

Anatomopathological and clinical evidence that has accumulated over the past 20 years has caused us to reevaluate our traditional concepts of the pathogenesis of atherosclerosis. In particular, these data have challenged our notions of the mechanisms by which chronic stable or asymptomatic disease transitions to the acute thrombotic manifestations of atherosclerosis that bring patients to our attention as practitioners most dramatically. This article updates our previous contribution to this journal on the subject of inflammation in plaque pathogenesis.[1]
Rather than a progressive stenosis to a chronically flow-limiting lesion, we now recognize that thrombotic complications result from physical disruption of the atherosclerotic plaque.[2][3][4] The major mechanisms that cause such disruption include rupture of the plaque's fibrous cap and superficial erosion of the endothelial monolayer (Figure 1).[5] The fibrous cap overlies the lipid core of the atheromatous, fatty plaque. The fracture of this structure exposes blood to procoagulants in the lipid core and triggers thrombosis. This mechanism accounts for some two-thirds to three-quarters of fatal acute myocardial infarction, based on autopsy studies. Superficial erosion accounts for fatal coronary thrombosis in about 20-25% of cases.
1.
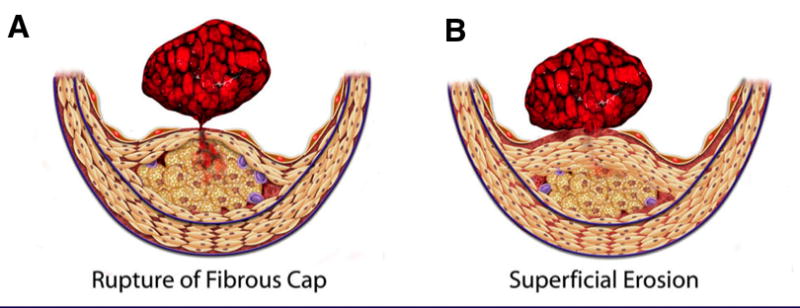
Fibrous cap rupture and superficial erosion. Rupture of the fibrous cap (left) triggers two-thirds to three-quarters of all cases of fatal coronary thromboses. Superficial erosion (right) takes place in one-fifth to one-quarter of fatal coronary thromboses. Certain populations, such as individuals with diabetes and women, appear to have superficial erosion more frequently as a mechanism of plaque disruption and thrombosis. (Libby P, Theroux P. Circulation 2005;111:3481-3488.)
Basic inflammation biology and mechanisms that regulate the integrity of the atherosclerotic plaque's fibrous cap
Because of the fundamental role of plaque disruption in the acute coronary syndromes, our laboratory sought to make the link between this aspect of plaque complication and the vascular biology of inflammation that had occupied our laboratory and others over the years. We reasoned that understanding the local control of the metabolism of collagen, i.e., the major structural component of the plaque's fibrous cap, would provide insights into thrombotic complications.
To make the link with inflammation biology, we hypothesized that inflammatory mediators would decrease the synthesis of interstitial collagen, a product of the smooth muscle cells (SMCs) in the artery wall that lends strength to the fibrous cap.[6] We performed metabolic labeling experiments that monitored de novo collagen synthesis by cultured human SMCs. We found that the inflammatory mediator gamma interferon markedly inhibited the ability of SMCs to manufacture new collagen. Even when stimulated by transforming growth factor-beta (TGF-β), the most potent stimulus of collagen synthesis, co-incubation with interferon gamma reduced the rate of new collagen synthesis to baseline or below. Even at the time we conducted these early experiments on the regulation of interstitial collagen synthesis by vascular SMCs, excellent evidence supported the operation of gamma interferon in the atherosclerotic plaque. For example, cells in the atheroma express major histocompatibility class II molecules, proteins whose expression depends for the most part on gamma interferon.[7][8] The major source of gamma-, or immune-, interferon, the T lymphocyte, also localizes in atheroma. Indeed, morphometric studies showed an inverse localization of T cells in plaques, and procollagen gene expression.[9] These results taken together provided support for the regulation by T cells involved in the adaptive immune response of the synthesis of collagen in the plaque needed to repair and maintain its all important protective fibrous cap.
The steady state level of interstitial collagen, like all macromolecules, depends not only on its rate of synthesis, but also its catabolism.[10] Interstitial collagen, a quite stable molecule under normal circumstances, resists the proteolytic attack of most enzymes. Interstitial collagenases, most of which belong to the matrix metalloproteinase (MMP) family, can initiate proteolytic cleavage on the intact triple helical collagen molecule. This cleavage performed by MMPs begins the catabolic cascade of collagen. Normal arteries do not express active collagenases. Macrophages in human atherosclerotic plaques, however, do express interstitial collagenases.[11][12] Over the years, our group demonstrated over-expression of MMP-1, the prototypical collagenase, in SMCs and in foam cells in plaques, and other MMP interstitial collagenases including MMP-13 and MMP-8.[13][14] Our studies showed that not only neutrophils express MMP-8, known as neutrophil collagenase, but also endothelial cells, SMCs, and macrophages in human atherosclerotic plaques.[14]
Plaques also overexpress active forms of the gelatinases MMP-2 and MMP-9, enzymes implicated in later steps of collagen catabolism after the collagenases make the initial attack on this critical constituent of the arterial extracellular matrix.[11] Plaques also contain other matrix-degrading enzymes including potent elastases (MMP-9, cathepsins S and K, neutrophil elastase) that may have important roles in arterial remodeling and aneurysm formation.[15][16] For detailed discussion of the role of these elastases in atherosclerosis, see elsewhere.[17]
Further studies sought to determine whether the immunostainable excess of collagenase actually reflects a stoichiometric excess of active enzyme over its endogenous inhibitors. The ubiquitous tissue inhibitors of matrix metalloproteinases (TIMPs) regulate their action in many tissues. Plaques contain TIMPs 1-4.[18][19][20] Therefore it was critical to determine if MMPs, even if in their biochemically activated form, could actually prevail over their endogenous inhibitors. We demonstrated immunochemical evidence of collegenolysis in situ in plaques by use of an antibody that selectively recognizes an intermediate in the degradation of interstitial collagen.[13] These areas of collagen degradation in the atherosclerotic plaque co-localized with the regions populated by macrophages over-expressing MMP-1 and MMP-13.(Figure 2)
2.
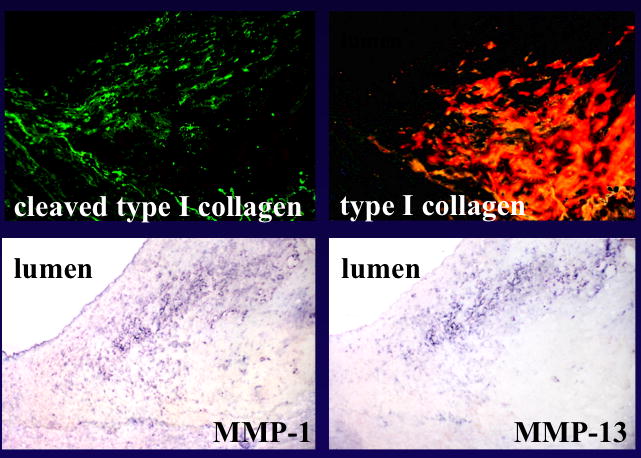
Collagenolysis in human atheromatous and fibrous plaques. Immunofluorescence for collagenase-cleaved type I collagen neoepitope (FITC, green; top left) and collagen type I (Texas red; top right) revealed cleaved collagen in the shoulder area. Adjacent sections were stained immunohistochemically for MMP-1 and -13, as well as for human Mϕ (bottom). (Sukhova GK et al. Circulation 1999;99:2503-2509.)
These various studies support the concept that inflammation in the intima places collagen in double jeopardy, i.e., decreased synthesis and increased breakdown, and thereby sets the stage for plaque rupture and the acute thrombotic complications of atherosclerosis. According to this model of molecular pathogenesis of acute plaque thrombosis, when inflammation prevails in the intima, T-cells can signal to the SMCs through gamma interferon. This T-cell-derived cytokine can restrict new collagen synthesis necessary to repair and maintain the interstitial collagen in the fibrous cap of the plaque. The T-cell can also send signals to the macrophage, elevating the production of interstitial collagenases that can cleave this collagen. Among these signals, CD40 ligand (CD154) appears prominent in its ability to stimulate proteinase production by macrophages (Figure 3).
3.

Inflammation regulates metabolism of fibrillar collagen, which may influence atherosclerotic plaque disruption. The T-lymphocyte releases proinflammatory cytokines such as IFN-γ (lower left) that inhibit SMCs from producing the new collagen required to lay down the collagenous matrix of the plaque's fibrous cap, which protects the plaque from rupture. The T-cell-derived cytokine CD40L stimulates mononuclear phagocytes (center) to elaborate interstitial collagenases including MMP-1, MMP-8, and MMP-13, which catalyze the initial proteolytic cleavage of the intact collagen fibril. The cleaved collagen can then undergo additional degradation by gelatinases such as MMP-9. In this way, inflammation can threaten the stability of atherosclerotic plaques; increase their tendency to rupture; and, hence, cause thromboses that trigger most acute coronary syndromes. TGFβ, transforming growth factor β. (Libby P., In: Morrow D, editor. Contemporary Cardiology, 2006)
Studies in genetically engineered mice support the role of matrix metalloproteinases in regulation of plaque structure
More recently we have obtained direct evidence that links interstitial collagenases with the regulation of collagen content of atheroma through work with genetically altered mice. In mice engineered to express a collagenase-resistant form of collagen by “knock-in” technology on an atherosclerosis-susceptible background (deficiency of apolipoprotein E), we showed accumulation of collagen in lesions produced by an atherogenic diet.[21] A converse experiment in compound mutant mice lacking both MMP-13, an important interstitial collagenase, and apolipoprotein E showed greater accumulation of collagen in the intimal lesions provoked by cholesterol feeding.[22] These experiments indicate that collagenase indeed influences importantly the economy of collagen in the experimental atherosclerotic plaque. Very recent work with mice engineered to lack another MMP (MMP-14) in cells derived from bone marrow showed a role for this membrane-associated MMP in plaque collagen accumulation, quite possibly due at least in part to an effect on the activation of the interstitial collagenases.[23]
Additional studies with advanced optical techniques showed that atheromata in the MMP-13–deficient mice display more organized collagen fibers.[22] The lesions in these mice had thicker collagen fibers, and tighter distribution of their angular dispersion than atheromata in mice wild type for MMP-13. Hence, the MMP-13–deficient mice had both more and better-organized collagen. Thus, the collagenases can contribute to both the accumulation and architecture of collagen in the atherosclerotic plaque of mice.
Basic inflammation biology and mechanisms that regulate the thrombogenicity of the plaque's lipid core
Fracture of the plaque's fibrous cap permits blood to contact the content of the compartment within the atheroma, notably the lipid-rich core, often a site of inflammation, and favors thrombus formation. What mechanisms trigger thrombosis when such plaque disruption occurs? The groups of Edgington, Nemerson, and Wilcox pointed to the expression of tissue factor in a subpopulation of plaque macrophages as a likely instigator of thrombosis when these inflammatory cells come in contact with the coagulation factors of blood.[24][25][26] The signals that induce tissue factor expression by macrophages, and thus regulate their thrombogenicity, include inflammatory mediators found in the plaque. Surprisingly, the usual soluble cytokines including interleukin-1, tumor necrosis factor, and interferon gamma have little effect on macrophage tissue factor expression. The exogenous inflammatory stimulus bacterial lipopolysaccharide, a mediator of innate immunity that signals through toll-like receptors (notably TLR-4) does augment tissue factor gene expression in mononuclear phagocytes.[27] Our group identified an endogenous signal CD40 ligand that can boost tissue factor expression by human monocyte/macrophages, an example of how inflammation can control not only the susceptibility of the plaque to rupture as described above, but also its thrombogenicity.[28]
Platelets can also exteriorize CD40 ligand upon activation, providing a potential amplification loop in thrombosis.[29] Such a pathway might operate when microvessels in plaques rupture, cause intraplaque hemorrhage, and thrombosis in situ. Platelet-derived CD40 ligand might stimulate local tissue factor production, and amplify the consequences of an eventual plaque disruption, exposing that region to the blood compartment. In superficial erosion (see below), the contact of platelets with the subendothelial extracellular matrix and other constituents of the basement membrane can activate platelets, and cause them to adhere. The CD40 ligand exposed by platelets in this circumstance could activate tissue factor on neighboring endothelial cells and propagate local formation of coagulum on the base of a platelet thrombus. Other links between thrombosis and inflammation have emerged, amply illustrating the principal that inflammation can beget thrombosis and vice versa (Figure 4).[30][31]
4.
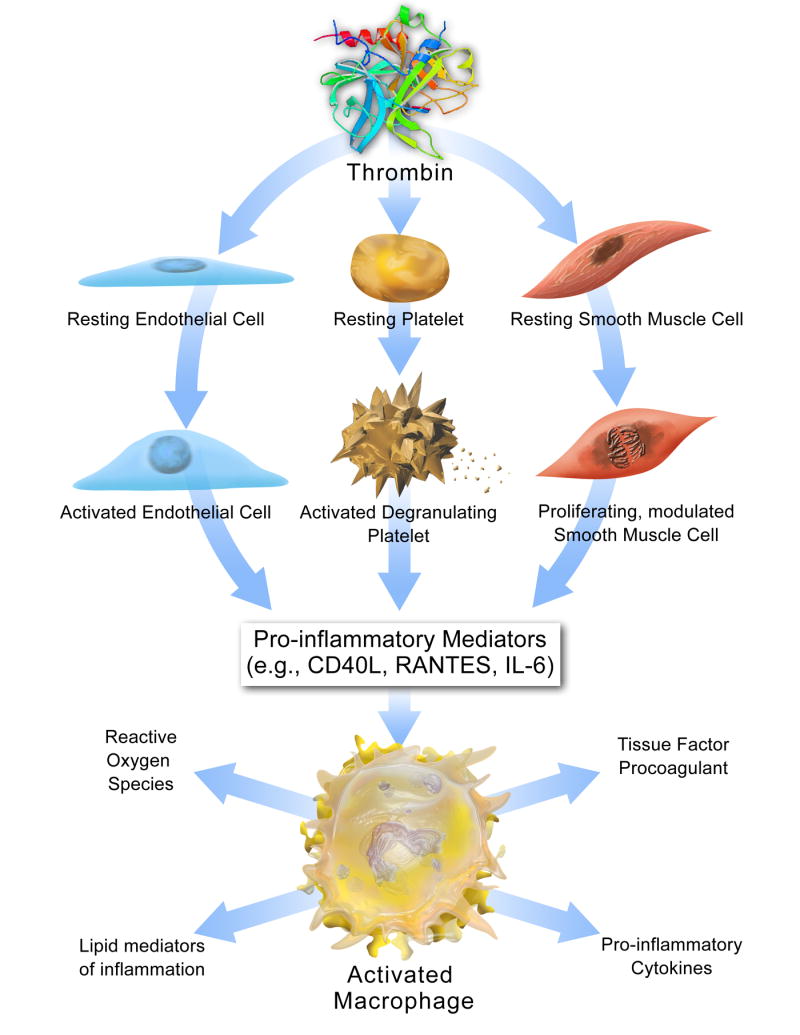
The relationship between thrombosis and inflammation. Thrombin stimulation prompts secretion of platelet, endothelial cell, and smooth muscle cell factors that trigger macrophages to produce tissue factor, pro-inflammatory cytokines, lipid mediators of inflammation, and reactive oxygen species. Via these mechanisms, thrombin inititates vascular inflammation, which promotes additional thrombin production through amplification loops that perpetuate atherogenic signals. (Croce K., Libby P. Curr Opin Hematol 2007;14:55)
Mechanisms of superficial erosion of endothelial monolayer
Superficial erosion of the endothelial monolayer provides another mechanism of plaque disruption, particularly in women and in persons with hypertriglyceridemia. We hypothesized that apoptosis of endothelial cells in response to inflammatory mediators could heighten the risk of this superficial erosion (Figure 5).[32] In particular, we wondered whether reactive oxygen species generated in response to inflammatory stimuli might promote endothelial cell apoptosis or impair the tethering of the endothelial cell to its subadjacent basement membrane.
5.
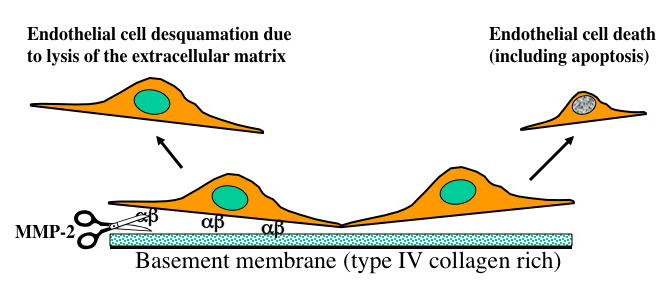
Potential mechanisms of superficial erosion leading to coronary thrombosis. Death of endothelial cells, for example by apoptosis, may cause desquamation and facilitate thrombosis as platelets encounter subendothelial pro-aggregatory collagen. Lysis of basement membrane (type IV) collagen by type IV collagenase (MMP-2) activated by inflammatory mediators may accelerate endothelial apoptosis and desquamation as well by severing attachment of the cells to the basement membrane. The endothelial cells attach to their subjacent matrix in part through membrane integrins, represented here by alpha and beta. (Libby P., In: Topol EJ, editor. Acute Coronary Syndromes, 2nd ed., 2000)
We found evidence for involvement of myeloperoxidase (MPO) in plaque disruption and thrombosis.[33] The enzyme MPO, released by activated granulocytes and in the atherosclerotic lesion by a monocyte subpopulation, can serve as a biomarker of risk for acute coronary events.[34][35][36] At sites of inflammation, MPO can bind to the extracellular matrix and convert chloride ion plus hydrogen peroxide to hypochlorous acid, a potent oxidant and chlorinating species. We and others have provided histochemical evidence of the expression of MPO and hypochlorous acid–modified proteins in plaques that have ruptured or undergone superficial erosion.[37][33] Further work has indicated that hypochlorous acid at concentrations relevant to sites of inflammation provokes programmed death of endothelial cells.[38] Increasing concentrations of hypochlorous acid caused laddering of the DNA in the endothelial cell consistent with oligonucleosomal fragmentation, characteristic of apoptosis, and increased caspase-3, the machinery enzyme involved in the apoptotic pathway. In this manner, oxidative stress, a process inextricably linked to inflammation, may sensitize endothelial monolayers to desquamation and thereby cause superficial erosion.
In addition, matrix-degrading proteinases may participate in the degradation of the basement membrane that furnishes the foundation on which endothelial cells reside in vessels. In particular, pro-inflammatory cytokines can augment the expression of gelatinases such as MMP-2 and MMP-9 that can attack the type IV collagen (a non-fibrillar form of collagen), a prominent component of the sub-endothelial basement membrane.[39][40] MMP-14, the prototypical membrane-bound MMP, can convert the inactive zymogen form of MMP-2 to its enzymatically active form.[41] We reported that oxidatively modified low-density lipoprotein, a putative driver of inflammation during atherogenesis, can augment endothelial expression of MMP-14.[42] Thus, inflammation may not only promote death of endothelial cells by augmenting apoptosis, but may also favor desquamation of the endothelium at site of inflammation via the overexpression and activation of type IV collagenases such as MMP-2. Aside from plaque disruption, our studies of matrix-degrading proteinases in atherosclerosis support their roles in angiogenesis, in ectasia including aneurysm formation, and potentially in other aspects of arterial remodeling including the compensatory enlargement of arteries that can conceal the disease from clinical symptoms or detection during much of its progression.[20]
Plaque thrombosis: a consequence of interaction of the “solid state” of the plaque with the “fluid phase” of blood
The sections above have focused on inflammation as a regulator of the features of the plaque itself that give rise to thrombosis. Such characteristics may include abundance of inflammatory leukocytes, paucity of collagen, heightened tissue factor expression, oxidative stress (including that mediated by MPO) and the action of matrix-degrading proteinases. These features determine the propensity of a plaque to rupture or erode and thus trigger thrombosis, according to much current thought and observation. I have referred to these aspects of the atheroma as its “solid state”(Figure 6).[4] Yet, plaque disruption by itself does not ineluctably determine the degree of thrombosis that may ensue.
6.
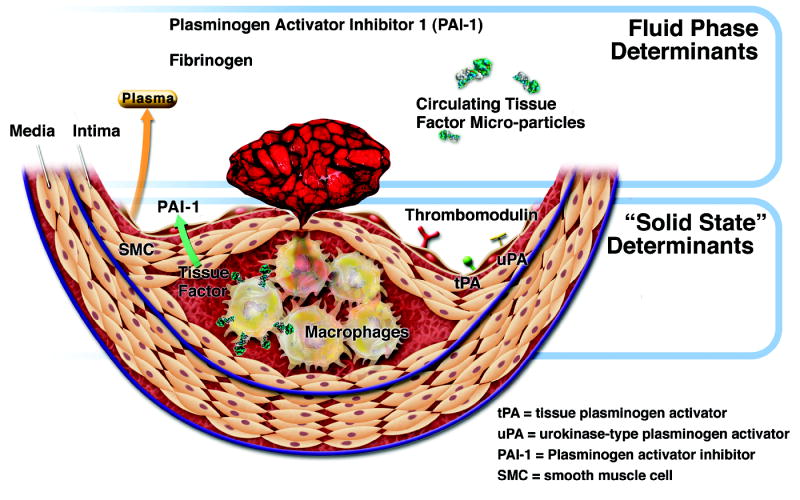
Thrombotic determinants in coronary atherosclerotic plaques. Formation, extenet, and duration of coronary thrombi produced by mechanisms such as those outlined in Figure 1 rely on both solid-state factors in the plaque itself and fluid-phase determinants in blood. See text for further explanation. (Libby P, Theroux P. Circulation 2005;111:3481-3488.)
The “fluid phase” of blood probably critically influences the consequences of any given plaque disruption. For example, the levels of fibrinogen and the endogenous inhibitor of fibrinolysis, plasminogen activator inhibitor-1 (PAI-1), can modulate coagulation and fibrinolysis respectively. These “fluid phase” factors can thus worsen or mitigate the formation and stability of a thrombus, depending on their levels.
Of note, inflammation can regulate the blood levels of these two “fluid phase” factors. Inflammatory signaling can alter the pattern of protein synthesis by the liver to favor production of the acute phase reactants. Some acute phase proteins such as CRP may serve as markers of risk of plaque complication, but others may actually promote coagulation (fibrinogen) or clot stability (PAI-1). Thus inflammation not only modulates the “solid state” features that influence plaque susceptibility to disruption, but also those of the “fluid phase” that determine the clinical fate of a particular plaque disturbance, ranging from a microscopic mural thrombus to a disastrous sustained and occlusive arterial thrombus.[4]
The healing of disrupted atheromata: a role for thrombosis-induced fibrosis in plaque progression
We have learned from the pathologists that plaque disruption probably usually occurs subclinically, and does not lead to a propagated, occlusive thrombus that causes ischemia and symptoms.[43][44] Autopsy studies of victims of non-cardiac death and studies of hearts explanted from transplantation recipients have shown mural thrombi in coronary arteries despite the apparent clinical stability. Our early studies of collagen synthesis by vascular SMCs revealed that multiple mediators released at sites of thrombosis could augment procollagen gene expression and protein production.[6] For example, platelet-derived growth factor (PDGF) and in particular transforming growth factor-beta, both constituents of platelets released upon activation, boost interstitial collagen synthesis by vascular SMC. Local elaboration of PDGF and generation of thrombin also likely recruits SMC by migration and mitogenesis, providing more cells capable of producing collagen. Thus, episodes of plaque disruption and mural thrombus formation well below the clinical threshold, and apparently quite common according to pathologists, may push plaque progression toward a fibrotic morphology. Such collagen-rich plaques may prove less prone to rupture, having thicker fibrous caps. Fibrous plaques have a greater ratio of extracellular matrix to lipid core, indeed many interpreted a “late stage” or “burned-out” lesion may have scant inflammatory cells, and substantial collagen accumulation and calcification. While not proven rigorously, the morphologic data, and the biological findings described above, support the notion that sub-clinical plaque disruption may play a role in the evolution of an atheromatous, lipid-rich, and highly inflamed plaque, to a fibrous lesion less likely to provoke an abrupt thrombosis. The fibrotic process may promote constrictive remodeling, a characteristic of plaques that cause stenosis. Thus, more “stable” stenotic lesions may reflect the product of a precursor inflamed or lipid-laden plaque that has disrupted, and undergone fibrosis as part of the healing response. While the proteinases associated with inflamed plaques may favor outward remodeling of the artery (“compensatory enlargement”), the healing response that ensues following rupture can cause constrictive remodeling consistent with flow-limiting plaques.
Applying molecular imaging to vascular biology
The proteolytic potential of plaque does not only predispose toward atheroma disruption and thrombosis. As a biological consequence of inflammatory activation of cells within atheroma of considerable function import, protease activity may serve as a “read-out” of plaque inflammation. We have collaborated with the group led by Professor Ralph Weissleder specializing in molecular imaging to study the possibility of monitoring proteinase activity in vivo and apply that modality to vascular biology.[45] We have taken steps toward imaging proteinase activity. For example, we have published early work with this technique in vivo and in vitro with the gelatinases MMP-2 and MMP-9, and performed pilot work with MMP-13, a collagenase for which we have demonstrated involvement in plaque collagen metabolism in plaques in mice and overexpression in human lesions.[46] Our mouse studies have also demonstrated the feasibility of imaging cathepsin K, an elastase of considerable interest in the pathogenesis of atherothrombosis.[47]
Current methodologies used by us for proteinase imaging involve near-infrared fluorescence. The penetration of the excitation and emission suffices for in vivo visualization of proteinases in mice using fluorescent tomographic techniques. While such methodology may prove applicable to superficial human arteries (e.g., the carotids and iliacs), catheter-based methods for intravascular detection might permit extension to coronary arteries. This extension to humans will require considerable further effort. With such development and validation, transfer of this technology to humans could provide at least a research tool. The ability to visualize proteinase activity in atheromata could furnish a method for testing the efficacy of interventions that modulate plaque inflammation that might evade evaluation by imaging modalities that detect anatomical features such as plaque volume or percent stenosis. Application to humans of molecular imaging could help validate pathophysiologic hypotheses and provide a more efficient way to develop and test therapeutic agents in the future.[45]
Conclusion
Our concepts of the thrombotic complication of atherosclerosis have undergone major revisions and deepening over the last decades. The story told above reflects a true translational tale, inspired by clinical and pathological findings, harnessing basic science to probe mechanisms suggested by clinical scenarios, and beginning to lead back to the bedside to improve knowledge of pathophysiology, diagnosis, and treatment. For example, our experimental studies have shown that lipid lowering can ameliorate features of experimental atheromata associated with thrombotic complications in humans, such as limiting inflammation, increasing collagen content, and reducing tissue factor levels.[48] Clinical studies have affirmed anti-inflammatory actions in humans of lipid-lowering treatments such as statin therapy, as gauged by reduction in C-reactive protein (CRP).[49] Current data suggest that some of the CRP lowering derives from lipoprotein-mediated effects, and some from direct anti-inflammatory actions.[50][51] The future use of molecular imaging and other biomarkers may continue to broaden the clinical validation of concepts that arise in the experimental laboratory. The bi-directional exchange of information from the laboratory to the clinic in unraveling the pathophysiology of the thrombotic complications of atherosclerosis has proven quite gratifying over the last few decades, and lays the foundation for further progress in this field in the future.
References
- 1.Libby P. Changing concepts of atherogenesis. J Intern Med. 2000;247:349–58. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 2.Falk E, Shah P, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–71. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- 3.Davies MJ. Stability and instability: the two faces of coronary atherosclerosis. The Paul Dudley White Lecture, 1995. Circulation. 1996;94:2013–20. doi: 10.1161/01.cir.94.8.2013. [DOI] [PubMed] [Google Scholar]
- 4.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–8. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 5.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–8. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 6.Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate intersitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1991;11:1223–30. doi: 10.1161/01.atv.11.5.1223. [DOI] [PubMed] [Google Scholar]
- 7.Hansson GK, Jonasson L, Holm J, Claesson-Welsh L. Class II MHC antigen expression in the atherosclerotic plaque: smooth muscle cells express HLA-DR, HLA-DQ and the invariant gamma chain. Clin Exp Immunol. 1986;64:261–8. [PMC free article] [PubMed] [Google Scholar]
- 8.Warner SJC, Friedman GB, Libby P. Regulation of major histocompatibility gene expression in cultured human vascular smooth muscle cells. Arteriosclerosis. 1989;9:279–88. doi: 10.1161/01.atv.9.3.279. [DOI] [PubMed] [Google Scholar]
- 9.Rekhter M, Zhang K, Narayanan A, Phan S, Schork M, Gordon D. Type I collagen gene expression in human atherosclerosis. Localization to specific plaque regions. Am J Pathol. 1993;143:1634–48. [PMC free article] [PubMed] [Google Scholar]
- 10.Libby P. The molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–50. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 11.Galis Z, Sukhova G, Lark M, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikkari ST, O'Brien KD, Ferguson M, Hatsukami T, Welgus HG, Alpers CE, Clowes AW. Interstitial collagenase (MMP-1) expression in human carotid atherosclerosis. Circulation. 1995;92:1393–8. doi: 10.1161/01.cir.92.6.1393. [DOI] [PubMed] [Google Scholar]
- 13.Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99:2503–9. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- 14.Herman MP, Sukhova GK, Libby P, et al. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104:1899–904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- 15.Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–83. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dollery CM, Owen CA, Sukhova GK, Krettek A, Shapiro SD, Libby P. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation. 2003;107:2829–36. doi: 10.1161/01.CIR.0000072792.65250.4A. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Sukhova GK, Sun JS, Xu WH, Libby P, Shi GP. Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1359–66. doi: 10.1161/01.ATV.0000134530.27208.41. [DOI] [PubMed] [Google Scholar]
- 18.Galis ZS, Muszynski M, Sukhova GK, Simon-Morrissey E, Libby P. Enhanced expression of vascular matrix metalloproteinases induced in vitro by cytokines and in regions of human atherosclerotic lesions. Annals of the New York Academy of Sciences. 1995;748:501–7. doi: 10.1111/j.1749-6632.1994.tb17348.x. [DOI] [PubMed] [Google Scholar]
- 19.Fabunmi RP, Sukhova GK, Sugiyama S, Libby P. Expression of tissue inhibitor of metalloproteinases-3 in human atheroma and regulation in lesion-associated cells: a potential protective mechanism in plaque stability. Circulation Research. 1998;83:270–8. doi: 10.1161/01.res.83.3.270. [DOI] [PubMed] [Google Scholar]
- 20.Dollery CM, Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. 2006;69:625–35. doi: 10.1016/j.cardiores.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Fukumoto Y, Deguchi JO, Libby P, et al. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation. 2004;110:1953–9. doi: 10.1161/01.CIR.0000143174.41810.10. [DOI] [PubMed] [Google Scholar]
- 22.Deguchi JO, Aikawa E, Libby P, et al. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–15. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- 23.Schneider F, Sukhova GK, Aikawa M, et al. Matrix metalloproteinase-14 deficiency in bone marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008 doi: 10.1161/CIRCULATIONAHA.107.707448. [DOI] [PubMed] [Google Scholar]
- 24.Wilcox JN, Smith KM, Schwartz SM, Gordon D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci U S A. 1989;86:2839–43. doi: 10.1073/pnas.86.8.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–97. [PMC free article] [PubMed] [Google Scholar]
- 26.Nemerson Y, Giesen PL. Some thoughts about localization and expression of tissue factor. Blood Coagulation & Fibrinolysis. 1998;9:S45–7. [PubMed] [Google Scholar]
- 27.Mackman N, Brand K, Edgington TS. Lipopolysaccharide-mediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor kappa B binding sites. Journal of Experimental Medicine. 1991;174:1517–26. doi: 10.1084/jem.174.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mach F, Schoenbeck U, Bonnefoy JY, Pober J, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40. Induction of collagenase, stromelysin, and tissue factor. Circulation. 1997;96:396–9. doi: 10.1161/01.cir.96.2.396. [DOI] [PubMed] [Google Scholar]
- 29.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–4. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 30.Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation. 2001;103:1718–20. doi: 10.1161/01.cir.103.13.1718. [DOI] [PubMed] [Google Scholar]
- 31.Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14:55–61. doi: 10.1097/00062752-200701000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Libby P, Ganz P, Schoen FJ, Lee RT. The vascular biology of the acute coronary syndromes. In: Topol EJ, editor. Acute Coronary Syndromes. New York: Marcel Dekker, Inc.; 2000. pp. 33–57. [Google Scholar]
- 33.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony- stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–91. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baldus S, Heeschen C, Meinertz T, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–5. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 35.Brennan ML, Penn MS, Van Lente F, et al. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 36.Meuwese MC, Stroes ES, Hazen SL, et al. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol. 2007;50:159–65. doi: 10.1016/j.jacc.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 37.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94:437–44. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H, Libby P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1309–14. doi: 10.1161/01.ATV.0000131784.50633.4f. [DOI] [PubMed] [Google Scholar]
- 39.Galis Z, Muszynski M, Sukhova G, et al. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res. 1994;75:181–9. doi: 10.1161/01.res.75.1.181. [DOI] [PubMed] [Google Scholar]
- 40.Saren P, Welgus HG, Kovanen PT. TNF-alpha and IL-1beta selectively induce expression of 92-kDa gelatinase by human macrophages. J Immunol. 1996;157:4159–65. [PubMed] [Google Scholar]
- 41.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–5. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 42.Rajavashisth TB, Liao JK, Galis ZS, et al. Inflammatory cytokines and oxidized low density lipoproteins increase endothelial cell expression of membrane type 1-matrix metalloproteinase. J Biol Chem. 1999;274:11924–9. doi: 10.1074/jbc.274.17.11924. [DOI] [PubMed] [Google Scholar]
- 43.Thomas AC, Knapman PA, Krikler DM, Davies MJ. Community study of the causes of “natural” sudden death. BMJ. 1988;297:1453–6. doi: 10.1136/bmj.297.6661.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davies MJ, Woolf N, Rowles PM, Pepper J. Morphology of the endothelium over atherosclerotic plaques in human coronary arteries. Br Heart J. 1988;60:459–64. doi: 10.1136/hrt.60.6.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaffer FA, Libby P, Weissleder R. Molecular imaging of cardiovascular disease. Circulation. 2007;116:1052–61. doi: 10.1161/CIRCULATIONAHA.106.647164. [DOI] [PubMed] [Google Scholar]
- 46.Deguchi J, Aikawa M, Tung CH, et al. Inflammation in atherosclerosis: visualizing matrix metalloproteinase action in macrophages in vivo. Circulation. 2006;114:55–62. doi: 10.1161/CIRCULATIONAHA.106.619056. [DOI] [PubMed] [Google Scholar]
- 47.Jaffer FA, Kim DE, Quinti L, et al. Optical visualization of cathepsin K activity in atherosclerosis with a novel, protease-activatable fluorescence sensor. Circulation. 2007;115:2292–8. doi: 10.1161/CIRCULATIONAHA.106.660340. [DOI] [PubMed] [Google Scholar]
- 48.Libby P, Aikawa M. Stabilization of atherosclerotic plaques: New mechanisms and clinical targets. Nat Med. 2002;8:1257–62. doi: 10.1038/nm1102-1257. [DOI] [PubMed] [Google Scholar]
- 49.Libby P, Ridker PM. Inflammation and atherothrombosis: from population biology and bench research to clinical practice. J Am Coll Cardiol. 2006;48:A33–46. [Google Scholar]
- 50.Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 51.Morrow DA, de Lemos JA, Sabatine MS, et al. Clinical relevance of C-reactive protein during follow-up of patients with acute coronary syndromes in the Aggrastat-to-Zocor Trial. Circulation. 2006;114:281–8. doi: 10.1161/CIRCULATIONAHA.106.628909. [DOI] [PubMed] [Google Scholar]