

Abstract
Amplification of the HER2 (ErbB2, c-Neu) proto-oncogene in breast cancer is associated with poor prognosis and high relapse rates. HER2/ErbB2, in conjunction with ErbB3, signals through the Akt/PI3-K pathway and leads to the activation of mTOR, a critical mRNA translation regulator that controls cell growth. Gene expression analysis of mammary tumors collected from MMTV-c-Neu transgenic mice revealed that mRNA levels of several mTOR pathway members were either up-regulated (p85/PI3-K and p70S6K) or down-regulated (eIF4E-BP1) in a manner expected to enhance signaling through this pathway. Treatment of MMTV-c-Neu transgenic mice with the mTOR inhibitor rapamycin caused growth arrest and regression of primary tumors with no evidence of weight loss or generalized toxicity. The treatment effects were due to decreased proliferation, associated with reduced cyclin D1 expression, and increased cell death in primary tumors. While many of the dead epithelial cells had the histopathologic characteristics of ischemic necrosis, rapamycin treatment was not associated with changes in microvascular density or apoptosis. Rapamycin also inhibited cellular proliferation in lung metastases. In summary, data from this preclinical model of ErbB2/Neu-induced breast cancer demonstrate that inhibition of the mTOR pathway with rapamycin blocks multiple stages of ErbB2/Neu-induced tumorigenic progression.
Keywords: Breast cancer, HER2/Neu/erbB2, transgenic mouse, rapamycin, metastasis
Introduction
Amplification of the HER2 (also called ErbB2 or c-Neu) proto-oncogene occurs in 25-30% of breast cancers and confers a poor prognosis due to refractoriness to conventional chemotherapies and high relapse rates (1,2). HER2/ErbB2 is a member of the Epidermal Growth Factor (EGF) family of receptor tyrosine kinases (3). In conjunction with ErbB3, HER2/ErbB2 activates a number of signaling pathways including the PI3-Kinase pathway (4). When PI3-K is activated, it signals through the Akt/PKB kinase which subsequently activates the growth regulator mTOR (mammalian Target of Rapamycin) (5). The mTOR kinase regulates cell proliferation and growth through translational control of an array of proteins (6). mTOR phosphorylates p70S6 kinase (p70S6K) which, in turn, phosphorylates the Ribosomal Protein S6 (S6), promoting translation of proteins bearing a 5’-oligopyrimidine tract. mTOR also phosphorylates eIF4E-BP1 (4EBP1), a negative regulator of the translational initiator eIF-4E. Phosphorylation of 4EBP1 induces its dissociation from eIF-4E, which enhances cap-dependent translation of numerous mRNAs including cell cycle regulators such as cyclin D1, an essential regulator of proliferation in HER2/ErbB2 cells (7,8).
Elevated levels of PI3-K, p70S6K and eIF4E as well as decreased levels of 4EBP1 are associated with tumor aggressiveness in vivo and in vitro (6,9,10). In human breast cancer, Zhou et. al. have linked mTOR activation to HER2-positivity; high levels of phosphorylated Akt, phosphorylated mTOR and phosphorylated 4EBP1 were all associated with HER2/ErbB2 expression as well as decreased disease-free survival (11). Rapamycin, an inhibitor of mTOR, decreases proliferation of a number of mammary epithelial cell lines, particularly those that overexpress HER2/ErbB2, have activated Akt or have upregulated p70S6K (12-14). In addition, forced expression of HER2/ErbB2 in MCF-7 cells promotes invasiveness and colony formation that can be blocked by rapamycin (11,14). In mice that develop mammary tumors in response to overexpression of the rat ErbB2 (c-Neu) gene, the rapamycin analog, RAD001 inhibits proliferation of tumor cells (15). In addition, growth of mouse mammary tumors induced by a constitutively active form of c-Neu (NeuYD) is also attenuated by rapamycin (16).
To date, the effects of rapamycin on multiple stages of ErbB2/Neu-induced tumor formation—ranging from early, hyperplastic changes through distal metastases--have not been assessed. In the present study we utilized MMTV-c-Neu mice (17) to determine if rapamycin has more global inhibitory effects on multiple stages of tumorigenic progression that extend beyond inhibiting proliferation of primary tumors. These mice express rat c-Neu/ErbB2 selectively in the mammary gland and have been used to study preneoplastic changes in the gland (18-20) and mechanisms of metastatic progression (21-23). Herein, we report that rapamycin induces regression of primary tumors that overexpress c-Neu/ErbB2 as well as hyperplastic epithelia. Rapamycin also attenuates proliferation of established lung metastases in these mice. Together, these data indicate that rapamycin can inhibit multiple stages of tumor progression, suggesting that inhibition of the mTOR pathway may be a useful chemopreventive strategy as well as an effective treatment for early and late stage HER2/ErbB2 positive breast cancers.
Materials and Methods
Materials
Primary antibodies used for immunoblotting were: P-mTOR (Ser2448) (Cell Signaling, #2971), mTOR (Cell Signaling, #2972), β-actin (Sigma, #A1978), cyclin D1 (BD Pharmingen, #554181), P-S6 Ribosomal Protein (Ser235/236) (Cell Signaling, #2211), Akt (Cell Signaling, #9272), P-Akt (Ser473) (Cell Signaling, #9271) and total caspase-3 (Cell Signaling, #9622). Secondary antibodies used for immunoblotting were horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit (Santa Cruz Biotechnology). Rapamycin was purchased from Eton Biosciences. Primary antibodies used for immunohistochemistry (IHC) were: P-S6 Ribosomal Protein (Cell Signaling, #2211), P-HER2/ErbB2 (Tyr877) (Cell Signaling, #2241), P-histone-H3 (Upstate Biotechnology, #06-570), cleaved caspase-3 (Asp175) (Cell Signaling, #9661), CD-31 (DakoCytomation, clone JC70A), Ki-67 (DakoCytomation, clone TEC-3). For TUNEL analysis, the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon International) was used.
Transgenic mice
FVB/N-TgN(MMTV-Neu)202Mul/J mice containing the rat proto-oncogene c-Neu targeted to mammary epithelium by the MMTV-LTR promoter (21) were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and bred with FVB/N wild type animals. Female progeny were housed in microisolator cages in pathogen-free conditions under a 12 h light/dark cycle and provided food and water ad libitum. Mice were genotyped as previously described (19). All animal studies were approved by the Case Western Reserve University Institutional Animal Care and Use Committee.
Gene expression microarray analysis
Gene expression microarray analysis of age-matched wild type glands (n=3 pools of 5 mice each), and primary tumors (n=7) using the Affymetrix (Santa Clara, CA, USA) Murine U74Av2 GeneChip Array have been previously described (19). Briefly, computational analyses were performed with Microarray Suite (v.5.0, Affymetrix), Data Mining Tool (DMT v.3.0, Affymetrix), MicroDB (v.3.0, Affymetrix), and GeneSpring (v.6.0, Silicon Genetics) software. Probes that received a call of “absent” by Affymetrix detection call on all GeneChips were not analyzed. Changed genes were identified by application of Welch’s approximate T-test using a Benjamini and Hochberg multiple testing correction to establish a false discovery rate of 5% (p<0.05).
Tumor studies
MMTV-c-Neu mice were randomly assigned to vehicle or rapamycin groups following palpable tumor detection. Mean tumor volumes at the start of the treatment paradigm were indistinguishable between the two groups (vehicle = 205 ± 58 mm3; rapamycin = 263 ± 77 mm3, p=0.56). Rapamycin-treated mice received 150 μg of rapamycin, made by diluting 3 μl of a 50 mg/ml stock (in 100% ethanol) into 100 μL of vehicle (5.2% Tween 80, 5.2% PEG 400 in 0.9% saline). Control mice received 3 μL of ethanol diluted in 100 μL of vehicle. Drug or vehicle was delivered by intraperitoneal injection every other day for 32 days. Tumor volumes, body weights and changes in grooming behavior were monitored during the treatment paradigm. Animals were euthanized if estimated tumor burden approached 10% of the total body weight. Tumor dimensions were measured by external calipers and volumes were estimated using the formula (L × W2)/2, where L is the largest diameter. Mice were killed by asphyxiation with CO2, mammary tumors and tissues were removed, and lungs were examined for metastases. For short term studies, mice were treated as described above, except that rapamycin or vehicle was given for either 5 (2 treatments) or 10 (5 treatments) days.
Histology
Tumors were collected and bisected. One half was frozen on dry ice and immediately stored at -80° C. The second half of the tumor as well as lungs were fixed overnight in 4% (w/v) paraformaldehyde in PBS. Fixed samples were paraffin embedded, sectioned at 5 μm, baked and either stained with hematoxylin and eosin (H&E) or used for IHC.
Viable tumor area was quantified by manually demarcating the tumor boundary and the boundary of the grossly necrotic portion of the tumor on an electronic image of an H&E stained section. These areas were quantified by using AxioVision (v4.1) software (Zeiss), and viable tumor area was defined as the proportion of total tumor area that was not grossly necrotic.
Immunoblotting
Whole tissues were homogenized in 1.5 mL of nondenaturing protein lysis buffer (20mM Tris-HCl, pH 7.5, 1% Triton X-100, 100mM NaCl, 40mM NaF, 1mM EDTA, 1mM EGTA) with Protease Inhibitor Cocktail (Sigma) and 1mM Na3VO4. Following lysis on ice for 30 min, homogenates were clarified by centrifugation (12 000 × g) for 10 min at 4C. Protein concentrations were quantified by Bradford Protein Assay (Biorad). 100 μg of protein was denatured in Laemmli Sample Buffer (BioRad), resolved by SDS-PAGE, and transferred to PVDF membranes (BioRad). Membranes were blocked with 5% (w/v) skim milk in either PBS or TBS with 0.05% (v/v) Tween-20 (PBS-T) (1h, RT). Membranes were incubated with primary antibody in 5% (w/v) bovine serum albumin (overnight, 4C) followed by incubation with the appropriate secondary antibody conjugated to horseradish peroxidase in 5% skim milk (1h, RT). Bound antibodies were detected by chemiluminescence (Amersham). Protein bands were quantified by densitometric analyses of photographic films using the BIORAD GS-710 densitometer. In this mouse model, hyperplastic tissue is largely restricted to glands that also harbor a tumor. Thus, all studies examining epithelial hyperplasia involved analyses of tissue surrounding tumors.
Immunohistochemistry
Boiling in 10mM citrate buffer, pH 6.0 for 15-20 min was used for antigen retrieval for the following antibodies; P-S6 (1:200), P-Histone-H3 (1:100), cleaved caspase-3 (1:50) and Ki-67 (1:25). A 1 min boil and 15 min sub-boil in 1mM EDTA, pH 8.0 was used for antigen retrieval for the p877 HER2/ErbB2 antibody (1:100) and a 30 min boil in 1mM EDTA and 10mM Tris, pH 9.0 followed by a 20 min cool was used for antigen retrieval for the CD-31 antibody (1:50). All primary antibodies were incubated overnight at 4°C. Primary antibodies were visualized by using either mouse or rabbit Envision Plus-HRP kits (DakoCytomation, Carpinteria, CA, USA)) or rabbit anti-rat biotinylated IgG (1:100) in combination with Streptavidin/HRP (1:200) (both from DakoCytomation) and bound antibody was detected by DAB reaction. All tissues were counterstained with Gill’s Hematoxylin #3 (Polysciences, Inc., Washington, PA, USA). As a negative control, blocking buffer was used without addition of primary antibody for each tissue.
Proliferation of tumor epithelia was quantified by counting the number of tumor cells staining positive for phosphorylated histone-H3 in a 20X field. At least 5 fields were counted per tumor—3 from the outer regions and 2 from viable tissue in the more central regions. The number of cells per field was identical for rapamycin and vehicle treated tumors. When lung metastases were immunostained for P-histone-H3, a very small proportion of cells showed positive staining (ranging from 5/1000 cells to 15/1000 cells), possibly indicative of low basal rates of proliferation. In addition, the small size of the metastases resulted in a low number of total cells available for assessment. Thus, proliferation in lung metastases was quantified by determining the percentage of mammary epithelial cells staining positively for Ki-67, which stained a more quantifiable proportion of cells (up to ~9% of cells), in all tumor metastases and emboli within a section of lung. Ki-67 expression occurs in multiple stages of the cell cycle; hence, antibodies to this protein inherently detect more proliferating cells than P-H3, a marker of mitosis. A minimum of 250 metastatic cells were counted per lung.
Tumor vascularity was quantified by measuring the average percentage of tumor area positively stained with an anti-CD31 antibody. Electronic images of tumor regions were quantified using Metamorph (v5.0) software. The number of tumor regions quantified varied, depending on tumor size, and ranged from 5 to 50 individual 20X fields.
Statistics
Statistical tests were performed using Microsoft EXCEL, or SAS (SAS Institute, Cary, NC). A general linear random-effects MIXED model incorporating repeated measures was used to test for differences in rates of tumor growth. For continuous data, a Student’s T-test assuming unequal variance was used to compare groups. A Fischer’s exact test was used to compare proportions. A p-value of less than 0.05 was considered statistically significant.
Results
Mammary tumors in MMTV-c-Neu mice have activated mTOR signaling
Previously, we characterized the progressive transcriptome changes associated with HER2/Neu-induced mammary cancer using microarray analyses of wild type mammary glands and mammary tumors in MMTV-c-Neu transgenic mice (19). Further analysis of this data revealed that message levels for several components of the PI3-Kinase (PI3-K) and mTOR pathways were altered. Specifically, mRNA levels for the p85 subunit of PI3-K and p70S6 kinase (p70S6K) were elevated approximately 3-4 fold in tumors compared to wild type glands (figure 1A). In contrast, mRNA levels for 4EBP1 were decreased approximately 3-fold in tumors compared to wild type controls (figure 1A). PI3-K is an upstream activator of mTOR while p70S6K is a downstream target of both mTOR and PI3-K. Elevated levels of these factors would be expected to promote activation of mTOR and its downstream targets. Specifically, p85, the regulatory domain of PI3K, initiates PI3K signaling by binding to phosphorylated sites on ErbB3, the preferred dimerization partner of ErbB2 (24), which is also highly expressed in these tumors (25). The concomitant upregulation of ErbB2, ErbB3 and p85 would be expected to promote PI3K activation, and this is supported by the activation of AKT observed in these tumors (26). Additional factors that can alter mTOR activity whose expression was changed include PTEN and RSK1 (27) (supplementary figure 1A). We also observed changes in a downstream target of mTOR; 4EBP1 is a negative regulator of the translational initiator eIF-4E that is inactivated following phosphorylation by mTOR. Lower levels of this protein would diminish eIF4E inhibition, promoting cap-dependent translation of mRNAs. Thus, the net changes in PI3-K, p70S6K, PTEN, RSK, and 4EBP1 mRNA levels are consistent with a cellular environment favoring enhanced signaling through the mTOR pathway.
Figure 1. Mammary tumors in MMTV-c-Neu mice have activated mTOR signaling.
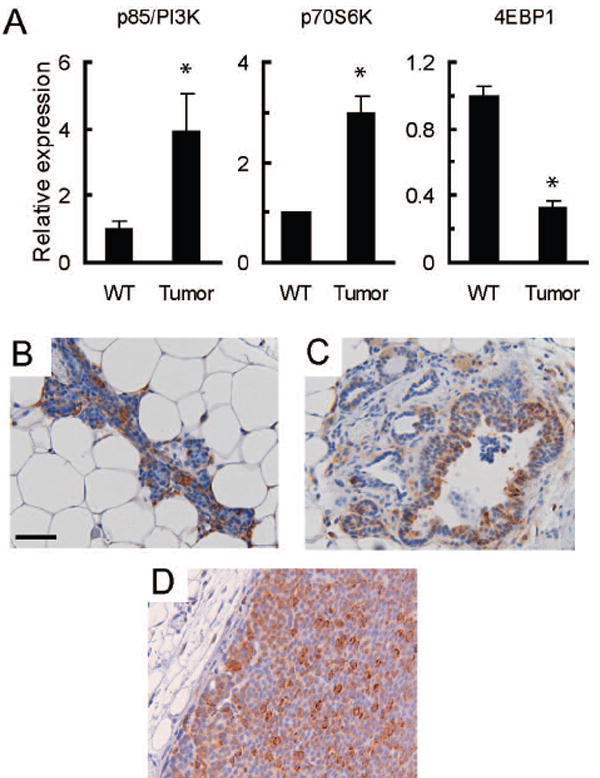
A. Gene expression microarray analysis indicates that mRNA levels for components of the PI3-Kinase and mTOR signaling pathways are altered in MMTV-c-Neu tumors. As previously described, age-matched wild type glands (WT, n=3 pools of 5 mice each) were collected and compared to primary tumors (n=7) (19). Mean signal intensities from microarrays were normalized to wild-type levels, and the normalized values are displayed on the y-axis with tissue type on the x-axis. The graphs show relative mRNA levels for p85/PI3-K, p70S6K and 4EBP1. Error bars represent standard errors, * = p<0.05. B/C/D. Ribosomal Protein S6 is phosphorylated in wild type tissue (B), hyperplastic peri-tumoral epithelium (C) and MMTV-c-Neu tumors (D). Control samples examined without primary antibody showed no staining (data not shown). Representative tissue sections (200X, bar 50μM) of immunohistochemical staining for phosphorylated ribosomal protein S6 (serine 235/236) are shown.
Human HER2-positive breast cancers have active signaling through the mTOR pathway (11). Consistent with this observation, we found that levels of phosphorylated and total mTOR were elevated in MMTV-c-Neu tumors, as compared to wild type glands (supplemental figure 1B). To determine if pathways downstream of mTOR were also activated in MMTV-c-Neu-induced mammary gland tumors, wild-type mammary glands and tumors were examined by immunohistochemical staining for phosphorylated S6, a downstream target of p70S6K. Strong cytoplasmic staining was observed throughout tumors (figure 1D) and in hyperplastic tissue surrounding tumors (figure 1C). Epithelial cells in wild-type tissues also had marked phosphorylation of S6, suggesting that activation of the mTOR pathway is a common component of mammary epithelial homeostasis in normal cells (figure 1B). These data confirm that mTOR signaling is active in MMTV-c-Neu tumors.
Rapamycin induces regression of primary tumors and peri-tumoral hyperplastic epithelium in MMTV-c-Neu mice
Activation of the mTOR pathway in c-Neu-induced tumors suggested that rapamycin, an inhibitor of mTOR, may be an efficacious chemotherapeutic for these tumors. To test this hypothesis directly, 5 MMTV-c-Neu mice bearing tumors were treated every other day with 150 μg rapamycin for 32 days and 5 additional tumor-bearing transgenic mice were treated with vehicle. Treated mice showed no significant changes in weight (supplemental figure 2A) or grooming behavior. In contrast, when this dose was administered daily, substantial weight loss was observed within 4 days (data not shown). In mice treated with the non-toxic dosing paradigm, we measured external tumor volumes with calipers every four days. Two of the rapamycin-treated mice had two tumors, which were individually monitored. Among vehicle treated mice, the external volumes of all tumors increased during treatment, with final volumes ranging from 2 to 7-fold greater than baseline (figure 2A). In sharp contrast, five of the seven tumors in rapamycin-treated mice were reduced between 1.2 to 3.3 fold in size after 32 days. Two rapamycin-treated tumors increased in external volume by 1.1 and 1.4 fold. The average final fold changes in tumor volume within the vehicle and rapamycin-treated groups were 4.0 and 0.8, respectively (p=0.001, MIXED model). Together, these data demonstrate that rapamycin inhibits or regresses primary tumor growth at doses that do not promote overt toxicity.
Figure 2. Rapamycin induces regression of primary tumors and peri-tumoral hyperplastic epithelium in MMTV-c-Neu mice.
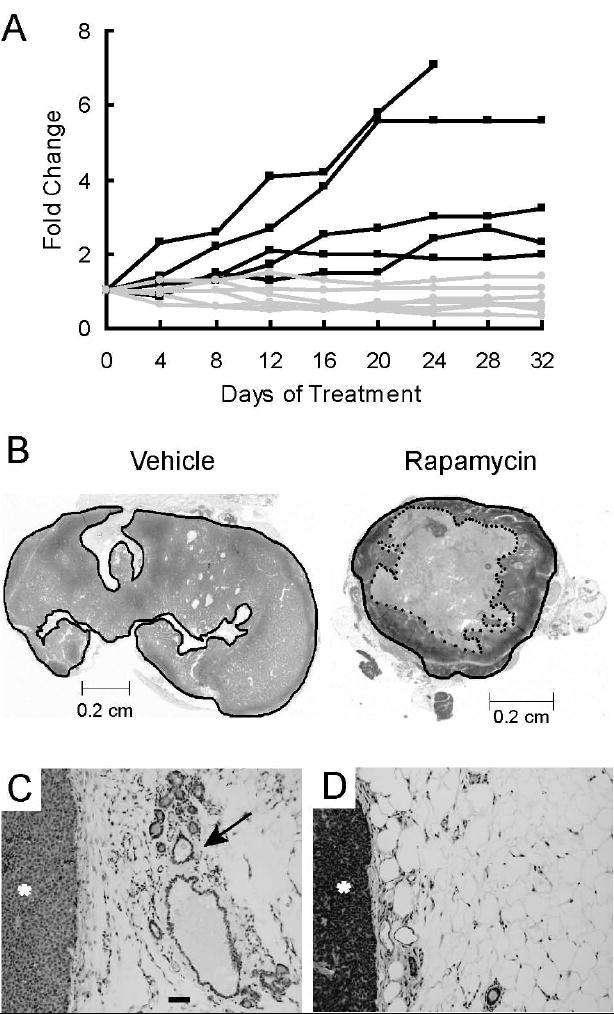
A. Rapamycin inhibits or regresses growth of primary tumors. Mice bearing mammary tumors were treated with 150 μg of rapamycin (n=5) or vehicle (n=5) every other day for 32 days. External tumor volumes were measured for rapamycin and vehicle treated mice every four days using external calipers. Volumes were computed as described in Materials and Methods. Fold changes in tumor volume, normalized to the initial volume, are shown for each tumor. Black lines represent vehicle-treated tumors and grey lines represent rapamycin-treated tumors. B. Rapamycin induces necrotic cavitation of primary tumors. Representative H&E stained sections of tumors taken from mice treated for 32 days with either vehicle or rapamycin. The dark line circumscribes the perimeter of the tumor. The dotted inner line in the rapamycin treated section circumscribes a grossly necrotic core. C/D. Representative H&E stained sections (100x, bar 50μM) of peri-tumoral regions collected from mice treated with either C) vehicle or D) rapamycin. The tumor is marked with an asterisk (*) and a hyperplastic, distended duct is marked with an arrow
H&E stained histological sections of vehicle-treated tumors collected at the end of the study revealed that the tumors were comprised predominantly of densely packed viable tumor epithelia (figure 2B). In contrast, rapamycin-treated tumors typically contained large, centrally-located regions of either acellular material or grossly necrotic epithelia (figure 2B). To quantify these histological differences, the cross-sectional area of tumor that was not grossly necrotic was expressed as a ratio of the total tumor area (supplemental figure 2B). Approximately 92% of the tumor area in vehicle-treated tumors was comprised of viable tissue, while the average proportion of viable tissue in rapamycin-treated tumors was 50% (Student’s T-test, p<0.05). Thus, changes in the external volumes in rapamycin-treated c-Neu/ErbB2 tumors were accompanied by significant necrotic cavitation. Of note, the rapamycin-treated tumor that had increased in external volume by 1.4 fold also had central necrosis.
The MMTV-c-Neu mice develop tumors in areas of hyperplasia. To determine if sustained activation of mTOR was essential for maintenance of hyperplastic lesions, the histopathology of mammary glands surrounding tumors was evaluated (figures 2C and 2D). For the vehicle treated tumors, 4 of 5 tumors (80%) were accompanied by hyperplastic mammary epithelium. In contrast, extensive depletion of this hyperplastic epithelium was observed in 5 of 5 (100%) rapamycin-treated mice (p<0.05, Fischer’s exact test).
Rapamycin blocks mTOR signaling in primary tumors without affecting c-Neu/ErbB2 activation
The c-Neu/ErbB2 receptor is activated by phosphorylation subsequent to dimerization with other ErbB receptors (4). To exclude the possibility that rapamycin-induced regression was due to altered transgene expression or activation, phosphorylated c-Neu/ErbB2 was examined by immunohistochemistry. Activation of c-Neu/ErbB2 was indistinguishable between vehicle and rapamycin treated tumor epithelia (supplemental figures 3A & 3B). Previous studies have reported that rapamycin increases phosphorylated Akt (P-Akt) levels with short term treatment and down-regulates P-Akt with long term exposure of tumor cells (28,29). In contrast to these reports, we found that P-Akt levels were unchanged in treated MMTV-c-Neu tumors (figure 3A). Thus, rapamycin does not block expression the MMTV-c-Neu transgene or alter activation of c-Neu/ErbB2 or its proximal target, Akt.
Figure 3. Rapamycin blocks mTOR signaling in primary tumors without affecting c-Neu/ErbB2 activation.
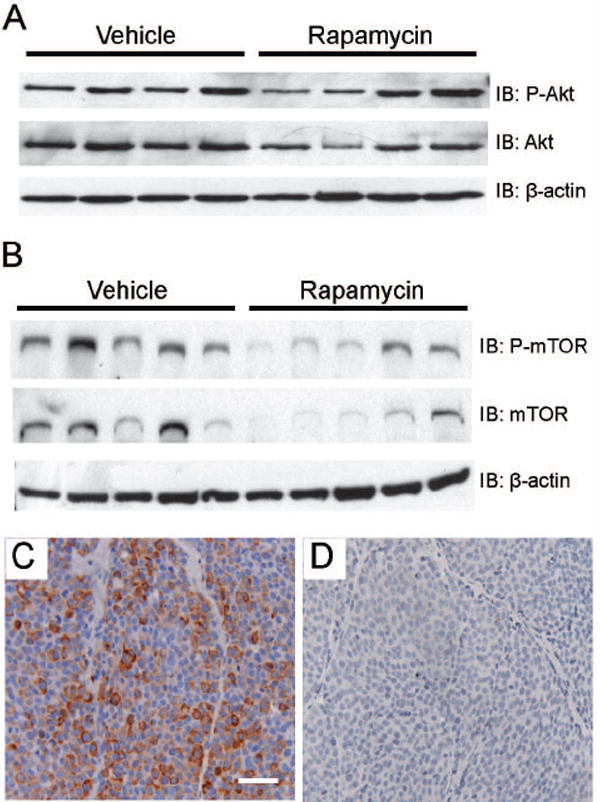
A. Western blot analysis indicates that rapamycin does not alter phosphorylation of Akt in tumors. Whole cell lysates (100 μg) of primary MMTV-c-Neu tumors were evaluated by immunoblots that were sequentially probed using antibodies directed against phospho-Akt and total Akt. B. Rapamycin does not affect the relative levels of phosphorylated mTOR in tumors. Western blots were sequentially probed using antibodies directed against phospho-mTOR (serine 2448) and total mTOR. C/D. Rapamycin decreases phosphorylation of Ribosomal Protein S6 in tumors. Representative sections (200X, bar 50μM) of immunohistochemical staining of cytoplasmic phosphorylated Ribosomal Protein S6 (serine 235/236) in primary tumors collected from vehicle (C) and rapamycin (D) treated mice.
To verify that rapamycin blocked mTOR activity, levels of phosphorylated mTOR and ribosomal subunit S6 were evaluated. Rapamycin did not alter the relative levels of phosphorylated mTOR (figure 3B), suggesting that upstream signaling cascades remained intact. While the relative levels of phosphorylated mTOR remained unchanged, the efficacy of rapamycin in blocking downstream mTOR signaling was clearly demonstrated by examining phosphorylated S6 in tumor sections. Phosphorylated S6 was present throughout tumor epithelia in vehicle-treated tumors, but was greatly diminished or absent in rapamycin-treated tumors (figures 3C and 3D). Thus, rapamycin-induced regression of tumors is associated with a loss of mTOR signaling, despite sustained activation of the c-Neu/ErbB2 receptor.
Rapamycin inhibits primary tumor cell proliferation and promotes focal cell death
To determine whether inhibition of tumor growth and regression was due to cytostatic drug effects, proliferation rates were measured by quantifying levels of phosphorylated histone-H3, a nuclear marker of mitosis, in primary tumors that had been treated for 32 days. Rapamycin-treated tumors had a 49% reduction in the number of proliferating cells within the viable regions of the tumor, as compared to vehicle-treated tumors (p<0.05, Student’s T-test) (figure 4A and supplemental figures 4A and 4B). One reported mechanism of rapamycin-induced inhibition of proliferation in cultured mammary epithelial cells is downregulation of cyclin D1 (12,13). This is particularly salient to c-Neu/ErbB2 tumors which are dependent upon cyclin D1 for growth (8). An analysis of whole cell lysates collected from tumors revealed that cyclin D1 expression was decreased in 4 of 5 rapamycin-treated tumors, with a mean reduction of 73% compared to untreated control tumors (figures 4B and 4C, p<0.05, Student’s T-test). Thus, the ability of rapamycin to block proliferation in primary tumors may be due, in part, to down-regulating cyclin D1 expression.
Figure 4. Rapamycin inhibits primary tumor cell proliferation and promotes focal cell death.
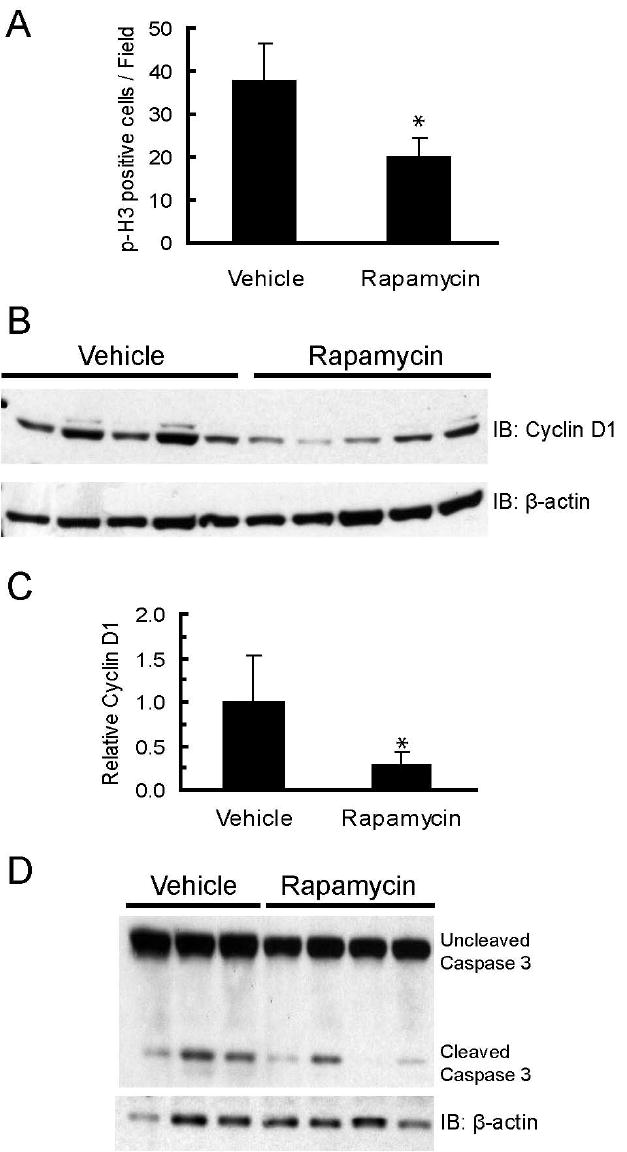
A. Rapamycin decreases the number of cells expressing phosphorylated histone-H3. The graph shows the average number of phosphorylated histone-H3 positive cells per field for primary tumors collected from vehicle (n=5) and rapamycin (n=6) treated mice. Five fields with equal numbers of cells/field were counted per tumor. Error bars represent standard deviations, * = p<0.05. B. Rapamycin decreases cyclin D1 levels in primary tumors. Whole cell lysates (100 μg) of primary MMTV-c-Neu tumors from vehicle and rapamycin treated mice were resolved by SDS-PAGE and transferred to PVDF membrane. Membranes were probed with an anti-cyclin D1 antibody. β-actin was used as a loading control. C. Bands were quantified by densitometry and the graph depicts mean cyclin D1 levels normalized to β-actin for vehicle and rapamycin treated mice. Error bars represent standard deviations, * = p<0.05. D. Western blot of uncleaved and cleaved caspase-3 levels in tumors collected from mice treated with vehicle or rapamycin for 5 days. β-actin serves as a loading control.
To characterize the progression of histological changes underlying tumor cavitation, tumors were collected from mice treated with rapamycin every other day for 5, 10 and 32 days (supplemental figures 4D-4F). At 5 days, some treated tumors had developed small foci of cells with pyknotic nuclei. By day 10, numerous foci, many of which had coalesced into larger bands of dying cells, were present throughout much of the central portion of the tumors. By 32 days, only small islands of viable tumor cells were found in the central regions of the tumors, with the majority of the central core being comprised of diffusely staining material. A closer examination of the tumor epithelia within the central necrotic core showed that they had features of ischemic necrosis (30). Specifically, “mummified” cells were observed; i.e., cells that were structurally intact, but had lost their ability to retain staining due to protein and nucleic acid degradation.
To characterize the mechanism of cell death in tumors, histological sections were examined for evidence of DNA fragmentation using TUNEL analysis. While “mummified” cells in the rapamycin treated tumors were not TUNEL positive, foci of TUNEL positivity were found along the perimeters of the necrotic zone as well as in the viable tissue regions. However, similar foci were also observed in vehicle-treated tumors (data not shown). Isolated TUNEL positive cells were rare in both vehicle and rapamycin treated tumors, indicating that cell death characterized by TUNEL staining only occurred within large groups of tumor cells. The percentage of isolated TUNEL positive cells was also not different between the two treatment groups (data not shown). Thus, TUNEL positivity could not be directly linked to rapamycin treatment. To ascertain whether dying cells were undergoing caspase-3 dependent apoptosis, levels of activated (cleaved) caspase-3 were measured in vehicle- and rapamycin-treated tumors by immunoblotting. There were no significant differences in the relative amount of activated caspase-3 after either 5 (figure 4D) or 32 days (data not shown) of treatment, suggesting that the extensive cell death in rapamycin-treated tumors was not associated with caspase-3 activation. These data demonstrate that rapamycin induced rapid, focal cell death and that the majority of the dead or dying cells in rapamycin treated tumors were TUNEL-negative, did not have activated caspase-3, and manifested the characteristic features of ischemic necrosis.
Rapamycin inhibits mTOR activity in tumor endothelial cells, but does not alter microvessel density
mTOR signaling is activated in tumor endothelia by growth factors such as VEGF, and its inhibition by rapamycin decreases proliferation of endothelial cells (31-34). Thus, tumor cell death and cavitation in MMTV-c-Neu mice could be the consequence of disrupted blood flow caused by the direct effects of rapamycin on vascular endothelia. Vehicle-treated tumor endothelial cells stained positively for phosphorylated S6 (figure 5A), indicating that the mTOR pathway is active in these cells. In contrast, S6 phosphorylation was considerably diminished with rapamycin treatment (figure 5B), indicating that this drug inhibits mTOR activity in endothelial cells within tumors.
Figure 5. Rapamycin inhibits mTOR activity in tumor endothelial cells, but does not alter microvessel density.
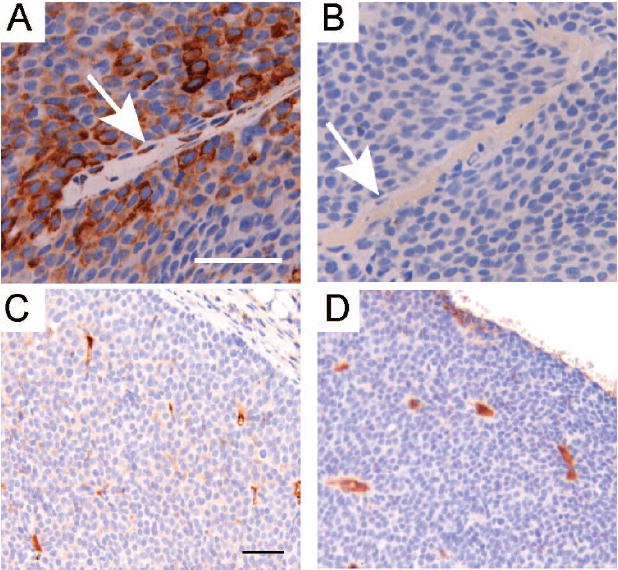
A/B. Representative sections (400X, bar 50μM) are shown that were immunohistochemically stained for phosphorylated Ribosomal Protein S6 (serine 235/236) in primary tumors collected from mice treated with either vehicle (A) or rapamycin (B) for 32 days. Arrows identify vascular endothelial cells. C/D. Representative sections (200X, bar 50μM) are shown that were immunohistochemically stained for CD31 in primary tumors collected from mice treated with either vehicle (C) or rapamycin (D) for 32 days.
One manifestation of disrupted endothelial signaling is decreased microvascular density due, in part, to an inability to form new vessels. To assess whether rapamycin treatment altered intratumoral vasculature in MMTV-c-Neu tumors, tumor sections from mice treated for either 5 or 32 days were immunostained for CD31, a cell adhesion and signaling molecule restricted to vascular endothelial cells (figures 5C and 5D). When average areas of CD31-postive staining were compared in vehicle and rapamycin-treated mice (1.9% vs. 1.2%, supplemental figure 5), there was no difference between treatment groups (Student’s T-test, p=0.25). Thus, although rapamycin blocked mTOR signaling in tumor endothelia, it did not induce a measurable change in microvessel density within the remaining areas of viable tumor tissue.
Rapamycin inhibits proliferation in lung metastases
Tumors in MMTV-c-Neu mice spontaneously metastasize to the lung (21). To assess whether rapamycin treatment affected metastases, lungs were collected from mice treated with vehicle or rapamycin for 32 days. Like the primary MMTV-c-Neu tumors, metastases had prominent phosphorylation of S6 that was blocked by rapamycin treatment (supplemental figures 6A and 6B). To determine whether rapamycin decreased proliferation of metastatic cells, these cells were immunostained for Ki-67, a nuclear protein present in proliferating cells (figures 6A and 6B). When compared to the vehicle controls, rapamycin caused a significant 3-fold decrease in proliferation of metastatic cells (p<0.05, figure 6C). Similar to the primary tumors, no change in apoptosis was detected between the two groups using TUNEL staining or immunohistochemistry for cleaved caspase-3 (data not shown). Unlike the primary tumors, rapamycin treatment did not promote gross necrotic cavitation of lung metastases. These data show that rapamycin blocks proliferation of metastatic tumor cells, but does not cause apoptotic cell death.
Figure 6. Rapamycin inhibits proliferation in lung metastases.
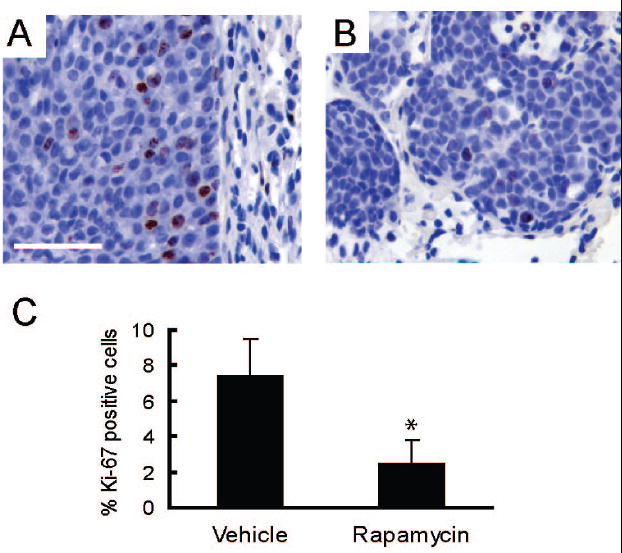
A/B. Representative sections (400X, bar 50μM) are shown for nuclear staining for Ki-67 in lung metastases collected from mice treated with either vehicle (A) or rapamycin (B) for 32 days. C) The graph shows the average percentage of Ki-67 positive metatstatic epithelial cells per mouse in lung metastases collected from vehicle (n=3) and rapamycin (n=4) treated mice. Error bars represent standard deviations, * = p<0.05.
Discussion
Breast cancer is the most common malignancy in women, comprising approximately a third of all newly diagnosed cancers (35). Critical to the advancement of effective treatment modalities for this disease is the development and utilization of biologically relevant cancer models to assess the impact and mechanisms of action of molecularly targeted anti-cancer therapies. Studies presented herein extend previous research on the effects of rapamycin in the c-Neu/ErbB2 mammary tumor models by assessing the impact of rapamycin on hyperplasia of the mammary gland as well as metastatic tumor cells (15,16).
Rapamycin induced regression of primary tumors as a result of extensive caspase-independent cell death accompanied by a decrease in proliferation. We have previously shown that tissues surrounding tumors have focal hyperplasia, express active c-Neu/ErbB2 and specific markers of human HER2/ErbB2 tumors, and have a unique mRNA expression signature that clusters between wild-type mammary glands and tumors (19). We found that rapamycin treatment causes regression of this peritumoral tissue, indicating that, like the primary tumors, hyperplastic cells may also require mTOR signaling for growth and survival. These results reveal that mTOR dependency may occur early in the c-Neu/ErbB2-induced tumorigenic process, suggesting that treatment of hyperplastic lesions with rapamycin could prevent or delay primary tumor formation. In support of this concept, rapamycin prevented primary tumor formation in MMTV-NeuYD mice (16) as well as secondary tumor growth in MMTV-c-Neu mice (15). The presence of phosphorylated S6 levels in normal epithelial cells was unexpected because rapamycin does not negatively impact mammary gland development in early pubertal mice (36). These observations suggest that mTOR signaling may play a more significant role in malignant c-Neu/ErbB2-driven epithelial growth than in the normal mammary gland.
Like human HER2/ErbB2 positive tumors, MMTV-c-Neu tumors are highly metastatic (21,37). Rapamycin treatment impeded mTOR signaling in lung metastases, and this blockade was associated with a significant reduction in proliferation. While previous studies have suggested that rapamycin inhibits metastatic progression, they focused on the use of experimental metastases (38-41). The results presented herein indicate that rapamycin is efficacious at inhibiting proliferation of spontaneous distant metastases arising in an autochthonous tumor model. Because this is a spontaneous lung metastasis model with considerable intrinsic variability in the onset and extent of metastatic progression, an assessment of the impact of rapamycin on the formation and growth of metastatic lesions following primary tumor formation will require both extended treatment paradigms on large numbers of mice and quantitative in vivo imaging of metastatic tumors.
The effects of rapamycin on proliferation of malignant epithelial cells from primary tumors and lung metastases were similar. In contrast, rapamycin-induced cell death was only seen in primary tumors and occurred in a focal pattern. The focal nature of the epithelial cell death suggests that it may be due to factors extrinsic to epithelial cells. This reasoning is supported by studies characterizing the intrinsic effects of rapamycin on HER2/ErbB2 expressing mammary epithelial cells in culture, which have shown that rapamycin blocks proliferation, but generally does not promote apoptosis (33).
Our findings related to rapamycin-induced cell death differ somewhat from those reported in a study of the effects of rapamycin treatment of transplanted polyoma middle T induced tumors (MMTV-PyV-mT) (42). In these mice, an early transient increase in apoptosis was observed in tumor cells after 3 hours of rapamycin treatment, but not after 7 days. These observations prompted the authors to conclude that apoptotic cell death is a transient event which only occurs early in the treatment paradigm. While this explanation is consistent with the mode of action of the drug in the PyV-mT transplant model, it would not explain our observation that the vast majority of cell death occurred after the fifth day of treatment when no changes in apoptotic levels were observed. This difference may be due to the fact that, while PyV-mT recapitulates some of the signaling of HER2/ErbB2, it is not identical (43) and their associated tumors have distinct histopathologies (44).
One explanation for the extensive, but focal, cell death associated with rapamycin treatment in the MMTV-c-Neu mice is vascular dysfunction. Rapamycin blocked mTOR signaling in vascular endothelia in vivo, an effect that would be expected to make these cells refractory to growth factor stimulation (33,34). Despite endothelial mTOR inhibition, however, rapamycin did not induce significant changes in tumor vessel density. This finding contrasts with an earlier study which showed that rapamycin decreased microvessel density in a Neu-YD transgenic mouse model (16). The courses of tumor progression in Neu-YD and MMTV-c-Neu mice are distinct (45), and the differences in signaling properties between the two Neu transgenes may underlie the differences in rapamycin regulation of microvessel density.
Rapamycin-induced cell death was observed as early as 5 days after initiation of treatment in the MMTV-c-Neu tumors and was pervasive by day 10. The rapid and focal nature of tumor cell death may reflect acute changes in established blood flow, rather than attenuation of vessel proliferation. Rapamycin can promote thrombotic occlusion of tumor vessels (46,47). We observed histological evidence of vessel occlusion, including clogged and dilated vessels and dying cells with features of ischemic necrosis, in tumors and peritumoral regions that were similar to those described for other types of tumors treated with rapamycin (data not shown). However, unlike these other studies, we could not definitively link this pathology to the effects of rapamycin because similar features were observed in vehicle-treated tumors. One explanation for this difference is that our study utilized a spontaneous tumor model that develops tumors with long latency and has intrinsic inter-tumor variability while previous studies examining rapamycin-induced vessel occlusion involved orthotopic transplant models that rapidly generate clonal, relatively homogenous tumors. Thus, while progressive vessel occlusion is a highly plausible mechanism accounting for tumor cell death, and could explain why cell death was not observed within the relatively avascular lung metastases, we cannot rule out the possibility that death was caused by other mechanisms.
In summary, rapamycin inhibits multiple stages of c-Neu/ErbB2 induced tumorigenesis in vivo. In addition to inducing regression of primary tumors, we found that rapamycin inhibits proliferation in spontaneous lung metastases and causes regression of peritumoral hyperplasia. These findings underscore the exquisite dependence of c-Neu/ErbB2 tumorigenic progression on intact mTOR signaling. This study employed a mouse model that develops spontaneous mammary gland tumors due to aberrant upregulation of a non-mutated c-Neu/ErbB2 oncogene, comparable to that found in human tumors. Consequently, our findings provide strong support for further evaluating the efficacy of rapamycin in patients with HER2/ErbB2 positive breast cancers, including those with early and late stage disease. Of note, clinical trials utilizing rapamycin for the treatment of breast cancer have yielded promising, but mixed results (48). This may be due to the heterogeneity of this disease. Our studies suggest that HER2/ErbB2 positive cancers may be particularly sensitive to the effects of rapamycin.
Supplementary Material
Acknowledgments
We thank Dr. Nick Ziats for providing reagents and assistance with blood vessel staining and David Danielpour for thoughtful discussions regarding mechanisms of rapamycin action. We would also like to thank Kristen Lozada for excellent technical assistance. These studies were facilitated by the Histology and Gene Expression Core Facilities of the Case Comprehensive Cancer Center (P30CA43703).
This work was supported by grants from the National Institutes of Health [RO1-CA090398 (to RAK) and training grant T32-GM07250 (MSTP/CWRU) (to JDM)], an Ohio Innovation Incentive Fellowship (to JDM), and by an Endocrine Society Summer Research Fellowship (to JTP).
References
- 1.Menard S, Tagliabue E, Campiglio M, Pupa SM. Role of HER2 gene overexpression in breast carcinoma. J Cell Physiol. 2000;182:150–162. doi: 10.1002/(SICI)1097-4652(200002)182:2<150::AID-JCP3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 2.Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 3.Citri A, Yarden Y. EGF–ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 4.Citri A, Skaria KB, Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 5.Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinase switching between life and death. Pharmacol Res. 2004;50:545–549. doi: 10.1016/j.phrs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Janus A, Robak T, Smolewski P. The mammalian target of the rapamycin (mTOR) kinase pathway: its role in tumourigenesis and targeted antitumour therapy. Cell Mol Biol Lett. 2005;10:479–498. [PubMed] [Google Scholar]
- 7.Hulit J, Lee RJ, Russell RG, Pestell RG. ErbB-2-induced mammary tumor growth: the role of cyclin D1 and p27Kip1. Biochem Pharmacol. 2002;64:827–836. doi: 10.1016/s0006-2952(02)01145-0. [DOI] [PubMed] [Google Scholar]
- 8.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 9.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–3199. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 10.Jiang H, Coleman J, Miskimins R, Miskimins WK. Expression of constitutively active 4EBP-1 enhances p27Kip1 expression and inhibits proliferation of MCF7 breast cancer cells. Cancer Cell Int. 2003;3:2. doi: 10.1186/1475-2867-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou X, Tan M, Stone HV, et al. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin Cancer Res. 2004;10:6779–6788. doi: 10.1158/1078-0432.CCR-04-0112. [DOI] [PubMed] [Google Scholar]
- 12.Yu K, Toral-Barza L, Discafani C, et al. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr Relat Cancer. 2001;8:249–258. doi: 10.1677/erc.0.0080249. [DOI] [PubMed] [Google Scholar]
- 13.Noh WC, Mondesire WH, Peng J, et al. Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res. 2004;10:1013–1023. doi: 10.1158/1078-0432.ccr-03-0043. [DOI] [PubMed] [Google Scholar]
- 14.Hermanto U, Zong CS, Wang LH. ErbB2-overexpressing human mammary carcinoma cells display an increased requirement for the phosphatidylinositol 3-kinase signaling pathway in anchorage-independent growth. Oncogene. 2001;20:7551–7562. doi: 10.1038/sj.onc.1204964. [DOI] [PubMed] [Google Scholar]
- 15.Law M, Forrester E, Chytil A, et al. Rapamycin disrupts cyclin/cyclin-dependent kinase/p21/proliferating cell nuclear antigen complexes and cyclin D1 reverses rapamycin action by stabilizing these complexes. Cancer Res. 2006;66:1070–1080. doi: 10.1158/0008-5472.CAN-05-1672. [DOI] [PubMed] [Google Scholar]
- 16.Liu M, Howes A, Lesperance J, et al. Antitumor activity of rapamycin in a transgenic mouse model of ErbB2-dependent human breast cancer. Cancer Res. 2005;65:5325–5336. doi: 10.1158/0008-5472.CAN-04-4589. [DOI] [PubMed] [Google Scholar]
- 17.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 18.Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–397. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 19.Landis MD, Seachrist DD, Montanez-Wiscovich ME, Danielpour D, Keri RA. Gene expression profiling of cancer progression reveals intrinsic regulation of transforming growth factor-beta signaling in ErbB2/Neu-induced tumors from transgenic mice. Oncogene. 2005;24:5173–5190. doi: 10.1038/sj.onc.1208712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niemeyer CC, Spencer-Dene B, Wu JX, Adamson ED. Preneoplastic mammary tumor markers: Cripto and Amphiregulin are overexpressed in hyperplastic stages of tumor progression in transgenic mice. Int J Cancer. 1999;81:588–591. doi: 10.1002/(sici)1097-0215(19990517)81:4<588::aid-ijc14>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 21.Guy CT, Webster MA, Schaller M, et al. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muraoka-Cook RS, Shin I, Yi JY, et al. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25:3408–3423. doi: 10.1038/sj.onc.1208964. [DOI] [PubMed] [Google Scholar]
- 23.Trimble MS, Xin JH, Guy CT, Muller WJ, Hassell JA. PEA3 is overexpressed in mouse metastatic mammary adenocarcinomas. Oncogene. 1993;8:3037–3042. [PubMed] [Google Scholar]
- 24.Roux PP, Shahbazian D, Vu H, et al. RAS/ERK Signaling Promotes Site-specific Ribosomal Protein S6 Phosphorylation via RSK and Stimulates Cap-dependent Translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007;67:167–177. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- 27.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101:13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarbassov dD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 30.Cotran RS, Vinay K, Collins T. Pathologic Basis of Disease. Sixth. W.B. Saunders Company; 1999. [Google Scholar]
- 31.Vinals F, Chambard JC, Pouyssegur J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J Biol Chem. 1999;274:26776–26782. doi: 10.1074/jbc.274.38.26776. [DOI] [PubMed] [Google Scholar]
- 32.Yu Y, Sato JD. MAP kinases, phosphatidylinositol 3-kinase, and p70 S6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J Cell Physiol. 1999;178:235–246. doi: 10.1002/(SICI)1097-4652(199902)178:2<235::AID-JCP13>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 33.Del Bufalo D, Ciuffreda L, Trisciuoglio D, et al. Antiangiogenic potential of the Mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006;66:5549–5554. doi: 10.1158/0008-5472.CAN-05-2825. [DOI] [PubMed] [Google Scholar]
- 34.Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16:771–780. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 35.Jemal A, Tiwari RC, Murray T, et al. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 36.Jankiewicz M, Groner B, Desrivieres S. Mammalian target of rapamycin regulates the growth of mammary epithelial cells through the inhibitor of deoxyribonucleic acid binding Id1 and their functional differentiation through Id2. Mol Endocrinol. 2006;20:2369–2381. doi: 10.1210/me.2006-0071. [DOI] [PubMed] [Google Scholar]
- 37.Aziz SA, Pervez S, Khan S, et al. Significance of immunohistochemical c-ErbB-2 product localisation pattern for prognosis in human breast cancer. Pathol Oncol Res. 2001;7:190–196. doi: 10.1007/BF03032348. [DOI] [PubMed] [Google Scholar]
- 38.Guba M, von Breitenbuch P, Steinbauer M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- 39.Klos KS, Wyszomierski SL, Sun M, et al. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res. 2006;66:2028–2037. doi: 10.1158/0008-5472.CAN-04-4559. [DOI] [PubMed] [Google Scholar]
- 40.Boffa DJ, Luan F, Thomas D, et al. Rapamycin inhibits the growth and metastatic progression of non-small cell lung cancer. Clin Cancer Res. 2004;10:293–300. doi: 10.1158/1078-0432.ccr-0629-3. [DOI] [PubMed] [Google Scholar]
- 41.Luan FL, Ding R, Sharma VK, et al. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney Int. 2003;63:917–926. doi: 10.1046/j.1523-1755.2003.00805.x. [DOI] [PubMed] [Google Scholar]
- 42.Namba R, Young LJ, Abbey CK, et al. Rapamycin inhibits growth of premalignant and malignant mammary lesions in a mouse model of ductal carcinoma in situ. Clin Cancer Res. 2006;12:2613–2621. doi: 10.1158/1078-0432.CCR-05-2170. [DOI] [PubMed] [Google Scholar]
- 43.Desai KV, Xiao N, Wang W, et al. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc Natl Acad Sci U S A. 2002;99:6967–6972. doi: 10.1073/pnas.102172399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cardiff RD, Wellings SR. The comparative pathology of human and mouse mammary glands. J Mammary Gland Biol Neoplasia. 1999;4:105–122. doi: 10.1023/a:1018712905244. [DOI] [PubMed] [Google Scholar]
- 45.Dankort D, Maslikowski B, Warner N, et al. Grb2 and Shc adapter proteins play distinct roles in Neu (ErbB-2)-induced mammary tumorigenesis: implications for human breast cancer. Mol Cell Biol. 2001;21:1540–1551. doi: 10.1128/MCB.21.5.1540-1551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruns CJ, Koehl GE, Guba M, et al. Rapamycin-induced endothelial cell death and tumor vessel thrombosis potentiate cytotoxic therapy against pancreatic cancer. Clin Cancer Res. 2004;10:2109–2119. doi: 10.1158/1078-0432.ccr-03-0502. [DOI] [PubMed] [Google Scholar]
- 47.Guba M, Yezhelyev M, Eichhorn ME, et al. Rapamycin induces tumor-specific thrombosis via tissue factor in the presence of VEGF. Blood. 2005;105:4463–4469. doi: 10.1182/blood-2004-09-3540. [DOI] [PubMed] [Google Scholar]
- 48.Chan S, Scheulen ME, Johnston S, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–5322. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.