

Summary
We recently demonstrated that the occupancy of endothelial protein C receptor (EPCR) by its natural ligand activated protein C (APC)/protein C switches the protease activated receptor 1 (PAR-1)-dependent signaling specificity of thrombin from a disruptive to a protective effect in cultured human umbilical vein endothelial cells. Given the phenotypic differences between endothelial cells in venular and arterial beds, in this study we evaluated the signaling function of thrombin in human pulmonary artery endothelial cells (HPAECs) before and after treating them with PC-S195A which lacks catalytic activity but exhibits a normal affinity for EPCR. As expected, both thrombin and thrombin receptor agonist peptide (TRAP) enhanced the permeability barrier of HPAECs, however, both PAR-1 agonists exhibited a potent barrier protective effect when the cells were treated with PC-S195A prior to stimulation by the agonists. Interestingly, similar to APC, thrombin exhibited a potent cytoprotective activity in the LPS-induced permeability and TNF-α-induced apoptosis and adhesion assays in the PC-S195A treated HPAECs. Treatment of HPAECs with the cholesterol depleting molecule methyl-β-cyclodextrin eliminated the protective effect of both APC and thrombin. These results suggest that the occupancy of EPCR by its natural ligand recruits PAR-1 to a protective signaling pathway within lipid rafts of HPAECs. Based on these results we conclude that the activation of PAR-1 by thrombin would initiate a protective response in intact arterial vascular cells expressing EPCR. These findings may have important ramifications for understanding the mechanism of the participation of the vascular PAR-1 in pathophysiology of the inflammatory disorders.
Keywords: APC, Thrombin, EPCR, PAR-1, Inflammation, Signaling
Introduction
Activated protein C (APC) is a trypsin-like vitamin K-dependent serine protease in plasma that down-regulates thrombin generation by degrading the procoagulant cofactors Va and VIIIa by limited proteolysis (1, 2). The anticoagulant activity of APC is markedly improved by the cofactor function of protein S (3, 4). In addition to its well-studied anticoagulant effect, APC also elicits potent cytoprotective and antiinflammatory responses in endothelial cells (5-10). Because of these properties, recombinant APC has been approved as a therapeutic drug for treating severe sepsis (11). The mechanism of the protective effect of APC in severe sepsis is poorly understood, however, it has been hypothesized that when APC forms a complex with endothelial protein C receptor (EPCR) it acquires a different specificity, thus activating protease activated receptor 1 (PAR-1), thereby initiating protective signaling events in endothelial cells (12, 13). However, this hypothesis is controversial because thrombin can cleave the same receptor with at least three orders of magnitude higher catalytic efficiency than APC to initiate proinflammatory responses in endothelial cells (14, 15). In a recent study, we provided some insight into the PAR-1-dependent signaling mechanism of thrombin and APC by demonstrating that both EPCR and PAR-1 are associated with caveolin-1 within lipid rafts of human umbilical vein endothelial cells (HUVECs) (16). We discovered that the occupancy of EPCR by either APC or the zymogen protein C leads to dissociation of EPCR from caveolin-1 and recruitment of PAR-1 to a protective signaling pathway (17). Thus, the activation of PAR-1 by either APC or thrombin initiated protective signaling responses in HUVECs activated with proinflammatory cytokines (17).
Noting the phenotypic differences between venous and arterial endothelial cells and the observation that PAR-1 activation by thrombin elicits potent proinflammatory responses in both cell types (18, 19), we decided to evaluate the PAR-1-dependent signaling function of thrombin in human pulmonary artery endothelial cells (HPAECs) under culture conditions in which EPCR has been occupied with a physiological concentration of the zymogen protein C. To make the analysis straightforward, we used the catalytically inactive S195A mutant of protein C (PC-S195A) in the experiments. Furthermore, we also used the thrombin receptor agonist peptide (TRAP) as the PAR-1 activator in HPAECs. The results indicate that the activation of PAR-1 by either thrombin or TRAP is protective when EPCR is occupied by its ligand. Further studies employing siRNA for PAR-3, PAR-4 and sphingosine 1-phosphate receptor 1 (S1P1) revealed that the proinflammatory activity of thrombin is primarily mediated through the activation of PAR-4, and similar to APC, the EPCR- and PAR-1-dependent protective activity of thrombin involves the transactivation of S1P1 in HPAECs.
Materials and Methods
Wild-type protein C and its Ser-195 → Ala (PC-S195A) (chymotrypsinogen numbering) (20) mutant were expressed in human embryonic kidney (HEK-293) cells and purified to homogeneity as described (17). Blocking anti-PAR-1 (H-111), non-blocking anti-PAR-1 (S-19), anti-PAR-3 (H-103), anti-PAR-4 (H-120) and anti- EDG-1 (H-60) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The blocking anti-EPCR antibody (clone RCR-252) was purchased from Cell Sciences (Canton, MA). The antibody concentrations in the blocking assays were 25 μg/mL in all experiments. Anti-caveolin-1 antibody was purchased from BD Biosciences (San Jose, CA). Pertussis toxin (PTX), staurosporine, bacterial lipopolysaccharide (LPS), methyl-β-cyclodextrin (MβCD) and hirudin were purchased from Sigma (St. Louis, MO). Thrombin receptor agonist peptide (TRAP) for PAR-1 (H-Ser-Phe-Leu-Leu-Arg-Asn-NH2) was purchased from Bachem Bioscience (Torrance, CA).
Cell culture
Human pulmonary artery endothelial cells (HPAECs) were purchased from Lonza Walkersville Inc. (Walkersville, MD) and maintained in EGM-2 medium containing 10% fetal bovine serum (FBS) as recommended by the same manufacturer. All of the experiments described in this study were performed with cells passages 3-5. Human monocytic leukemia (THP-1) cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in RPMI-1640 supplemented with 10% FBS as recommended by the supplier.
Permeability assay
HPAEC permeability in response to thrombin (5 nM for 10 min) or LPS (10 ng/mL for 4 h) following treatment of cells with increasing concentrations of APC (0-100 nM plus 4 units/mL hirudin for 3 h) or thrombin (0-10 nM for 3 h) was quantitated by spectrophotometric measurement of the flux of Evans blue-bound albumin across functional HPAE cell monolayers using a modified 2-compartment chamber model as described (17). In experiments with thrombin or TRAP (0-2 mM for 3 h), cells were also pretreated with PC-S195A (0-80 nM) for 15 min. The cell permeability assays were also carried out in presence of pertussis toxin (PTX). In this case, confluent HPAE cell monolayers were pretreated with the toxin (100 ng/mL) for 16 h prior to their incubation with PAR-1 agonists. The extent of permeability was also evaluated in the presence of the cholesterol depleting molecule MβCD. In this case, cells were incubated with different concentrations of MβCD (0-10 mM for 1 h) before treating them with APC (50 nM), thrombin (5 nM ± 50 nM PC-S195A) for 3 h. For the function-blocking antibody treatments of the monolayers, medium was removed and antibodies were added for 30 min in serum-free medium followed by analysis of the permeability as described (17). Results are expressed as mean ±SEM and all experiments were repeated at least three times.
Apoptosis assay
The signaling effect of either thrombin or APC (0-50 nM for both proteases) in HPAECs in the absence or presence of PC-S195A (50 nM) was evaluated in a TNF-α-induced apoptosis assay as described (17). The number of apoptotic cells was expressed as the percentage of TUNEL-positive cells of the total number of nuclei determined by Hoechst staining as described (17). Results are expressed as mean ±SEM and all experiments were repeated three times. The APC assays contained 4 units/mL hirudin.
Adhesion assay
The adhesion of THP-1 cells to TNF-α-activated endothelial cells was evaluated by fluorescent labeling of THP-1 cells as described (21). Briefly, THP-1 cells were labeled with 5 μM Vybrant DiD (Molecular Probes) for 20 min at 37 °C in phenol red-free RPMI containing 5% fetal bovine serum. Following twice washing of cells (1.5 × 106/ml, 200 μl/well), they were resuspended in adhesion medium (RPMI containing 2% fetal bovine serum and 20 mM HEPES) and added to confluent monolayers of HPAEC cells in 96-well plates which were treated for 24 h with APC (10-100 nM) and 4 h with either thrombin (2-50 nM) or thrombin + PC-S195A (50 nM) followed by TNF-α (10 ng/mL for 4 h). In blocking experiments, the monolayers were preincubated for 30 min at 37 °C with appropriates antibodies. The fluorescence of labeled cells was measured (total signal) using a fluorescence microplate reader (Molecular Device). After incubation for 60 min at 37 °C, non-adherent cells were removed by washing four times with pre-warmed RPMI and the fluorescent signals of adherent cells were measured by the same methods. The percentage of adherent THP-1 cells was calculated by the formula: % adherence = (adherent signal/total signal) × 100 as described (21).
RNA interference
HPAEC permeability in response to thrombin (0-10 nM) or APC (0-100 nM) was evaluated following the knockdown of PAR-3, PAR-4 or S1P1 expression by pools of three target-specific 20-25 nucleotide siRNAs (Santa Cruz Biotechnology, Santa Cruz, CA) according to the manufacturer’s instruction as described (17). A non-targeting 20-25 nucleotide siRNA obtained from the same company was used as a negative control.
Isolation of lipid rafts/caveolae
Lipid rafts/caveolae proteins were isolated by slight modification of procedures as described (22). Briefly, confluent HPAE cells in 150-mm Petri dishes were washed with ice-cold PBS and scraped into Triton X-100 containing Tris lysis buffer (25 mM Tris-HCl, 250 mM sucrose, 2 mM EDTA and 1% Triton X-100). Cell pellets were then homogenized with a tight-fitting Dounce homogenizer followed by 3 × 20 sec bursts of ultrasonic sonicator on ice. The lysate was adjusted to 45% sucrose by the addition of an equal volume of 90% sucrose in 25 mM MES-buffered saline (pH 6.5) and placed at the bottom of an ultracentrifuge tube. Two solutions (1.7 mL each) of 35% and 5% sucrose were laid sequentially on the top of the 45% sucrose solution. After ultracentrifugation at 35,000 rpm with Beckman SW Ti55 rotor for 16–20 h, 10 × 0.5-mL fractions were collected from the top of tubes, and a portion of each fraction was analyzed by SDS-PAGE, transferred to nitrocellulose membranes and subjected to Western- blotting with appropriate primary and secondary antibodies as described (16).
Immunoprecipitation, SDS-PAGE and Western-blotting
Total cellular proteins were extracted by sonication with PBS containing 25 μM proteosome inhibitor MG132 (Sigma-Aldrich) and complete protease inhibitor cocktail (Roche Molecular Systems, Summerville, NJ) as described (17). Lysates were combined with 3 μg of each specific antibody (H-111 for PAR-1, H-103 for PAR-3, H-120 for PAR-4 and H-60 for EDG-1 (S1P receptor, S1P1), and incubated for 2 h at 4°C. Immunoprecipitates were collected with protein A/G Agarose (Santa Cruz, CA), fractionated on a 10% SDS-PAGE, transferred to membranes, and subjected to Western-blotting with appropriate primary and horseradish peroxidase-conjugated secondary antibodies as described (17). The immunoreactive protein bands were visualized by SuperSignal West Pico (Pierce, Rockford, IL).
Results
Protective effect of APC and thrombin + PC-S195A in HPAECs
Previous studies have indicated that, unlike the barrier protective activity of APC, thrombin and proinflammatory cytokines enhance permeability by cleaving PAR-1 on the surface of endothelial cells (19, 23). However, we recently demonstrated that the occupancy of EPCR by its natural ligand protein C switches the PAR-1-dependent signaling specificity of thrombin from a barrier disruptive to a barrier protective effect in HUVECs (17). The results presented in Fig. 1 suggest that this is also true for HPAECs. Thus, in agreement with previous results (17, 23), thrombin enhanced the permeability of HPAECs, showing only a partial protective effect at 50 pM thrombin (Fig. 1A). The partial protective effect of thrombin is mediated through PAR-1 since the function-blocking anti-PAR-1 antibody eliminated this response (Fig. 1A). On the other hand, prior treatment of cells with PC-S195A resulted in thrombin exhibiting a potent protective effect in a concentration dependent manner with optimal effect occurring at 5 nM thrombin thereafter, decreasing and reverting to a fully disruptive effect at 50 nM. A similar disruptive effect was observed for APC at concentrations above 150 nM (Fig. 1A). As shown below, the disruptive effects at higher concentrations of both proteases are independent of PAR-1. A similar barrier protective effect for thrombin in the presence of PC-S195A was also observed when the permeability in HPAECs was induced by LPS, suggesting that the PAR-1-dependent protective response is independent of the proinflammatory stimuli (Fig. 1B). The barrier protective effect of APC and thrombin + PC-S195A required both EPCR and PAR-1 since function-blocking antibodies to either receptor abrogated the response (data not shown). The concentration dependence of the protective effect of PC-S195A suggest that the protective effect of 5 nM thrombin reaches saturation (~40 nM) at far below the physiological concentration of protein C in circulation (~70-80 nM) (Fig. 1C). It is known that factor VIIa can also bind EPCR with an affinity comparable to that of APC/protein C (24, 25). Nevertheless, the EPCR-dependent protective effect of thrombin was specific for protein C since thrombin did not exhibit a protective activity in the presence of factor VIIa (Fig. 1D).
Figure 1.

Effect of APC and thrombin on permeability of HPAECs treated or not treated with PC-S195A. (A) HPAE cells were incubated with increasing concentrations of thrombin (○), thrombin + blocking anti-PAR-1 antibody (●), thrombin + PC-S195A (50 nM) (■), and APC (□) 3 h before inducing permeability with 5 nM thrombin for 10 min. (B) The same as (A) except that following incubation of endothelial cells with increasing concentrations of thrombin (○), thrombin + PC-S195A (■) and APC (□), the permeability was induced with LPS (10 ng/mL) for 4 h. (C) The same as (A) with the exception that the concentration dependence of the protective effect of PC-S195A in the presence of 5 nM thrombin (○) is measured in the permeability assay. The extent of the protective effect of 50 nM APC (■) is also presented. (D) HPAE cells were incubated with APC (100 nM) and thrombin (5 nM) in the absence and presence of either PC-S195A or factor VIIa (50 nM) for 3 h before inducing permeability with 5 nM thrombin for 10 min. Baseline represents the permeability of untreated control cells not stimulated with thrombin.
It is known that thrombin, proinflammatory cytokines and the kinase inhibitor staurosporine can induce apoptosis in endothelial cells that can be reversed by APC by an EPCR and PAR-1-dependent mechanism (7, 16). As presented in Fig. 2, the treatment of HPAECs with TNF-α induced apoptosis and APC inhibited cell death by a concentration dependent manner. As with the permeability assay, at a low concentration of 50 pM, thrombin exhibited a partial cytoprotective effect, however, in the presence of PC-S195A, the signaling effect by thrombin was fully protective for up to 5 nM protease thereafter decreasing and completely disappearing at 50 nM (Fig. 2).
Figure 2.

The anti-apoptotic activity of APC and thrombin + PC-S195A in HPAE cells. Confluent monolayers were treated with increasing concentrations of thrombin (○), thrombin + PC-S195A (■) and APC (□) for 24 h followed by induction of apoptosis with TNF-α (10 ng/mL) for 4 h. The number of apoptotic cells is expressed as the percentage of TUNEL-positive cells of the total number of nuclei. The number of TUNEL-positive cells in the absence of TNF-α was 10-15%. The concentration of PC-S195A was 50 nM for this and all other figures described below.
PC-S195A switches the signaling specificity of TRAP
It is known that the PAR-1-dependent proinflammatory effect of thrombin in endothelial cells can be recapitulated by TRAP (23, 26, 27). To determine whether the occupancy of EPCR by its ligand protein C changes the signaling specificity of TRAP in HPAECs, the effect of increasing concentrations of the agonist peptide on the cellular permeability was evaluated before and after treating confluent cell monolayers with PC-S195A. As demonstrated in Fig. 3, TRAP potently enhanced the permeability of HPAE cells, eliciting a maximum response at 10-20 μM. On the other hand, prior treatment of cells with PC-S195A protected them from the barrier disruptive effect of TRAP. Interestingly, unlike thrombin which exhibits a partial protective effect at a low concentration of 50 pM, the barrier disruptive effect of TRAP was mono-phasic, suggesting that the protective effect of a low concentration of thrombin, as has been observed in this and previous studies (17, 23) may not be mediated though the activation of PAR-1. Furthermore, increasing the concentration of TRAP up to 0.5 mM, which is sufficient to activate all available cell surface PAR-1, did not abolish the barrier protective effect of the agonist peptide in cells pretreated with PC-S195A as observed with high concentrations of both thrombin and APC. These results suggest that the hyperpermeability effect of higher concentrations of these proteases may not be mediated through the activation of PAR-1.
Figure 3.
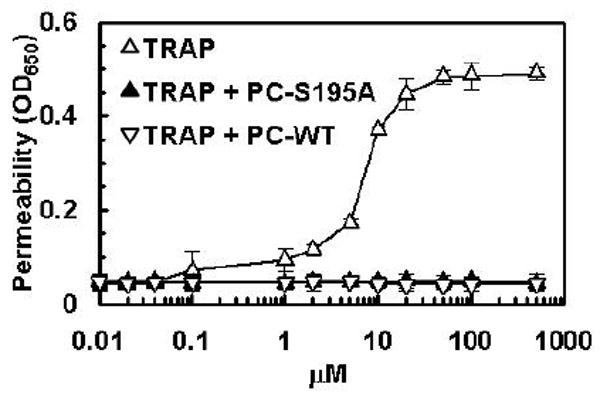
The barrier disruptive effect of TRAP and its reversal by the ligand occupancy of EPCR. (A) HPAE cells were incubated with increasing concentrations TRAP (△), TRAP + wild-type protein C (▽), TRAP + PC-S195A (▲) for 3 h. The first data point on the y-axis represents the baseline permeability level.
Thrombin + PC-S195A inhibits adhesion of THP-1 cells to TNF-α-activated HPAECs
TNF-α treatment of vascular endothelial cells is associated with up-regulation of several cell surface adhesion molecules, thereby mediating the interaction of leukocytes with activated endothelial cells under inflammatory conditions. APC has been demonstrated to inhibit the interaction of leukocytes with the activated endothelium (6, 7). To confirm that the occupancy of EPCR by its natural ligand protein C switches the signaling specificity of thrombin from a proinflammatory to an antiinflammatory response, we monitored the adhesion of the monocytic THP-1 cell line to TNF-α-activated HPAECs which were treated with PAR-1 agonists. As demonstrated in Fig. 4A, APC inhibited the adherence of THP-1 cells to activated HPAEC monolayers by a concentration dependent manner. On the other hand, thrombin by itself (20 nM) did not inhibit the adherence of THP-1 cells to HPAECs, however, in the presence of PC-S195A thrombin inhibited the interaction with an optimal concentration of 20 nM thrombin (Fig. 4B). The protective effects were PAR-1 and EPCR dependent since the function-blocking antibodies to either receptor abrogated the protective response of APC as well as thrombin + PC-S195A (Figs. 4A and B). These results clearly suggest that the cleavage of PAR-1 by thrombin elicits protective responses if EPCR is occupied by protein C.
Figure 4.
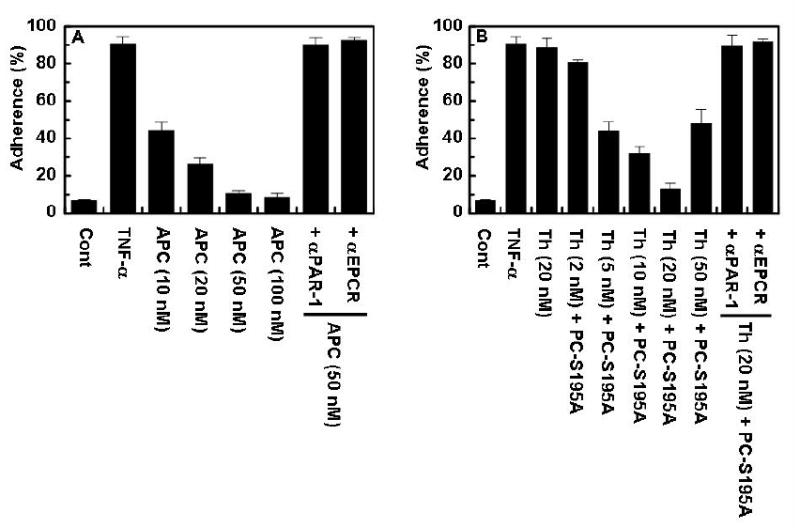
The ligand occupancy of EPCR inhibits the adhesion of THP-1 cells to TNF-α-activated HPAE cells. (A) TNF-α-mediated adherence of THP-1 cells to HPAE cell monolayers was analyzed after treating monolayers with different concentration of APC in the absence or presence of function-blocking anti-EPCR (RCR-252) and anti-PAR-1 (H-111) antibodies as described under “Materials and Methods”. (B) The same as (A) except that the HPAEC monolayers were treated with either thrombin or different concentrations of thrombin + PC-S195A (50 nM).
siRNA knockdown of PAR-3, PAR-4 and S1P1 in HPAECs
To determine whether the partial protective effect of pM concentrations of thrombin and the permeability-enhancing effect of a higher concentration of thrombin and APC is mediated through activation of other PARs, we employed siRNA for either PAR-3 or PAR-4 to knockdown or down-regulate their expression in HPAECs. As shown in Fig. 5A, while the siRNA for PAR-3 markedly down-regulated the expression of this receptor, the siRNA for PAR-4 completely inhibited the expression of PAR-4 in HPAECs. The analysis of the signaling activity of thrombin in cells treated with siRNA for PAR-3 revealed that the partial protective effect of a very low concentration of thrombin may be mediated through the cleavage of PAR-3 since thrombin showed a disruptive effect in the siRNA treated cells at all concentrations (Fig. 5B). On the other hand, the knockdown of PAR-4 abrogated the barrier disruptive effect of a higher concentration of thrombin, suggesting than this effect is mediated through the cleavage of PAR-4. This hypothesis is also true for APC since the disruptive effect of high concentrations of APC also disappeared in HPAECs treated with PAR-4 siRNA (Fig. 5C). Previous results have indicated that the EPCR and PAR-1 dependent protective activity of APC is mediated through the transactivation of sphingosine 1-phosphate receptor S1P1 (19, 23). In this study, we used siRNA to S1P1 in order to determine whether the EPCR-dependent protective activity of thrombin is also mediated through the same signaling pathway. The siRNA markedly down-regulated the expression of S1P1 in HPAECs (Fig. 5A) and results presented in Fig. 5D suggest that, similar to APC, the protective activity of thrombin in the presence of PC-S195A is mediated through the same signaling pathways. The control siRNA did not influence the signaling activity of either protease in any one of the assays described above.
Figure 5.

The effect of thrombin on the permeability of HPAECs in the absence and presence of PC-S195A and siRNA for PAR-3, PAR-4 and S1P1. (A) HPAE cells were transiently transfected with each siRNA or control siRNA (1 μg each). Following three days of incubation at 37 °C, total cellular proteins were extracted, separated on 10% SDS-PAGE and Western-blotted with antibodies directed to indicated receptors. An antibody against actin was used as internal control. (B) HPAE cells were incubated with increasing concentrations of thrombin (○), thrombin with prior treatment of cells for three days with control siRNA (●), thrombin with prior treatment of cells with siRNA for PAR-3 (□), thrombin with prior treatment of cells with siRNA for PAR-4 (■), thrombin + PC-S195A (△), thrombin + PC-S195A with prior treatment of cells with control siRNA (▲), thrombin + PC-S195A with prior treatment of cells with siRNA for PAR-3 (▽) and thrombin + PC-S195A with prior treatment of cells with siRNA for PAR-4 (▼) for 3 h before inducing permeability with 5 nM thrombin for 10 min. (C) The same as (B) with the exception that the barrier protective effect of APC was monitored in the absence (○) and presence of control siRNA (●), siRNA for PAR-3 (□) or siRNA for PAR-4 (■). (D) The same as (B) with the exception that the barrier protective effect was monitored for thrombin (○), thrombin with prior treatment of cells for three days with control siRNA (●), thrombin with prior treatment of cells with siRNA for S1P1 (□), thrombin + PC-S195A (△), thrombin + PC-S195A with prior treatment of cells with control siRNA (▲), thrombin + PC-S195A with prior treatment of cells with siRNA for S1P1 (■).
EPCR and PAR-1 are associated with Caveolin-1 in lipid rafts of HPAECs
In a recent study, we demonstrated that EPCR and PAR-1 are associated with caveolin-1 within the lipid rafts of HUVECs (17). Moreover, we showed that the occupancy of EPCR by its ligand results in dissociation of EPCR from caveolin-1, thereby recruiting PAR-1 to a protective pathway by coupling it to the pertussis toxin (PTX) sensitive Gi protein (17). To determine whether similar events are responsible for the protective activity of thrombin + PC-S195A in HPAECs, we isolated lipid rafts of non-treated and PC-S195A treated cells and analyzed the protein contents of different low and high buoyancy fractions by immunoblotting using antibodies against EPCR, PAR-1 and caveolin-1. Similar to results with HUVECs, all three proteins were found to be colocalized within the cholesterol rich lipid rafts/caveolae membrane microdomains in both the absence and presence of PC-S195A (data not shown). Furthermore, co-immunoprecipitation studies using anti-caveolin-1, anti-PAR-1 and anti-EPCR antibodies indicated that, similar to HUVECs, both PAR-1 and EPCR are associated with caveolin-1, however, in endothelial cells treated with PC-S195A, EPCR is dissociated from caveolin-1 (Fig. 6A). The analysis of the activity of APC and thrombin + PC-S195A in the cell permeability assay in the presence of PTX suggests that the protective activity of both proteases is mediated through Gi-protein since this activity was lost in cells treated with the toxin (Fig. 6B). Furthermore, the cholesterol depleting molecule MβCD eliminated the barrier protective activity of APC and thrombin + PC-S195A, thus reverting it to a disruptive effect with both proteases (Fig. 6C). These results suggest that the cleavage of PAR-1 by either protease outside the lipid rafts initiates a proinflammatory response in endothelial cells.
Figure 6.

SDS-PAGE and co-immunoprecipitation of lipid raft/caveolae preparations derived from HPAE cells with and without treatment with PC-S195A and permeability assays in the presence of PTX and MβCD. (A) Total cellular proteins from non-treated and PC-S195A treated HPAE cells were immunoprecipitated with anti-caveolin-1, anti-PAR-1 and anti-EPCR antibodies, separated on SDS-PAGE and immunoblotted with different pairs of the same antibodies. (B) HPAE cells were incubated with increasing concentrations of thrombin (○), thrombin with prior treatment of cells with 100 ng/mL PTX for 16 h (●), APC (□), APC with prior treatment of cells with 100 ng/mL PTX for 16 h (■), thrombin + PC-S195A (△), and thrombin + PC-S195A with prior treatment of cells with PTX (▲) for 3 h before inducing permeability with 5 nM thrombin. (C) The same as (B) with the exception that the effect on permeability for thrombin (5 nM) (○), thrombin + S195A (■), APC (□) was monitored after incubation of cells with MβCD (0-10 mM) for 1 h. The symbols (▲) represent the baseline permeability in the absence of an agonist.
Discussion
The cleavage of the PAR family of G-protein coupled receptors by coagulation proteases plays an important role in modulating cellular responses in endothelial and other cells (27). The common view is that the activation of PAR-1 by either thrombin or TRAP elicits proinflammatory responses in vascular endothelial cells (19, 23, 27, 28). However, in a recent study we discovered that the occupancy of EPCR by its natural ligand APC/protein C switches the PAR-1-dependent signaling specificity of thrombin from a disruptive to a protective response in HUVECs (17). Our results in this study show that an EPCR-dependent signaling switch for thrombin also occurs in the arterial endothelial cells as evidenced by thrombin exhibiting barrier protective and cytoprotective activities in HPAE cells treated with PC-S195A prior to activation of the cells by either thrombin or TRAP. The EPCR and PAR-1-dependent protective activity of thrombin requires the localization of both receptors to the lipid rafts of endothelial cells since the treatment of HPAECs with MβCD reversed the effect of thrombin to a proinflammatory response even when EPCR was occupied by its ligand. The co-immunoprecipitation studies revealed that, similar to HUVECs (17), both PAR-1 and EPCR are associated with caveolin-1 in the lipid rafts of HPAECs. However, the occupancy of EPCR with PC-S195A resulted in dissociation of EPCR from caveolin-1, thereby changing the PAR-1-dependent signaling specificity of thrombin. The protective signaling activity of thrombin was PTX sensitive, suggesting that Gi/o member of the G-protein family is involved in mediating the response. It is known that the protective activity of APC is mediated through transactivation of S1P1 (19, 23). The observation that siRNA to S1P1 eliminated the EPCR-dependent protective effect of thrombin suggests that this effect is also mediated through the same pathway.
The observation that, in the presence of PC-S195A, a high concentration of TRAP elicits only protective responses in HPAECs (this study) and HUVECs (17) reinforces our hypothesis that the activation of PAR-1 in intact vascular endothelial cells expressing EPCR would be protective under physiological conditions. The permeability studies in the presence of PAR-4 siRNA clearly suggest that it is the activation of PAR-4 by thrombin that is responsible for the proinflammatory activity of the protease. Since unlike PAR-1 and PAR-3, PAR-4 lacks the hirudin-like sequence at its C-terminus site of the scissile bond, its cleavage requires a relatively higher concentration of thrombin. The ramification of our findings is that thrombin at low concentrations (<5 nM) may primarily play a protective role under normal physiological conditions by both TM- and EPCR-dependent activation of protein C and EPCR-dependent cleavage of PAR-1 in endothelial cells of both arterial and venular beds. However, during injury or trauma, which may lead to denudation of the endothelium or down-regulation of the cell surface EPCR, thrombin can initiate proinflammatory responses through the activation of PAR-1. This hypothesis is consistent with the observation that the infusion of a low concentration of thrombin to dogs conferred protection in response to a lethal dose of endotoxin (29). The threshold thrombin concentration required to observe an EPCR-dependent barrier protective effect was 2 nM in HUVECs (17) and 5 nM for HPAECs (Fig. 1), which is consistent with previous findings that endothelial cells of arterial beds are more restrictive to liquid and albumin flux than those in the venular beds (28, 30). The observation that the activation of PAR-1 by either thrombin or TRAP elicits only a protective response in both types of endothelial cells when EPCR is occupied by its ligand challenges the relevance of previous in vitro studies which have concluded a proinflammatory role resulting from the PAR-1 activation in endothelial cells. Nevertheless, several in vivo studies have also observed a disruptive and proinflammatory effect for PAR-1 activation in acute long injury in mouse models (31-33). Though our studies cannot address the in vivo data, nevertheless, in light of established species differences between human and mouse in the expression and function of PAR-1, we believe that further investigation in human systems is required to understand the exact signaling effect of PAR-1 activation in intact vascular endothelial cells.
Previous results have indicated that a low concentration of thrombin (50 pM) elicits a partial protective response in HUVECs by an unknown mechanism (17, 23). It was interesting to note that siRNA for PAR-3 eliminated the EPCR-independent barrier protective effect of a low pM concentration of thrombin and also reduced the extent of the EPCR-dependent response with both thrombin (Fig. 5B) and APC (Fig. 5C). PAR-3 may not be directly involved in signal transduction in endothelial cells since it lacks a cytosolic domain. Thus, the mechanism by which PAR-3 alters the signaling activities of thrombin and APC is not known. Nevertheless, it was recently reported that PAR-1 can form heterodimers with PAR-3 and that the dimerization modulates the signaling specificity of PAR-1 in endothelial cells (34). The result of this previous study showed that thrombin primarily signals through PAR-1, but PAR-1 interaction with PAR-3 is required for a maximal response (34). Furthermore, it was found that dimerization with PAR-3 alters the G-protein coupling specificity of PAR-1. The observation that the optimal EPCR-dependent protective effect of both thrombin and APC requires PAR-3 is consistent with previous findings and this mechanism also appears to account for the EPCR-independent partial protective effect of a pM concentration of thrombin.
In summary, our results both in this and a previous study showed that EPCR switches the signaling specificity of thrombin from a cytotoxic and barrier disruptive to a protective effect in endothelial cells of both arterial and venular beds. Our results suggest that in the absence of the occupancy of EPCR by its natural ligand protein C, both EPCR and PAR-1 are associated with caveolin-1 within the lipid rafts of endothelial cells (Fig. 7). However, when EPCR is bound by protein C/APC, EPCR is dissociated from caveolin-1, a process which directly or indirectly (through S1P1, not shown in Fig. 7) alters the G-protein mediated signaling specificity of PAR-1 from a proinflammatory (Gq and/or G12/13) to an antiinflammatory (Gi) response (Fig. 7). Thus, the activation of PAR-1 by both thrombin and APC initiate protective signaling responses in endothelial cells when EPCR is occupied. This important finding is another example of the interaction that exists between the players of the procoagulant, anticoagulant and antiinflammatory pathways (35). Thus, similar to thrombomodulin (TM), EPCR plays a critical role in regulating the physiological function of thrombin. In the case of TM, the direct interaction of thrombin with TM switches the specificity of thrombin in the clotting cascade and the occupancy of EPCR by protein C indirectly switches the signaling specificity of thrombin in the inflammatory pathways. In light of these observations, the inhibition of thrombin may not be a good strategy for treating inflammatory disorders. The observation that both thrombin and TRAP inhibited the adherence of the monocytic THP-1 cells to TNF-α-activated HPAE cells in the presence of PC-S195A further supports this contention.
Figure 7.

Simplified models of PAR-1 activation by either APC or thrombin when EPCR is occupied by its ligand protein C. (A) The unoccupied EPCR is associated with caveolin-1 (Cav-1) within lipid rafts of endothelial cells. Thrombin cleavage of PAR-1 elicits a proinflammatory signal through G12/13 and Gq under these conditions. (B) The occupancy of EPCR by protein C (PC) results in dissociation of EPCR from caveolin-1. This process is linked with coupling of PAR-1 to Gi. Thrombin cleavage of PAR-1 initiates an antiinflammatory response under these conditions. (C) The same as (B) except that the EPCR and PAR-1 dependent protective signaling response is mediated by APC.
Acknowledgments
We thank Audrey Rezaie for proofreading the manuscript.
The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institute of Health HL 68571 and HL 62565 to ARR.
References
- 1.Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70:1–5. [PubMed] [Google Scholar]
- 2.Stenflo J. Structure and function of protein C. Sem Thromb Hemost. 1984;10:109–21. doi: 10.1055/s-2007-1004413. [DOI] [PubMed] [Google Scholar]
- 3.Walker FJ, Fay PJ. Regulation of blood coagulation by the protein C system. FASEB J. 1992;6:2561–7. doi: 10.1096/fasebj.6.8.1317308. [DOI] [PubMed] [Google Scholar]
- 4.Dahlbäck B. Protein S and C4b-binding protein: Components involved in the regulation of the protein C anticoagulant system. Thromb Haemostas. 1991;66:49–61. [PubMed] [Google Scholar]
- 5.Esmon CT, Taylor FB, Jr, Snow TR. Inflammation and coagulation: Linked processes potentially regulated through a common pathway mediated by protein C. Thromb Haemost. 1991;66:160–5. [PubMed] [Google Scholar]
- 6.Joyce DE, Gelbert L, Ciaccia A, et al. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 7.Mosnier LO, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 8.Cheng T, Liu D, Griffin JH, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nature Medicine. 2003;9:338–42. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 9.Brueckman M, Horn S, Lang S, et al. Recombinant human activated protein C upregulates cyclooxygenase-2 expression in endothelial cells via binding to endothelial cell protein C receptor and activation of protease-activated receptor-1. Thromb Haemost. 2005;93:743–50. doi: 10.1160/TH04-08-0511. [DOI] [PubMed] [Google Scholar]
- 10.Nold MF, Nold-Petry CA, Fischer D, et al. Activated protein C downregulates p38 mitogen-activated protein kinase and improves clinical parameters in an in-vivo model of septic shock. Thromb Haemost. 2007;98:1118–1126. doi: 10.1160/th07-01-0052. [DOI] [PubMed] [Google Scholar]
- 11.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Eng J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 12.Ruf W, Dorfleutner A, Riewald M. Specificity of coagulation factor signaling. J Thromb Haemost. 2003;1:1495–503. doi: 10.1046/j.1538-7836.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 13.Riewald M, Petrovan RJ, Donner A, et al. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–2. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 14.Ludeman MJ, Kataoka H, Srinivasan Y, et al. PAR1 cleavage and signaling in response to activated protein C and thrombin. J Biol Chem. 2005;280:13122–8. doi: 10.1074/jbc.M410381200. [DOI] [PubMed] [Google Scholar]
- 15.Esmon CT. Is APC activation of endothelial cell PAR1 important in severe sepsis?: No. J Thromb Haemost. 2005;3:1910–1. doi: 10.1111/j.1538-7836.2005.01573.x. [DOI] [PubMed] [Google Scholar]
- 16.Bae J-S, Yang L, Rezaie AR. Receptors of the protein C activation and activated protein C signaling pathways are colocalized in lipid rafts of endothelial cells. Proc Natl Acad Sci (USA) 2007;104:2867–72. doi: 10.1073/pnas.0611493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bae J-S, Yang L, Manithody C, et al. The ligand occupancy of endothelial protein C receptor switches the PAR-1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007;110:3909–16. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia JGN, Pavalko FM, Patterson CE. Vascular endothelial cell activation and permeability responses to thrombin. Blood Coag Fibrinol. 1995;6:609–26. doi: 10.1097/00001721-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Finigan JH, Dudek SM, Singleton PA, et al. Activated protein C mediates novel lung endothelial barrier enhancement. J Biol Chem. 2005;280:17286–93. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- 20.Bode W, Mayr I, Baumann U, et al. The refined 1.9 Å crystal structure of human α-thrombin: interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–75. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim I, Moon SO, Kim SH, et al. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276:7614–20. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Casey L, Pike LJ. Compartmentalization of phosphatidylinositol 4,5-bisphosphate in low-density membrane domains in the absence of caveolin. Biochem Biophys Res Commun. 1998;245:684–90. doi: 10.1006/bbrc.1998.8329. [DOI] [PubMed] [Google Scholar]
- 23.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–84. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 24.Preston RJS, Ajzner E, Razzari C, et al. Multifunctional specificity of the protein C/activated protein C Gla domain. J Biol Chem. 2006;281:28850–7. doi: 10.1074/jbc.M604966200. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh S, Pendurthi UR, Esmon CT, et al. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007;282:11849–57. doi: 10.1074/jbc.M609283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaughlin JN, Shen L, Holinstat M, et al. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem. 2005;280:25048–59. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- 27.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–14. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 28.Komarova YA, Mehta D, Malik AB. Dual regulation of endothelial junctional permeability. Sci STKE. 2007;re8:1–9. doi: 10.1126/stke.4122007re8. [DOI] [PubMed] [Google Scholar]
- 29.Taylor FB, Jr, Chang A, Hinshaw LB, et al. A model for thrombin protection against endotoxin. Thromb Res. 1984;36:177–85. doi: 10.1016/0049-3848(84)90339-6. [DOI] [PubMed] [Google Scholar]
- 30.Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: An electron microscopic study. J Biophys Biochem Cytol. 1961;11:571–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenkins RG, Su X, Su G, et al. Ligation of protease-activated receptor 1 enhances αvβ6 integrin-dependent TGF-β activation and promotes acute lung injury. J Clin Invest. 2006;116:1606–14. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jesmin S, Gando S, Zaedi S, et al. Differential expression, time course and distribution of four PARs in rats with endotoxin-induced acute lung injury. Inflammation. 2007;30:14–27. doi: 10.1007/s10753-006-9017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel SM, Gao X, Mehta D, et al. Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol Genomics. 4:137–45. doi: 10.1152/physiolgenomics.2000.4.2.137. [DOI] [PubMed] [Google Scholar]
- 34.McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci (USA) 2007;104:5662–7. doi: 10.1073/pnas.0700763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esmon CT. Role of coagulation inhibitors in inflammation. Thromb Haemostas. 2001;86:51–6. [PubMed] [Google Scholar]