with lipid bilayers. Biochemistry. 1990;29:3883–3888. doi: 10.1021/bi00468a013. [DOI] [PubMed] [Google Scholar]
- 56.Noy N. Retinoid-binding proteins: mediators of retinoid action. Biochem J. 2000;348:481–495. [PMC free article] [PubMed] [Google Scholar]
- 57.Noy N, Blaner WS. Interactions of retinol with binding proteins: studies with rat cellular retinol-binding protein and with rat retinol-binding protein. Biochemistry. 1991;30:6380–6386. doi: 10.1021/bi00240a005. [DOI] [PubMed] [Google Scholar]
- 58.Heller J. Interactions of plasma retinol-binding protein with its receptor. Specific binding of bovine and human retinol-binding protein to pigment epithelium cells from bovine eyes. J Biol Chem. 1975;250:3613–3619. [PubMed] [Google Scholar]
- 59.Rask L, Peterson PA. In vitro uptake of vitamin A from the retinol-binding plasma protein to mucosal epithelial cells from the monkey’s small intestine. J Biol Chem. 1976;251:6360–6366. [PubMed] [Google Scholar]
- 60.International Agency for Research on Cancer. IARC Handbooks of Cancer Prevention, Vol. 4. Retinoids. World Health Organization Press; Geneva: 2000. [Google Scholar]
- 61.Sundaram M, Sivaprasadarao A, DeSousa MM, Findlay JB. The transfer of retinol from serum retinol-binding protein to cellular retinol-binding protein is mediated by a membrane receptor. J Biol Chem. 1998;273:3336–3342. doi: 10.1074/jbc.273.6.3336. [DOI] [PubMed] [Google Scholar]
- 62.Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 63.Bouillet P, Oulad-Abdelghani M, Vicaire S, Garnier JM, Schuhbaur B, Dolle P, Chambon P. Efficient cloning of cDNAs of retinoic acid-responsive genes in P19 embryonal carcinoma cells and characterization of a novel mouse gene, Stra1 (mouse LERK-2/Eplg2) Dev Biol. 1995;170:420–433. doi: 10.1006/dbio.1995.1226. [DOI] [PubMed] [Google Scholar]
- 64.Bouillet P, Sapin V, Chazaud C, Messaddeq N, Decimo D, Dolle P, Chambon P. Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech Dev. 1997;63:173–186. doi: 10.1016/s0925-4773(97)00039-7. [DOI] [PubMed] [Google Scholar]
- 65.Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nurnberg G, Brasch F, Schirmer-Zimmermann H, Tolmie JL, Chitayat D, Houge G, Fernandez-Martinez L, Keating S, Mortier G, Hennekam RC, von der Wense A, Slavotinek A, Meinecke P, Bitoun P, Becker C, Nurnberg P, Reis A, Rauch A. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. 2007;80:550–560. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seeliger MW, Biesalski HK, Wissinger B, Gollnick H, Gielen S, Frank J, Beck S, Zrenner E. Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Invest Ophthalmol Visual Sci. 1999;40:3–11. [PubMed] [Google Scholar]
- 67.Blaner WS. STRA6, a cell-surface receptor for retinol-binding protein: the plot thickens. Cell Metab. 2007;5:164–166. doi: 10.1016/j.cmet.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 68.Noy N, Slosberg E, Scarlata S. Interactions of retinol with binding proteins: studies with retinol-binding protein and with transthyretin. Biochemistry. 1992;31:11118–11124. doi: 10.1021/bi00160a023. [DOI] [PubMed] [Google Scholar]
- 69.Retinoic Acid. Vol. 4 2000. IARC Handbooks of Cancer Prevention. [Google Scholar]
- 70.Blaner WS, Olson JA. The Retinoids: Biology, Chemistry, and Medicine. 2. Raven Press; New York: 1994. Retinol and Retinoic acid Metabolism. [Google Scholar]
- 71.Kurlandsky SB, Gamble MV, Ramakrishnan R, Blaner WS. Plasma delivery of retinoic acid to tissues in the rat. J Biol Chem. 1995;270:17850–17857. doi: 10.1074/jbc.270.30.17850. [DOI] [PubMed] [Google Scholar]
- 72.Krasinski SD, Cohn JS, Russell RM, Schaefer EJ. Postprandial plasma vitamin A metabolism in humans: a reassessment of the use of plasma retinyl esters as markers for intestinally derived chylomicrons and their remnants. Metabolism. 1990;39:357–365. doi: 10.1016/0026-0495(90)90249-c. [DOI] [PubMed] [Google Scholar]
- 73.Goswami BC, Reida AK, Ivanoff KD, Barua AB, Olson JA. Intestinal absorption and metabolism of retinoyl beta-glucuronide in humans, and of 15-[14C]-retinoyl beta-glucuronide in rats of different vitamin A status. J Nutr Biochem. 2003;14:703–709. doi: 10.1016/j.jnutbio.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 74.Ueltschy A, Gunning DB, Barua AB, Olson JA. Effects of subcutaneously injected graded doses of all-trans retinoic acid and all-trans retinoyl beta-glucuronide on the outcome of pregnancy in Sprague-Dawley rats. Int J Vitam Nutr Res. 2002;72:229–235. doi: 10.1024/0300-9831.72.4.229. [DOI] [PubMed] [Google Scholar]
- 75.Liu L, Gudas LJ. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J Biol Chem. 2005;280:40226–40234. doi: 10.1074/jbc.M509643200. [DOI] [PubMed] [Google Scholar]
- 76.Tsutsumi C, Okuno M, Tannous L, Piantedosi R, Allan M, Goodman DS, Blaner WS. Retinoids and retinoid-binding protein expression in rat adipocytes. J Biol Chem. 1992;267:1805–1810. [PubMed] [Google Scholar]
- 77.Zovich DC, Orologa A, Okuno M, Kong LW, Talmage DA, Piantedosi R, Goodman DS, Blaner WS. Differentiation-dependent expression of retinoid-binding proteins in BFC-1 beta adipocytes. J Biol Chem. 1992;267:13884–13889. [PubMed] [Google Scholar]
- 78.Blaner WS, Obunike JC, Kurlandsky SB, al-Haideri M, Piantedosi R, Deckelbaum RJ, Goldberg IJ. Lipoprotein lipase hydrolysis of retinyl ester. Possible implications for retinoid uptake by cells. J Biol Chem. 1994;269:16559–16565. [PubMed] [Google Scholar]
- 79.Okuno M, Caraveo VE, Goodman DS, Blaner WS. Regulation of adipocyte gene expression by retinoic acid and hormones: effects on the gene encoding cellular retinol-binding protein. J Lipid Res. 1995;36:137–147. [PubMed] [Google Scholar]
- 80.Wei S, Lai K, Patel S, Piantedosi R, Shen H, Colantuoni V, Kraemer FB, Blaner WS. Retinyl ester hydrolysis and retinol efflux from BFC-1beta adipocytes. J Biol Chem. 1997;272:14159–14165. doi: 10.1074/jbc.272.22.14159. [DOI] [PubMed] [Google Scholar]
- 81.Yen CL, Monetti M, Burri BJ, Farese RV., Jr The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J Lipid Res. 2005;46:1502–1511. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- 82.Orland MD, Anwar K, Cromley D, Chu CH, Chen L, Billheimer JT, Hussain MM, Cheng D. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: diacylglycerol acyltransferase 1. Biochim Biophys Acta. 2005;1737:76–82. doi: 10.1016/j.bbalip.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 83.Isken A, Holzschuh J, Lampert JM, Fischer L, Oberhauser V, Palczewski K, von Lintig J. Sequestration of retinyl esters is essential for retinoid signaling in the zebrafish embryo. J Biol Chem. 2006;282:1144–1151. doi: 10.1074/jbc.M609109200. [DOI] [PubMed] [Google Scholar]
- 84.Molotkov A, Ghyselinck NB, Chambon P, Duester G. Opposing actions of cellular retinol-binding protein and alcohol dehydrogenase control the balance between retinol storage and retinoic acid synthesis. Biochem J. 2004;383:295–302. doi: 10.1042/BJ20040621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003;3:136–158. doi: 10.1038/sj.tpj.6500171. [DOI] [PubMed] [Google Scholar]
- 86.Brierley CH, Senafi SB, Clarke D, Hsu MH, Johnson EF, Burchell B. Regulation of the human bilirubin UDP-glucuronosyltransferase gene. Adv Enzyme Regul. 1996;36:85–97. doi: 10.1016/0065-2571(95)00006-2. [DOI] [PubMed] [Google Scholar]
- 87.Zile MH, Schnoes HK, DeLuca HF. Characterization of retinoyl beta-glucuronide as a minor metabolite of retinoic acid in bile. Proc Natl Acad Sci USA. 1980;77:3230–3233. doi: 10.1073/pnas.77.6.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frolik CA, Swanson BN, Dart LL, Sporn MB. Metabolism of 13-cis-retinoic acid: identification of 13-cis-retinoyl- and 13-cis-4-oxoretinoyl-beta-glucuronides in the bile of vitamin A-normal rats. Arch Biochem Biophys. 1981;208:344–352. doi: 10.1016/0003-9861(81)90518-x. [DOI] [PubMed] [Google Scholar]
- 89.Napoli JL, Khalil H, McCormick AM. Metabolism of 5,6-epoxyretinoic acid in vivo: isolation of a major intestinal metabolite. Biochemistry. 1982;21:1942–1949. doi: 10.1021/bi00537a038. [DOI] [PubMed] [Google Scholar]
- 90.Golczak M, Imanishi Y, Kuksa V, Maeda T, Kubota R, Palczewski K. Lecithin:retinol acyltransferase is responsible for amidation of retinylamine, a potent inhibitor of the retinoid cycle. J Biol Chem. 2005;280:42263–42273. doi: 10.1074/jbc.M509351200. [DOI] [PubMed] [Google Scholar]
- 91.Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, Dierich A, Nilsson CB, Hakansson H, Sauvant P, Azais-Braesco V, Frasson M, Picaud S, Chambon P. Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J. 1999;18:4903–4914. doi: 10.1093/emboj/18.18.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.E X, Zhang L, Lu J, Tso P, Blaner WS, Levin MS, Li E. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. J Biol Chem. 2002;277:36617–36623. doi: 10.1074/jbc.M205519200. [DOI] [PubMed] [Google Scholar]
- 93.Vogel S, Mendelsohn CL, Mertz JR, Piantedosi R, Waldburger C, Gottesman ME, Blaner WS. Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. J Biol Chem. 2001;276:1353–1360. doi: 10.1074/jbc.M005118200. [DOI] [PubMed] [Google Scholar]
- 94.Piantedosi R, Ghyselinck N, Blaner WS, Vogel S. Cellular retinol-binding protein type III is needed for retinoid incorporation into milk. J Biol Chem. 2005;280:24286–24292. doi: 10.1074/jbc.M503906200. [DOI] [PubMed] [Google Scholar]
- 95.Randolph RK, Ross AC. Vitamin A status regulates hepatic lecithin:retinol acyltransferase activity in rats. J Biol Chem. 1991;266:16453–16457. [PubMed] [Google Scholar]
- 96.Zolfaghari R, Ross AC. Lecithin:retinol acyltransferase expression is regulated by dietary vitamin A and exogenous retinoic acid in the lung of adult rats. J Nutr. 2002;132:1160–1164. doi: 10.1093/jn/132.6.1160. [DOI] [PubMed] [Google Scholar]
- 97.Ross AC, Ambalavanan N, Zolfaghari R, Li NQ. Vitamin A combined with retinoic acid increases retinol uptake and lung retinyl ester formation in a synergistic manner in neonatal rats. J Lipid Res. 2006;47:1844–1851. doi: 10.1194/jlr.M600061-JLR200. [DOI] [PubMed] [Google Scholar]
- 98.Ross AC, Li NQ, Wu L. The components of VARA, a nutrient-metabolite combination of vitamin A and retinoic acid, act efficiently together and separately to increase retinyl esters in the lungs of neonatal rats. J Nutr. 2006;136:2803–2807. doi: 10.1093/jn/136.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vogel S, Piantedosi R, Frank J, Lalazar A, Rockey DC, Friedman SL, Blaner WS. An immortalized rat liver stellate cell line (HSC-T6): a new cell model for the study of retinoid metabolism in vitro. J Lipid Res. 2000;41:882–893. [PubMed] [Google Scholar]
- 100.Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong ES, Evans RM. A direct repeat in the cellular retinol-binding protein type II gene confers differential regulation by RXR and RAR. Cell. 1991;66:555–561. doi: 10.1016/0092-8674(81)90018-0. [DOI] [PubMed] [Google Scholar]
- 101.Tang XH, Suh MJ, Li R, Gudas LJ. Cell proliferation inhibition and alterations in retinol esterification induced by phytanic acid and docosahexaenoic acid. J Lipid Res. 2007;48:165–176. doi: 10.1194/jlr.M600419-JLR200. [DOI] [PubMed] [Google Scholar]
- 102.Guo X, Ruiz A, Rando RR, Bok D, Gudas LJ. Esterification of all-trans-retinol in normal human epithelial cell strains and carcinoma lines from oral cavity, skin and breast: reduced expression of lecithin:retinol acyltransferase in carcinoma lines. Carcinogenesis. 2000;21:1925–1933. doi: 10.1093/carcin/21.11.1925. [DOI] [PubMed] [Google Scholar]
- 103.Guo X, Nanus DM, Ruiz A, Rando RR, Bok D, Gudas LJ. Reduced levels of retinyl esters and vitamin A in human renal cancers. Cancer Res. 2001;61:2774–2781. [PubMed] [Google Scholar]
- 104.Guo X, Knudsen BS, Peehl DM, Ruiz A, Bok D, Rando RR, Rhim JS, Nanus DM, Gudas LJ. Retinol metabolism and lecithin:retinol acyltransferase levels are reduced in cultured human prostate cancer cells and tissue specimens. Cancer Res. 2002;62:1654–1661. [PubMed] [Google Scholar]
- 105.Simmons DP, Andreola F, De Luca LM. Human melanomas of fibroblast and epithelial morphology differ widely in their ability to synthesize retinyl esters. Carcinogenesis. 2002;23:1821–1830. doi: 10.1093/carcin/23.11.1821. [DOI] [PubMed] [Google Scholar]
- 106.Zhan HC, Gudas LJ, Bok D, Rando R, Nanus DM, Tickoo SK. Differential expression of the enzyme that esterifies retinol, lecithin:retinol acyltransferase, in subtypes of human renal cancer and normal kidney. Clin Cancer Res. 2003;9:4897–4905. [PubMed] [Google Scholar]
- 107.Boorjian S, Tickoo SK, Mongan NP, Yu H, Bok D, Rando RR, Nanus DM, Scherr DS, Gudas LJ. Reduced lecithin: retinol acyltransferase expression correlates with increased pathologic tumor stage in bladder cancer. Clin Cancer Res. 2004;10:3429–3437. doi: 10.1158/1078-0432.CCR-03-0756. [DOI] [PubMed] [Google Scholar]
- 108.Sheren-Manoff M, Shin SJ, Su D, Bok D, Rando RR, Gudas LJ. Reduced lecithin:retinol acyltransferase expression in human breast cancer. Int J Oncol. 2006;29:1193–1199. [PubMed] [Google Scholar]
- 109.Lotan R, Ong DE, Chytil F. Comparison of the level of cellular retinoid-binding proteins and susceptibility to retinoid-induced growth inhibition of various neoplastic cell lines. J Natl Cancer Inst. 1980;64:1259–1262. [PubMed] [Google Scholar]
- 110.Fex G, Ekelund G, Leandoer L, Sternby NH. Cellular retinol-binding protein (CRBP) in human colorectal adenocarcinoma. Br J Cancer. 1986;53:687–690. doi: 10.1038/bjc.1986.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fex G, Wahlberg P, Biorklund A, Wennerberg J, Willen R. Studies of cellular retinol-binding protein (CRBP) in squamous-cell carcinomas of the head and neck region. Int J Cancer. 1986;37:217–221. doi: 10.1002/ijc.2910370208. [DOI] [PubMed] [Google Scholar]
- 112.Fex G, Linell F, Ljungberg O. Cellular retinol-binding protein in normal and neoplastic human mammary gland. Breast Cancer Res Treat. 1985;6:131–136. doi: 10.1007/BF02235744. [DOI] [PubMed] [Google Scholar]
- 113.Kuppumbatti YS, Bleiweiss IJ, Mandeli JP, Waxman S, Mira YLR. Cellular retinol-binding protein expression and breast cancer. J Natl Cancer Inst. 2000;92:475–480. doi: 10.1093/jnci/92.6.475. [DOI] [PubMed] [Google Scholar]
- 114.Kuppumbatti YS, Rexer B, Nakajo S, Nakaya K, Mira-y-Lopez R. CRBP suppresses breast cancer cell survival and anchorage-independent growth. Oncogene. 2001;20:7413–7419. doi: 10.1038/sj.onc.1204749. [DOI] [PubMed] [Google Scholar]
- 115.Arapshian A, Bertran S, Kuppumbatti YS, Nakajo S, Mira-y-Lopez R. Epigenetic CRBP downregulation appears to be an evolutionarily conserved (human and mouse) and oncogene-specific phenomenon in breast cancer. Mol Cancer. 2004;3:13. doi: 10.1186/1476-4598-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lotan R. A crucial role for cellular retinol-binding protein I in retinoid signaling. J Natl Cancer Inst. 2005;97:3–4. doi: 10.1093/jnci/dji031. [DOI] [PubMed] [Google Scholar]
- 117.Farias EF, Ong DE, Ghyselinck NB, Nakajo S, Kuppumbatti YS, Mira y Lopez R. Cellular retinol-binding protein I, a regulator of breast epithelial retinoic acid receptor activity, cell differentiation, and tumorigenicity. J Natl Cancer Inst. 2005;97:21–29. doi: 10.1093/jnci/dji004. [DOI] [PubMed] [Google Scholar]
- 118.Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, Curry CJ, Fernhoff PM, Grix AW, Jr, Lott IT, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313:837–841. doi: 10.1056/NEJM198510033131401. [DOI] [PubMed] [Google Scholar]
- 119.Soprano DR, Soprano KJ. Retinoids as teratogens. Annu Rev Nutr. 1995;15:111–132. doi: 10.1146/annurev.nu.15.070195.000551. [DOI] [PubMed] [Google Scholar]
- 120.Collins MD, Mao GE. Teratology of retinoids. Annu Rev Pharmacol Toxicol. 1999;39:399–430. doi: 10.1146/annurev.pharmtox.39.1.399. [DOI] [PubMed] [Google Scholar]
- 121.Rothman KJ, Moore LL, Singer MR, Nguyen US, Mannino S, Milunsky A. Teratogenicity of high vitamin A intake. N Engl J Med. 1995;333:1369–1373. doi: 10.1056/NEJM199511233332101. [DOI] [PubMed] [Google Scholar]
- 122.Cohlan SQ. Excessive intake of vitamin A as a cause of congenital anomalies in the rat. Science. 1953;117:535–536. doi: 10.1126/science.117.3046.535. [DOI] [PubMed] [Google Scholar]
- 123.Cohlan SQ. Congenital anomalies in the rat produced by excessive intake of vitamin A during pregnancy. Pediatrics. 1954;13:556–567. [PubMed] [Google Scholar]
- 124.Geelen JA, Langman J, Lowdon JD. The influence of excess vitamin A on neural tube closure in the mouse embryo. Anat Embryol. 1980;159:223–234. doi: 10.1007/BF00304980. [DOI] [PubMed] [Google Scholar]
- 125.Collins MD, Tzimas G, Hummler H, Burgin H, Nau H. Comparative teratology and transplacental pharmaco-kinetics of all-trans-retinoic acid, 13-cis-retinoic acid, and retinyl palmitate following daily administrations in rats. Toxicol Appl Pharmacol. 1994;127:132–144. doi: 10.1006/taap.1994.1147. [DOI] [PubMed] [Google Scholar]
- 126.Abroms L, Maibach E, Lyon-Daniel K, Feldman SR. What is the best approach to reducing birth defects associated with isotretinoin? PLoS Med. 2006;3:e483. doi: 10.1371/journal.pmed.0030483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Standeven AM, Davies PJ, Chandraratna RA, Mader DR, Johnson AT, Thomazy VA. Retinoid-induced epiphyseal plate closure in guinea pigs. Fundam Appl Toxicol. 1996;34:91–98. doi: 10.1006/faat.1996.0179. [DOI] [PubMed] [Google Scholar]
- 128.Prendiville J, Bingham EA, Burrows D. Premature epiphyseal closure–a complication of etretinate therapy in children. J Am Acad Dermatol. 1986;15:1259–1262. doi: 10.1016/s0190-9622(86)70300-9. [DOI] [PubMed] [Google Scholar]
- 129.Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111:761–772. doi: 10.1001/archopht.1993.01090060049022. [DOI] [PubMed] [Google Scholar]
- 130.Sibulesky L, Hayes KC, Pronczuk A, Weigel-DiFranco C, Rosner B, Berson EL. Safety of <7500 RE (<25000 IU) vitamin A daily in adults with retinitis pigmentosa. Am J Clin Nutr. 1999;69:656–663. doi: 10.1093/ajcn/69.4.656. [DOI] [PubMed] [Google Scholar]
- 131.Nagai K, Hosaka H, Kubo S, Nakabayashi T, Amagasaki Y, Nakamura N. Vitamin A toxicity secondary to excessive intake of yellow-green vegetables, liver and laver. J Hepatol. 1999;31:142–148. doi: 10.1016/s0168-8278(99)80174-3. [DOI] [PubMed] [Google Scholar]
- 132.Guarascio P, Portmann B, Visco G, Williams R. Liver damage with reversible portal hypertension from vitamin A intoxication: demonstration of Ito cells. J Clin Pathol. 1983;36:769–771. doi: 10.1136/jcp.36.7.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nollevaux MC, Guiot Y, Horsmans Y, Leclercq I, Rahier J, Geubel AP, Sempoux C. Hypervitaminosis A-induced liver fibrosis: stellate cell activation and daily dose consumption. Liver Int. 2006;26:182–186. doi: 10.1111/j.1478-3231.2005.01207.x. [DOI] [PubMed] [Google Scholar]
- 134.Hautekeete ML, Geerts A. The hepatic stellate (Ito) cell: its role in human liver disease. Virchows Arch. 1997;430:195–207. doi: 10.1007/BF01324802. [DOI] [PubMed] [Google Scholar]
- 135.Scheven BA, Hamilton NJ. Retinoic acid and 1,-25-dihydroxyvitamin D3 stimulate osteoclast formation by different mechanisms. Bone. 1990;11:53–59. doi: 10.1016/8756-3282(90)90072-7. [DOI] [PubMed] [Google Scholar]
- 136.Baxi SC, Dailey GE., 3rd Hypervitaminosis A. A cause of hypercalcemia. West J Med. 1982;137:429–431. [PMC free article] [PubMed] [Google Scholar]
- 137.Genaro P, de S, Martini LA. Vitamin A supplementation and risk of skeletal fracture. Nutr Rev. 2004;62:65–7. doi: 10.1111/j.1753-4887.2004.tb00026.x. [DOI] [PubMed] [Google Scholar]
- 138.Melhus H, Michaelsson K, Kindmark A, Bergstrom R, Holmberg L, Mallmin H, Wolk A, Ljunghall S. Excessive dietary intake of vitamin A is associated with reduced bone mineral density and increased risk for hip fracture. Ann Intern Med. 1998;129:770–778. doi: 10.7326/0003-4819-129-10-199811150-00003. [DOI] [PubMed] [Google Scholar]
- 139.Maeda A, Maeda T, Golczak M, Imanishi Y, Leahy P, Kubota R, Palczewski K. Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol Pharmacol. 2006;70:1220–1229. doi: 10.1124/mol.106.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]