

Abstract
The chemical reactivity, toxicology and pharmacological responses to nitroxyl (HNO) are often distinctly different from those of nitric oxide (NO). The discovery that HNO donors may have pharmacological utility for treatment of cardiovascular disorders such as heart failure and ischemia reperfusion has led to increased speculation of potential endogenous pathways for HNO biosynthesis. Here, the ability of heme proteins to utilize H2O2 to oxidize hydroxylamine (NH2OH) or N-hydroxy-L-arginine (NOHA) to HNO was examined. Formation of HNO was evaluated with a recently developed selective assay in which the reaction products in the presence of reduced glutathione (GSH) were quantified by HPLC. Release of HNO from the heme pocket was indicated by formation of sulfinamide (GS(O)NH2), while the yields of nitrite and nitrate signified the degree of intramolecular recombination of HNO with the heme. Formation of GS(O)NH2 was observed upon oxidation of NH2OH, whereas NOHA, the primary intermediate in oxidation of L-arginine by NO synthase, was apparently resistant to oxidation by the heme proteins utilized. In the presence of NH2OH, the highest yields of GS(O)NH2 were observed with proteins in which the heme was coordinated to a histidine (horseradish peroxidase, lactoperoxidase, myeloperoxidase, myoglobin and hemoglobin) in contrast to a tyrosine (catalase) or cysteine (cytochrome P450). That peroxidation of NH2OH by horseradish peroxidase produced free HNO, which was able to affect intracellular targets, was verified by conversion of 4,5-diaminofluorescein to the corresponding fluorophore within intact cells.
Keywords: heme, peroxidase, hydroxylamine, N-hydroxy-L-arginine, nitroxyl, sulfinamide
Introduction
Nitroxyl (HNO) has been shown to exhibit unique biological and pharmacological properties compared to those of other nitrogen oxides. In the cardiovascular system, HNO donors increase cardiac function (e.g., inotropy and lusitropy) in an additive and independent manner to β-adrenergic signaling [1, 2]. While HNO can induce early preconditioning-like effects that protect heart tissue against ischemia-reperfusion injury [3], the outcome is dependent upon the timing of administration [4]. In addition, HNO regulates calcium channels, both in the cardiovascular and nervous systems [5–10]. These responses to HNO are often discrete from those of its redox cousin nitric oxide (NO) [11, 12]. Such findings have led recently to extensive investigation of the chemical, biochemical and pharmacological properties of HNO, particularly in comparison to NO (reviewed in [12, 13]). Moreover, the potential for positive cardiovascular impacts has intensified interest both in the production of HNO donors as pharmacological agents [14] and in the elucidation of biosynthetic routes to HNO production.
HNO donors have been used clinically since the 1950s [15]. For instance, HNO is the active metabolite of the alcohol sensitizing drug cyanamide (NH2CN) [16], which is used for pharmacotherapy of alcohol abuse. Additionally, hydroxyurea, which is a chemotherapeutic used to reduce the complications of sickle cell disease, was found to be oxidatively degraded to HNO [17, 18]. Although endogenous production of HNO has yet to be demonstrated in vivo, numerous in vitro assays have indicated the existence of several potential biosynthetic mechanisms. For instance HNO can be produced by the enzyme NO synthase (NOS) under conditions where cofactor concentrations are limited [19–24] or by association of an S-nitrosothiol and a thiol [25–28]. Furthermore, formation of HNO during the enzymatic oxidation of cyanamide and hydroxyurea suggests that endogenous reduced nitrogen species may be similarly converted to HNO.
Many heme proteins function as peroxidases in the catalyzed oxidation of a wide range of substrates. Despite the protective and functional utility of peroxidases, an increase in peroxidase activity has been implicated in the pathology of a number of diseases [29, 30]. Here, we examined the ability of heme proteins to produce HNO from peroxidation of hydroxylamine (NH2OH) or N-hydroxy-L-arginine (NOHA), which are both produced during catalytic turnover of NOS [20, 31]. Hydroxylamine and derivatives such as NOHA and hydroxyurea are logical substrates for formation of HNO by peroxidation since their nitrogens are two electrons more reduced than that in HNO. Although in vivo concentrations of NH2OH and NOHA are not well established, the pharmokinetics of hydroxyurea are better understood. At a standard 20 mg/kg dose of hydroxyurea, maximum serum levels can exceed 100 mM [32]. Although such levels are expected only from exogenous sources, the demonstrated ability to accumulate hydroxylamines suggests the possibility that this class of compound may function as endogenous precursors of HNO.
Production of HNO from oxidation of NH2OH and NOHA was evaluated with a recently developed, selective assay in which formation of sulfinamide (GS(O)NH2) from the association of HNO with reduced glutathione (GSH) was quantified by HPLC [33]. The reaction of HNO with GSH was further investigated in real-time by both chemiluminescence and fluorometric assays.
Experimental Procedures
Materials
Hydroxylamine hydrochloride (NH2OH), reduced and oxidized glutathione (GSH and GSSG, respectively), sodium nitrite (NO2−), sodium nitrate (NO3−), diethylenetriaminepentaacetic acid (DTPA), horseradish peroxidase (HRP), horse heart myoglobin (metMb), human hemoglobin (metHb), hemin, catalase and lactoperoxidase (LPO) were obtained from Sigma-Aldrich. 4,5-Diaminofluorescein diacetate (DAF) was purchased from Calbiochem (San Diego, CA). Human leukocytes myeloperoxidase (MPO) [34] was purchased from Alexis while human cytochrome P450 (P450) was acquired from Oxford Biomedical Research. All proteins were used without further purification, and stock solutions were prepared fresh daily at 100× in Milli-Q filtered water unless otherwise specified. NOHA was a generous gift from Prof. Jon Fukuto (UCLA School of Medicine). 1-[2-(Carboxylato)pyrrolidin-1-yl]diazem-1-ium-1,2-diolate (PROLI/NO) was a generous gift from Drs. Joseph Saavedra and Larry Keefer (Laboratory of Comparative Carcinogenesis, National Cancer Institute at Frederick). Sulfinamide was synthesized as previously described [33, 35]. Stock solutions of NO were prepared by sparging argon-deaerated 100 mM phosphate buffer (pH 7.4) with hydroxide scrubbed NO (>99%; Matheson, Montgomeryville, PA).
Heme protein- and hemin-mediated peroxidation of NH2OH or NOHA
Enzymes, metMb, metHb or hemin (5 μM) were dissolved in sodium phosphate buffer (10 mM, pH 7.4) containing the metal chelator DTPA (50 μM), GSH (100 μM) and either NH2OH or NOHA (500 μM). This level of substrate was assumed to be biologically feasible given the high millimolar concentrations of hydroxyurea that can established [32]. Furthermore, these conditions were chosen to optimize evaluation of the reaction given the concentration of GSH (100 μM) required for the assay. The reaction was initiated by addition of H2O2 (100 μM) and was allowed to proceed at 37°C for 10 min. The reaction was terminated by removal of protein by centrifugal filtration (14,000 rpm for 30 min at 4°C, Microcon 3 kDa centrifugal filter device, Millipore). The deproteinized ultrafiltrate was injected directly onto a C-18 HPLC column to characterize the reaction products as previously described [33].
Fluorescent analysis
Human breast carcinoma (MCF-7) cells were cultured as attached cells to 80% confluence in T-75 flasks (Nalge Nunc International, Rochester, NY) containing RPMI 1640 medium at 37°C in a humidified incubator with 5% CO2 in air. A suspension of cells (106) was incubated with DAF (5 μM) at 37°C for 15 min. Cells were rinsed with PBS three times by a cycle of suspension and centrifugation. Fluorescence signal from cell suspensions in PBS containing DTPA (50 μM) were analyzed on a Perkin Elmer LS50B fluorometer using an excitation at 495 and emission at 515 nm with 5.0 mm slit widths in a 2 mL reaction volume held at 37°C while stirring with a water-jacketed cuvette holder [36].
Chemiluminescence analysis
Production of NO is commonly quantified from the chemiluminescence signal resulting from reaction of NO with ozone [37]. Recently, a commercially available chemiluminescence system (Sievers NO Analyzer, Ionics, Boulder, CO) was shown to also detect HNO in a GSH-dependent manner [38]. Therefore, production of HNO during peroxidation of NH2OH or NOHA was examined by performing the reaction in the helium-purged reaction vessel of the analyzer.
Gas chromatographic and electron paramagnetic resonance analysis
Peroxidation of NH2OH (100 mM) by HRP (500 μM) was initiated by addition of H2O2 (50 mM) to the deaerated solution in phosphate buffer (50 mM, pH 7.4, 1.0 mL) contained in a septum-stoppered flask. After two hours at room temperature, the yield of nitrous oxide (N2O), as a marker of free HNO, was analyzed by injecting an aliquot of the reaction headspace (250 μL) onto a 6890 Hewlett Packard gas chromatograph equipped with a thermal conductivity detector and a 6 ft × 1/8 in Porapak Q column at an operating oven temperature of 50°C (injector and detector 150°C) with a flow rate of 16.67 mL/min (He carrier gas). The retention time of N2O of 2.78 min was identical to authentic N2O (Aldrich), and the yield was calculated from an N2O standard curve. An aliquot (300 μL) of the reaction solution was also transferred to an EPR tube and frozen in liquid nitrogen. EPR spectra were recorded on a Bruker ER200D spectrometer using 8.5 mW microwave power, 5.0 G modulation amplitude and 9.32 GHz microwave frequency.
Results
Exposure of solutions of NH2OH (500 μM), H2O2 (100 μM) and GSH (100 μM) to hemin or a selection of heme proteins induced production of GS(O)NH2, GSSG, nitrite and nitrate in varying yields (Table 1). Product yields and composition were dependent upon the concentrations of NH2OH and GSH, and the reaction did not proceed in the absence of H2O2 (data not shown). We have previously shown that GS(O)NH2, formed from the association of HNO with GSH (Eq. 1), is a reproducible and selective marker for HNO [33].
Table 1.
Micromolar product formation from heme-mediated peroxidation of NH2OH in the presence of GSH.
| GS(O)NH2 | GSH | GSSG | NO2− | NO3− | |
|---|---|---|---|---|---|
| HRP | 20.8 ± 1.3 | 51 ± 2.2 | 4.5 ± 0.4 | ND | 3 ± 0.3 |
| metMb | 29.4 ± 1.47 | 23.6 ± 2.4 | 11.5 ± 2.3 | 6.8 ± 0.7 | 5.2 ± 1.3 |
| metHb | 14.6 ± 1.8 | 39.5 ± 6.4 | 6.9 ± 1.1 | 5.6 ± 0.6 | 3.5 ± 1.1 |
| LPO | 7.6 ± 0.5 | 72 ± 11.3 | 9.4 ± 0.9 | 0.5 ± 0.07 | 5.0 ± 0.5 |
| MPO | 43.4 ± 5 | ND | 7.0 ± 0.7 | ND | ND |
| catalase | 2.1 ± 1 | 48.6 ± 11 | 17.1 ± 2 | ND | 13.2 ± 2 |
| hemin | 7.3 ± 1.3 | 46.0 ± 1.6 | 11.0 ± 1.3 | 4.3 ± 0.2 | 6.0 ± 1.2 |
Heme protein or hemin (5 μM) was incubated in 10 mM phosphate buffer (pH 7.4) containing DTPA (50 μM), GSH (100 μM) and NH2OH (500 μM). The reaction was initiated by addition of H2O2 (100 μM), and was terminated following a 10 min incubation at 37°C by centrifugation through a 3-kDa cut-off filter. The deproteinized ultrafiltrate was analyzed by HPLC or spectrophotometrically. Each product concentration (μM) is reported as the mean ± SEM of at least three experiments with the exception of MPO. The balance of the products is assumed to be oxidized sulfur compounds (e.g., sulfenic acid), which are not detectable by the methods utilized. ND = not detectable.
| (1) |
Based on generation of GS(O)NH2, metMb, HRP, MPO, and to a lesser extent metHb, were most efficient at generating HNO in this system. HRP was used in subsequent studies since it was among the most efficient producers of HNO.
In the absence of NH2OH, the oxidation of GSH to GSSG was enhanced 5-fold (22 ± 0.3 μM) by addition of HRP compared to control, and formation of GS(O)NH2 was not observed. Conversely, oxidation of NH2OH by HRP in the absence of GSH resulted in three- and twofold increases in nitrite and nitrate, respectively. These results suggest that under the experimental conditions utilized, GSSG represents oxidation of GSH, GS(O)NH2 indicates the amount of free HNO that can be trapped by GSH, and nitrite/nitrate suggests autoxidation of either HNO or NO, which is most likely produced by reductive nitrosylation of heme by HNO (Eq. 2).
| (2) |
Interestingly, HRP, MPO and LPO produced high yields of GS(O)NH2 and nitrate (with the exception of MPO) relative to nitrite. The yields of GS(O)NH2 and nitrate where similar for LPO and hemin, but hemin also produced significant nitrite as did the globins. Catalase primarily produced GSSG and nitrate. Lastly, preliminary data indicated that cytochrome P450 produced little or no product, although the protein concentrations were low due to solubility issues of this membrane protein.
EPR and gas chromatographic analyses provided further evidence of formation of HNO from the reaction of NH2OH with HRP in the presence of H2O2. Reductive nitrosylation of the HRP heme by HNO (Eq. 2) was indicated by the characteristic three-line EPR pattern of an Fe(II)NO complex (Figure 1A). Gas chromatographic headspace analysis [16] confirmed a 13% yield of N2O, which forms upon dehydration of the HNO dimer (Eq. 3) [39, 40].
Figure 1. Generation of Fe(II)NO and NO/HNO by HRP-mediated peroxidation of NH2OH.
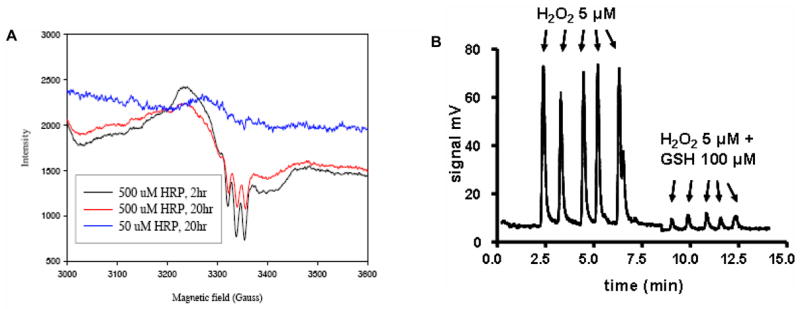
A) EPR spectra of HRP (50 or 500 μM) in the presence of NH2OH (100 mM) and H2O2 (100 mM). The spectrum shows the characteristic three line spectrum of a ferrous nitrosyl heme complex. B) Chemiluminescence signal intensity during peroxidation of NH2OH by HRP. H2O2 (final concentration 5 μM) was injected (arrows) into a sealed glass reaction chamber containing 10 mM phosphate buffer (pH 7.4, 2 mL) with DTPA (50 μM), NH2OH (500 μM) and HRP (5 μM) in either the absence or presence of GSH (100 μM) as indicated.
| (3) |
Production of free NO and HNO was detected by a chemiluminescence analyzer. Repeated additions of H2O2 (5 μM) to a solution of NH2OH (500 μM) and HRP (5 μM) contained in the analyzer reaction led to rapid but transient signal increases (Figure 1B). The consistent maximum signal intensity upon each injection of stoichiometric aliquots of H2O2 compared to HRP suggests that the measured product(s) is produced catalytically. Substantiation of a catalytic mechanism will require future detailed analysis of kinetic parameters.
The magnitude of the chemiluminescent signal decreased by 95% when GSH (100 μM) was present in the buffer. Since the signal intensity of repeated injections of aliquots of an NO solution (~100 mV) was insensitive to GSH (100 μM) (data not shown), these data suggest that the chemiluminescence signal could result entirely from HNO or perhaps arise from a combination of HNO and NO produced via reductive nitrosylation (Eq. 1). Alternatively, GSH could quench the signal by reacting with an oxidative intermediate of HRP. Formation of GS(O)NH2 as the primary product (Table 1) suggests that GSH reacts predominantly with HNO rather than HRP intermediates.
NOHA is an intermediate of NOS catalysis and a potential source of HNO [31]. In contrast to NH2OH (Table 1), addition of NOHA (500 μM) to solutions of HRP, metMb or metHb (5 μM) and H2O2 (100 μM) predominantly produced GSSG without observable GS(O)NH2 (Table 2), suggesting that free HNO is not produced by peroxidation of NOHA.
Table 2.
Micromolar product formation from heme-mediated peroxidation of NOHA in the presence of GSH.
| GS(O)NH2 | GSH | GSSG | NO2− | NO3− | |
|---|---|---|---|---|---|
| HRP | ND | 2 ± 3 | 43.3 ± 0.6 | 4.0 ± 1.0 | 2.3 ± 0.6 |
| metMb | ND | 42 ± 9.2 | 29 ± 8.5 | 1.6 ± 1.3 | 2 ±1.4 |
| metHb | ND | 79.5 ± 2.1 | 7.8 ± 0.4 | 0.4 ± 0.5 | 1 ± 0.7 |
Heme proteins (5 μM) were incubated and processed as described for in Table 1 with the substitution of NOHA (500 μM) for NH2OH (500 μM). Each product concentration (μM) is reported as the mean ± SEM of at least three experiments. The balance of the products is assumed to be oxidized sulfur compounds (e.g., sulfenic acid), which are not detectable by the methods utilized. ND = not detectable.
In previous studies, diffusion of HNO, releases upon decomposition of the donor Angeli’s salt, into human cells was observed with the fluorophore DAF as the intracellular reporter [36]. The species generated during decomposition of Angeli’s salt under aerobic conditions are distinguishable from NO or peroxynitrite (ONOO−) [41–43]. For instance, Angeli’s salt produced cardiac effects in vivo and in vitro that were different from those elicited by NO or ONOO− [1, 44, 45]. In addition, nitrosation of DAF by NO occurred on a much longer time scale [46] than triazole formation by HNO [36], due to the third order kinetics of NO autoxidation. To confirm that peroxidatively generated HNO could migrate from the extracellular medium into cells, MCF7 cells preloaded with DAF were exposed to HRP (5 μM), H2O2 (1–10 μM) and NH2OH (5–25 μM). The fluorescence intensity was enhanced dose-dependently by both NH2OH (Figure 2A) and H2O2 (Figure 2B). Exposure of DAF-loaded cells to only HRP, H2O2 or NH2OH or substitution of NOHA (100 μM) for NH2OH did not induce fluorescent signal within cells (data not shown). When cells were exposed to HRP, washed three times, then exposed to NH2OH and H2O2, the signal intensity was consistent with HRP-null controls. Therefore, the cells did not appreciably internalize HRP, and peroxidation primarily led to extracellular formation of a nitrogen oxide capable of oxidizing DAF upon migration into the cell.
Fig. 2. Fluorometric analysis of formation of HNO from peroxidation of NH2OH by HRP.
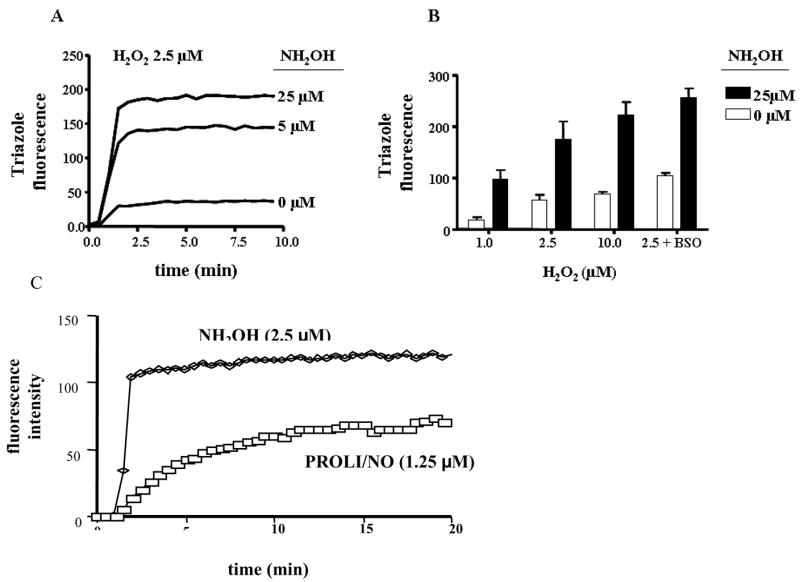
DAF (5 μM) was incorporated into MCF-7 cells during a 15 min incubation period at 37°C. Following centrifugation and wash steps to remove unincorporated DAF, signal intensity was monitored in a suspension of 106 cells/mL in 10 mM phosphate buffer (pH 7.4) containing DTPA (50 μM), HRP (5 μM) and H2O2 (2.5 μM) at 37°C. Intracellular triazole fluorescence was detected at 515 nm following 488 nm excitation. A) Dose response to NH2OH; B) intensity after 5 min following addition of H2O2 as indicated in the absence or presence of NH2OH. The last panel shows the effect of depletion of GSH by overnight incubation with BSO (5 mM). Data are representative A) or the mean B) of at least four individual experiments. C) Comparison of intracellular fluorescence intensity during peroxidation of NH2OH or decomposition of PROLI/NO (1.25 μM) in phosphate buffer alone.
To distinguish between NO and HNO, a comparison was performed between PROLI/NO, an NO donor with a two second half-life [47], and HRP peroxidation of NH2OH. Since PROLI/NO releases two equivalents of NO, 1.25 μM PROLI/NO was utilized assuming 100% efficiency of H2O2 (2.5 μM) consumption by HRP. The slower rate and lower final intensity of the triazole signal from PROLI/NO compared to the peroxidative conditions (Figure 2C), suggests that NO is not primarily responsible for the increase in intracellular fluorescence as a consequence of peroxidation of NH2OH. Furthermore, the traces are kinetically distinct, with the NO donor inducing an apparently first order increase and the HRP system presumably following saturable Michaelis-Menten kinetics that suggests enzymatic generation.
Discussion
Heme-containing proteins are crucial to both the basal functions of cellular metabolism and the responses to stress or disease conditions. When activated by H2O2, hemes peroxidatively modify a wide variety of substrates. Of importance here is that reduced nitrogen compounds, such as the hydroxylamine-derivative hydroxyurea, can be peroxidatively converted to HNO [48]. Such pharmacological production of HNO raises the issue of whether endogenous substrates can similarly function as biosynthetic precursors to HNO [49–51].
NH2OH and NOHA are both produced by NOS catalysis [20, 52, 53]. In the presence of excess thiol, NH2OH can also be formed from S-nitrosothiols (RSNO) under nitrosative stress [27] through the intermediacy of an HNO/RSH adduct (Eqs. 4–6).
| (4) |
| (5) |
| (6) |
Here, production of HNO from peroxidation of NH2OH or NOHA was investigated by analysis of GS(O)NH2 formation, by detection of chemiluminescence signal and by intracellular reactivity with DAF. Using this approach, the present data (Table 1) demonstrate that hemin and a variety of heme proteins are able to induce the oxidation of NH2OH to HNO, most likely via the intermediacy of high valent species (Eqs. 7 and 8).
| (7) |
| (8) |
Under the experimental conditions utilized, GS(O)NH2 indicates the amount of free HNO that can be trapped by GSH, GSSG represents oxidation of GSH, most likely by high-valent heme centers (Eq. 9), and nitrite/nitrate represent the fraction of HNO that recombines with heme to ultimately form a nitrosyl complex (Eq. 2) (Scheme 1).
Scheme 1.
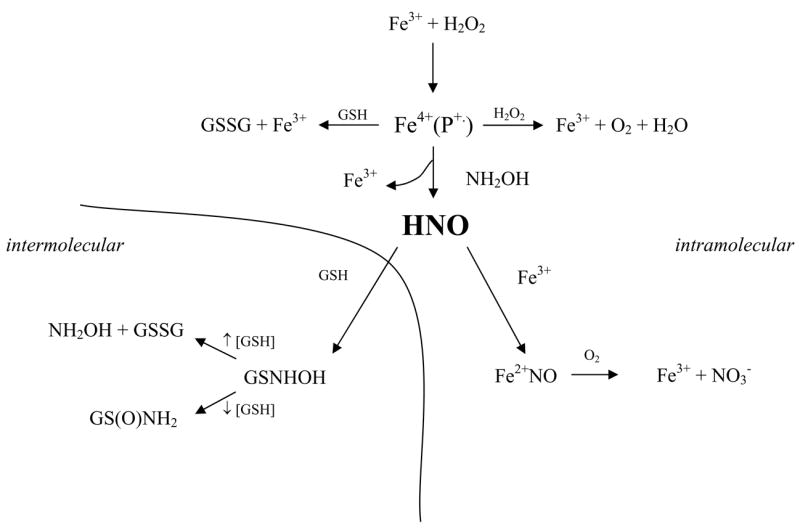
Competitive pathways in the peroxidation of NH2OH to HNO.
| (9) |
The yield of GSSG can also be influenced by the ratio of HNO to GSH (Eqs. 5 and 6) [33, 35], but experimental conditions (100 μM GSH) were chosen to minimize this pathway. That addition of NOHA did not result in formation of GS(O)NH2, nitrite or nitrate (Table 2) suggests that NOHA is either not competitive with GSH (Eq. 9) for the ferryl-π cation intermediate (Eq. 7) or that the peroxidases utilized here were incapable of using NOHA as a substrate.
The highest yields of GS(O)NH2 were observed with heme proteins that contain a proximal histidine (HRP, MPO, metMb and metHb, and to a lesser extent LPO; Table 1). In contrast, GS(O)NH2 was not generated by proteins in which the heme was coordinated proximally by either a tyrosine (catalase; Table 1) or a cysteine (P450; preliminary data). The product yield from P540 may be a function of solubility issues, but catalase is known to oxidize NH2OH to NO [54], rather than HNO. The relatively high yield of GSSG suggests either that GSH is a suitable substrate for catalase (Eq. 9) or that production of NO leads to formation of an intermediate capable of oxidizing NO. Interestingly, catalase oxidizes both cyanamide [16] and hydroxyurea [48] to HNO. Further studies examining the effects of heme axial ligation on HNO formation are required.
The yields of GS(O)NH2, signifying the presence of free HNO, and the varied ratios to other products indicate the existence of competing pathways (Scheme 1). The ratio of GS(O)NH2 to nitrate can be envisioned to indicate the probability that HNO escapes the pocket to associate with GSH (Eqs. 1, 5) rather than reductively nitrosylates the ferric heme to produce a ferrous nitrosyl complex (Eq. 2). Autoxidation could then subsequently convert bound NO to nitrate (Eq. 10)
| (10) |
These competing pathways appear to be kinetically similar for hemin and LPO, while the other proteins with the exception of catalase are biased toward diffusion and reaction of HNO with distal targets such as GSH. These results demonstrate that although heme proteins are capable of peroxidizing NH2OH to HNO, the ultimate fate of HNO is protein dependent.
The ratio of GS(O)NH2 to GSSG suggests that the kinetic relevance of GSH as a substrate (Eq. 9) compared to NH2OH (Eq. 8) is low for HRP and MPO and intermediate for meMb and metHb. Similarly, the prevalence for a futile cycle (Eq. 11) is indicated by the ratio of GS(O)NH2 to GSH.
| (11) |
In summary, MPO utilizes NH2OH and releases HNO most efficiently. HRP should also be considered as an effective generator of HNO, but with perhaps a higher propensity for futile cycles. LPO was utilized the substrates inefficiently and nonspecifically and had a similar propensity for release and binding of HNO. The globins and hemin appeared to function similarly except in the production of free HNO as depicted by the relative amounts of GS(O)NH2 and GSH.
In contrast to the other proteins, the globins and hemin also produced significant nitrite. There are many possible sources of nitrite. Likely possibilities include the reaction of HNO with NO [55] or a more significant contribution of compound II. The unfavorability of dissociation of free NO from the globin nitrosyls (Eq. 2) renders production of nitrite from autoxidation of NO unlikely. The mechanism and molecular basis for nitrite formation requires further study.
Rather than reacting with the ferric heme, HNO efficiently escapes from the site of initial oxidation in the heme pockets of MPO, HRP and metMb. Four factors would be predictive of formation of free HNO: the redox chemistry of HNO, the off-rate of heme associated ligands such as water, the acid-base chemistry of HNO, and the accessibility of the heme to the bulk solution. Oxidation of HNO can occur via either inner-sphere or outer-sphere electron transfer. The rate constant for inner sphere-electron transfer from HNO to heme proteins (~106 M− 1 s− 1 [11]) is two orders of magnitude higher than that of outer-sphere electron transfer to common oxidants such as Tempol, ferricytochrome c and ferricyanide [11, 56]. For the proteins studied, inner sphere-electron transfer would be expected to rapidly produce nitrosyl complexes (Eq. 2). However, two-electron transfer from NH2OH to compound I both produces HNO and regenerates the ferric heme with an associated water (Eq. 7). Ford and Lorkovic [57] have suggested that the rate of addition of NO to ferric hemes is determined by the dissociative rate of water or similar ligands to provide an open coordination site. A dissociative step is likely to be rate limiting for HNO as well.
Free NO could conceivably be produced upon deprotonation of HNO to NO− followed by outer-sphere electron transfer with the heme. However, due to differing ground states (singlet for HNO and triplet for NO−), acid-conjugate base conversion is extraordinarily slow [13] and likely not kinetically significant.
The heme pocket structure may be a major determinant in formation of biologically active HNO from peroxidases. Accessibility of the heme to extra-molecular targets or an open structure that facilitates migration of HNO out of the active site pocket would enhance the kinetic feasibility of diffusion compared to association with the heme. Ultimately, the ratio of HNO to NO produced during peroxidation of NH2OH is likely determined by both heme accessibility and the rate of dissociation of heme ligands.
In this study, proteins with proximal histidine ligands favored formation and migration of HNO from the heme site while catalase (Table 1) and P450 (preliminary data not shown), with tyrosine and cysteine proximal ligands, respectively, did not generate significant free HNO. Since hemin alone produced similar levels of GS(O)NH2 and nitrite/nitrate, the proximal ligand cannot be the sole factor in either production or release of HNO. Rather, protein structure may affect the efficiency of the peroxidation reaction or the kinetics of competing pathways for HNO. The lack of detectable production of GS(O)NH2 by P450 may also simply be a function of restricted diffusion of the substrate through the hydrophobic heme-binding pocket. Alternatively, NH2OH may bind strongly to the P450 heme, and thus restrict heme reactivity with peroxide. Furthermore, differences in product yields and ratios may result from varied lifetimes of oxidative intermediates as a function of protein structure. The current results provide the initial indication of the ability of heme proteins to biosynthesize HNO from reduced substrates; detailed mechanistic and kinetic analyses as a function of protein structure will be the subject of future studies.
It is likely that in the case of physiological production, the sensitivity to HNO produced peroxidatively would be higher than that of available chemical measurements. Prior analyses demonstrated that release of HNO from the donor Angeli’s salt resulted in conversion of intracellular DAF to the fluorescent triazole [36]. The rapid increase in intracellular fluorescence intensity upon incubation with extracellular HRP, NH2OH and H2O2 (Figure 2) corroborated the HPLC (Table 1) and chemiluminescence data (Figure 1) indicating formation of free HNO. The cellular assay also demonstrated that HNO was capable of diffusing from the extracellular medium into cells, even in the presence of high GSH concentrations (e.g., a 3000-fold ratio of extracellular GSH to NH2OH was required to quench HRP-mediated intracellular triazole signal). These findings are consistent with prior studies demonstrating that cells efficiently sequester HNO [36]. Furthermore, that the cellular fluorescence signal was augmented by 40% upon a twofold decrease in cellular GSH content (Figure 2B) suggests that while GSH is an important target for HNO, cellular compartmentalization may enhance the life-time of HNO sufficiently to facilitate cellular signaling [13, 36]. Certainly, HNO donors have been shown to be effective pharmacology agents both in vitro and in vivo for a variety of conditions ranging from treatment of congestive heart failure to alcoholism (e.g. [1–9, 58–65]), despite high levels of cytoplasmic GSH.
That intracellular fluorescence was enhanced beyond control intensities with HRP and H2O2 (Figure 2A) suggests the presence of endogenous nitrogen substrates. However, production of HNO required addition of an exogenous peroxidase system, and internalization of HRP was not observed. Therefore, it is expected that such nitrogen endogenous substrates would have to diffuse from the cell prior to peroxidation. Identification of such species requires further investigation.
In summary, heme proteins and hemin have been shown to peroxidatively convert NH2OH into HNO, which can escape the protein pocket to interact with other targets. End product ratio analysis suggests that protein structure influences the relative rate of release of HNO, potentially for cellular signaling, compared to consumption of HNO by recombination with the heme followed by oxidative degradation. Interestingly, proteins such as hemoglobin and myoglobin, which are not traditionally labeled as peroxidases, may be able to generate HNO under pathophysiological conditions, particularly involving significant hemolysis. Under the experimental conditions utilized, NOHA was not a suitable substrate for peroxidative production of HNO. Nonetheless, the results may be physiologically relevant given that NH2OH is endogenously available and can induce significant cardiovascular effects, including vasodilation [66, 67] and protection of cardiomyocytes against oxidative stress [68].
Acknowledgments
The authors would like to acknowledge the technical assistance of D. Janie Salmon and Derek Hollman (UA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc Natl Acad Sci USA. 2001;98:10463–10468. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paolocci N, Katori T, Champion HC, StJohn ME, Miranda KM, Fukuto JM, Wink DA, Kass DA. Positive inotropic and lusitropic effects of HNO/NO− in failing hearts: independence from b-adrenergic signaling. Proc Natl Acad Sci USA. 2003;100:5537–5542. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolocci N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic Biol Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 4.Ma XL, Gao F, Liu GL, Lopez BL, Christopher TA, Fukuto JM, Wink DA, Feelisch M. Opposite effects of nitric oxide and nitroxyl on postischemic myocardial injury. Proc Natl Acad Sci USA. 1999;96:14617–14622. doi: 10.1073/pnas.96.25.14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irvine JC, Favaloro JL, Kemp-Harper BK. NO− activates soluble guanylate cyclase and K+ channels to vasodilate resistance arteries. Hypertension. 2003;41:1301–1307. doi: 10.1161/01.HYP.0000072010.54901.DE. [DOI] [PubMed] [Google Scholar]
- 6.Cheong E, Tumbev V, Abramson J, Salama G, Stoyanovsky DA. Nitroxyl triggers Ca2+ release from skeletal and cardiac sarcoplasmic reticulum by oxidizing ryanodine receptors. Cell Calcium. 2005;37:87–96. doi: 10.1016/j.ceca.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Kim WK, Choi YB, Rayudu PV, Das P, Asaad W, Arnelle DR, Stamler JS, Lipton SA. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO−. Neuron. 1999;24:461–469. doi: 10.1016/s0896-6273(00)80859-4. [DOI] [PubMed] [Google Scholar]
- 8.Colton CA, Gbadegesin M, Wink DA, Miranda KM, Espey MG, Vicini S. Nitroxyl anion regulation of the NMDA receptor. J Neurochem. 2001;78:1126–1134. doi: 10.1046/j.1471-4159.2001.00509.x. [DOI] [PubMed] [Google Scholar]
- 9.Hewett SJ, Espey MG, Uliasz TF, Wink DA. Neurotoxicity of nitroxyl: insights into HNO and NO biochemical imbalance. Free Radic Biol Med. 2005;39:1478–1488. doi: 10.1016/j.freeradbiomed.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Tocchetti CG, Wang W, Froehlich JP, Huke S, Aon MA, Wilson GM, Di Benedetto G, O’Rourke B, Gao WD, Wink DA, Toscano JP, Zaccolo M, Bers DM, Valdivia HH, Cheng H, Kass DA, Paolocci N. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res. 2007;100:96–104. doi: 10.1161/01.RES.0000253904.53601.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc Natl Acad Sci USA. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wink DA, Miranda KM, Katori T, Mancardi D, Thomas DD, Ridnour LA, Espey MG, Feelisch M, Colton CA, Fukuto JM, Pagliaro P, Kass DA, Paolocci N. Orthogonal properties of the redox siblings nitroxyl and nitric oxide in the cardiovascular system: a novel redox paradigm. Am J Physiol-Heart Circul Physiol. 2003;285:H2264–H2276. doi: 10.1152/ajpheart.00531.2003. [DOI] [PubMed] [Google Scholar]
- 13.Miranda KM. The chemistry of nitroxyl (HNO) and implications in biology. Coordin Chem Rev. 2005;249:433–455. [Google Scholar]
- 14.Feelisch M. Nitroxyl gets to the heart of the matter. Proc Natl Acad Sci USA. 2003;100:4978–4980. doi: 10.1073/pnas.1031571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson JKW. A new drug for alcoholism treatment. Can Med Assoc J. 1956;74:793–795. [PMC free article] [PubMed] [Google Scholar]
- 16.Nagasawa HT, DeMaster EG, Redfern B, Shirota FN, Goon DJ. Evidence for nitroxyl in the catalase-mediated bioactivation of the alcohol deterrent agent cyanamide. J Med Chem. 1990;33:3120–3122. doi: 10.1021/jm00174a001. [DOI] [PubMed] [Google Scholar]
- 17.Kim-Shapiro DB, King SB, Bonifant CL, Kolibash CP, Ballas SK. Time resolved absorption study of the reaction of hydroxyurea with sickle cell hemoglobin. Biochim Biophys Acta. 1998;1380:64–74. doi: 10.1016/s0304-4165(97)00132-3. [DOI] [PubMed] [Google Scholar]
- 18.King SB. The nitric oxide producing reactions of hydroxyurea. Curr Med Chem. 2003;10:437–452. doi: 10.2174/0929867033368213. [DOI] [PubMed] [Google Scholar]
- 19.Pufahl RA, Wishnok JS, Marletta MA. Hydrogen peroxide-supported oxidation of N-G-hydroxy-L-arginine by nitric oxide synthase. Biochemistry. 1995;34:1930–1941. doi: 10.1021/bi00006a014. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt HHHW, Hofmann H, Schindler U, Shutenko ZS, Cunningham DD, Feelisch M. No NO from NO synthase. Proc Natl Acad Sci USA. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clague MJ, Wishnok JS, Marletta MA. Formation of N-delta-cyanoornithine from NG-hydroxy-L-arginine and hydrogen peroxide by neuronal nitric oxide synthase: implications for mechanism. Biochemistry. 1997;36:14465–14473. doi: 10.1021/bi971024u. [DOI] [PubMed] [Google Scholar]
- 22.Rusche KM, Spiering MM, Marletta MA. Reactions catalyzed by tetrahydrobiopterin-free nitric oxide synthase. Biochemistry. 1998;37:15503–15512. doi: 10.1021/bi9813936. [DOI] [PubMed] [Google Scholar]
- 23.Adak S, Wang Q, Stuehr DJ. Arginine conversion to nitroxide by tetrahydrobiopterin-free neuronal nitric-oxide synthase - implications for mechanism. J Biol Chem. 2000;275:33554–33561. doi: 10.1074/jbc.M004337200. [DOI] [PubMed] [Google Scholar]
- 24.Hobbs AJ, Fukuto JM, Ignarro LJ. Formation of free nitric oxide from L-arginine by nitric oxide synthase - direct enhancement of generation by superoxide dismutase. Proc Natl Acad Sci USA. 1994;91:10992–10996. doi: 10.1073/pnas.91.23.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oae S, Kim YH, Fukushima D, Shinhama K. New syntheses of thionitrites and their chemical reactivities. J Chem Soc Perkin Trans. 1978;1:913–917. [Google Scholar]
- 26.Schulz U, McCalla DR. Reactions of cysteine with N-methyl-N-nitroso-P-toluenesulfonamide and N-methyl-N′-nitro-N-nitrosoguanidine. Can J Chem. 1969;47:2021–2027. [Google Scholar]
- 27.Arnelle DR, Stamler JS. NO+, NO, and NO− donation by S-nitrosothiols - implications for regulation of physiological functions by S- nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 28.Hogg N, Singh RJ, Kalyanaraman B. The role of glutathione in the transport and catabolism of nitric oxide. FEBS Lett. 1996;382:223–228. doi: 10.1016/0014-5793(96)00086-5. [DOI] [PubMed] [Google Scholar]
- 29.Reeder BJ, Wilson MT. Hemoglobin and myoglobin associated oxidative stress: from molecular mechanisms to disease states. Curr Med Chem. 2005;12:2741–2751. doi: 10.2174/092986705774463021. [DOI] [PubMed] [Google Scholar]
- 30.Osawa Y, Williams MS. Covalent crosslinking of the heme prosthetic group to myoglobin by H2O2: toxicological implications. Free Radic Biol Med. 1996;21:35–41. doi: 10.1016/0891-5849(95)02215-5. [DOI] [PubMed] [Google Scholar]
- 31.Buga GM, Singh R, Pervin S, Rogers NE, Schmitz DA, Jenkinson CP, Cederbaum SD, Ignarro LJ. Arginase activity in endothelial cells: inhibition by NG-hydroxy-L-arginine during high-output NO production. Am J Physiol. 1996;271:H1988–H1998. doi: 10.1152/ajpheart.1996.271.5.H1988. [DOI] [PubMed] [Google Scholar]
- 32.Gwilt PR, Tracewell WG. Pharmacokinetics and pharmacodynamics of hydroxyurea. Clin Pharmacokinet. 1998;34:347–358. doi: 10.2165/00003088-199834050-00002. [DOI] [PubMed] [Google Scholar]
- 33.Donzelli S, Espey MG, Thomas DD, Mancardi D, Tocchetti CG, Ridnour LA, Paolocci N, King SB, Miranda KM, Lazzarino G, Fukuto JM, Wink DA. Discriminating HNO formation from that of other reactive nitrogen oxide species. Free Radic Biol Med. 2006;40:1056–1066. doi: 10.1016/j.freeradbiomed.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 34.Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprusside infusion. Circulation. 1994;89:2070–2207. 2078. doi: 10.1161/01.cir.89.5.2070. [DOI] [PubMed] [Google Scholar]
- 35.Wong PSY, Hyun J, Fukuto JM, Shirota FN, DeMaster EG, Shoeman DW, Nagasawa HT. Reaction between S-nitrosothiols and thiols: generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 36.Espey MG, Miranda KM, Thomas DD, Wink DA. Ingress and reactive chemistry of nitroxyl-derived species within human cells. Free Radic Biol Med. 2002;33:827–834. doi: 10.1016/s0891-5849(02)00978-4. [DOI] [PubMed] [Google Scholar]
- 37.Archer S. Measurement of nitric oxide in biological models. FASEB J. 1993;7:349–360. doi: 10.1096/fasebj.7.2.8440411. [DOI] [PubMed] [Google Scholar]
- 38.Miranda KM, Katori T, Torres de Holding CL, Thomas L, Ridnour LA, McLendon WJ, Dutton AS, Champion HC, Mancardi D, Tocchetti CG, Saavedra JE, Keefer LK, Houk KN, Fukuto JM, Kass DA, Paolocci N, Wink DA. Comparison of the NO and HNO donating properties of diazeniumdiolates: primary amine adducts release HNO in vivo. J Med Chem. 2005;48:8220–8228. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- 39.Smith PAS, Hein GE. The alleged role of nitroxyl in certain reactions of aldehydes and alkyl halides. J Am Chem Soc. 1960;82:5731–5740. [Google Scholar]
- 40.Kohout FC, Lampe FW. On the role of the nitroxyl molecule in the reaction of hydrogen atoms with nitric oxide. J Am Chem Soc. 1965;87:5795–5796. [Google Scholar]
- 41.Miranda KM, Espey MG, Yamada K, Krishna M, Ludwick N, Kim S, Jourd’heuil D, Grisham MB, Feelisch M, Fukuto JM, Wink DA. Unique oxidative mechanisms for the reactive nitrogen oxide species, nitroxyl anion. J Biol Chem. 2001;276:1720–1727. doi: 10.1074/jbc.M006174200. [DOI] [PubMed] [Google Scholar]
- 42.Miranda KM, Yamada K, Espey MG, Thomas DD, DeGraff W, Mitchell JB, Krishna MC, Colton CA, Wink DA. Further evidence for distinct reactive intermediates from nitroxyl and peroxynitrite: effects of buffer composition on the chemistry of Angeli’s salt and synthetic peroxynitrite. Arch Biochem Biophys. 2002;401:134–144. doi: 10.1016/S0003-9861(02)00031-0. [DOI] [PubMed] [Google Scholar]
- 43.Miranda KM, Dutton AS, Ridnour LA, Foreman CA, Ford E, Paolocci N, Katori T, Tocchetti CG, Mancardi D, Thomas DD, Espey MG, Houk KN, Fukuto JM, Wink DA. Mechanism of aerobic decomposition of Angeli’s salt (sodium trioxodinitrate) at physiological pH. J Am Chem Soc. 2005;127:722–731. doi: 10.1021/ja045480z. [DOI] [PubMed] [Google Scholar]
- 44.Katori T, Hoover DB, Ardell JL, Helm RH, Belardi DF, Tocchetti CG, Forfia PR, Kass DA, Paolocci N. Calcitonin gene-related peptide in vivo positive inotropy is attributable to regional sympatho-stimulation and is blunted in congestive heart failure. Circ Res. 2005;96:234–243. doi: 10.1161/01.RES.0000152969.42117.ca. [DOI] [PubMed] [Google Scholar]
- 45.Katori T, Donzelli S, Tocchetti CG, Miranda KM, Cormaci G, Thomas DD, Ketner EA, Lee MJ, Mancardi D, Wink DA, Kass DA, Paolocci N. Peroxynitrite and myocardial contractility: in vivo versus in vitro effects. Free Radic Biol Med. 2006;41:1606–1618. doi: 10.1016/j.freeradbiomed.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 46.Espey MG, Miranda KM, Thomas DD, Wink DA. Distinction between nitrosating mechanisms within human cells and aqueous solution. J Biol Chem. 2001;276:30085–30091. doi: 10.1074/jbc.M101723200. [DOI] [PubMed] [Google Scholar]
- 47.Saavedra JE, Southan GJ, Davies KM, Lundell A, Markou C, Hanson SR, Adrie C, Hurford WE, Zapol WM, Keefer LK. Localizing antithrombotic and vasodilatory activity with a novel, ultrafast nitric oxide donor. J Med Chem. 1996;39:4361–4365. doi: 10.1021/jm960616s. [DOI] [PubMed] [Google Scholar]
- 48.King SB. N-hydroxyurea and acyl nitroso compounds as nitroxyl (HNO) and nitric oxide (NO) donors. Curr Top Med Chem. 2005;5:665–673. doi: 10.2174/1568026054679362. [DOI] [PubMed] [Google Scholar]
- 49.Fukuto JM, Wallace GC, Hszieh R, Chaudhuri G. Chemical oxidation of N-hydroxyguanidine compounds: release of nitric oxide, nitroxyl and possible relationship to the mechanism of biological nitric oxide generation. Biochem Pharmacol. 1992;43:607–613. doi: 10.1016/0006-2952(92)90584-6. [DOI] [PubMed] [Google Scholar]
- 50.Fukuto JM, Stuehr DJ, Feldman PL, Bova MP, Wong P. Peracid oxidation of an N-hydroxyguanidine compound - a chemical model for the oxidation of N-omega-hydroxy-L-arginine by nitric oxide synthase. J Med Chem. 1993;36:2666–2670. doi: 10.1021/jm00070a010. [DOI] [PubMed] [Google Scholar]
- 51.Fukuto JM, Switzer CH, Miranda KM, Wink DA. Nitroxyl (HNO): chemistry, biochemistry, and pharmacology. Annu Rev Pharmacol Toxicol. 2005;45:335–355. doi: 10.1146/annurev.pharmtox.45.120403.095959. [DOI] [PubMed] [Google Scholar]
- 52.Stuehr DJ, Kwon NS, Nathan CF, Griffith OW, Feldman PL, Wiseman J. N-omega-hydroxy-L-arginine is an intermediate in the biosynthesis of nitric oxide from L-arginine. J Biol Chem. 1991;266:6259–6263. [PubMed] [Google Scholar]
- 53.Wallace GC, Gulati P, Fukuto JM. N-omega-Hydroxy-L-arginine: a novel arginine analog capable of causing vasorelaxation in bovine intrapulmonary artery. Biochem Biophys Res Commun. 1991;176:528–534. doi: 10.1016/0006-291x(91)90957-9. [DOI] [PubMed] [Google Scholar]
- 54.Katsuki S, Arnold W, Mittal CK, Murad F. Stimulation of guanylate cyclase by sodium nitroprusside, nitroglycerin and nitric oxide in various tissue preparations and comparison to the effects of sodium azide and hydroxylamine. J Cyclic Nucleotide Res. 1977;3:23–35. [PubMed] [Google Scholar]
- 55.Shafirovich V, Lymar SV. Nitroxyl and its anion in aqueous solutions: spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc Natl Acad Sci USA. 2002;99:7340–7345. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liochev SI, Fridovich I. The mode of decomposition of Angeli’s salt (Na2N2O3) and the effects thereon of oxygen, nitrite, superoxide dismutase, and glutathione. Free Radic Biol Med. 2003;34:1399–1404. doi: 10.1016/s0891-5849(03)00111-4. [DOI] [PubMed] [Google Scholar]
- 57.Ford PC, Lorkovic IM. Mechanistic aspects of the reactions of nitric oxide with transition-metal complexes. Chem Rev. 2002;102:993–1018. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- 58.DeMaster EG, Shirota FN, Nagasawa HT. The metabolic activation of cyanamide to an inhibitor of aldehyde dehydrogenase is catalyzed by catalase. Biochem Biophys Res Commun. 1984;122:358–365. doi: 10.1016/0006-291x(84)90483-2. [DOI] [PubMed] [Google Scholar]
- 59.Fukuto JM, Chiang K, Hszieh R, Wong P, Chaudhuri G. The pharmacological activity of nitroxyl: a potent vasodilator with activity similar to nitric oxide and/or endothelium-derived relaxing factor. J Pharmacol Exp Ther. 1992;263:546–551. [PubMed] [Google Scholar]
- 60.De Witt BJ, Marrone JR, Kaye AD, Keefer LK, Kadowitz PJ. Comparison of responses to novel nitric oxide donors in the feline pulmonary vascular bed. Eur J Pharmacol. 2001;430:311–315. doi: 10.1016/s0014-2999(01)01289-4. [DOI] [PubMed] [Google Scholar]
- 61.Cook NM, Shinyashiki M, Jackson MI, Leal FA, Fukuto JM. Nitroxyl-mediated disruption of thiol proteins: inhibition of the yeast transcription factor Ace1. Arch Biochem Biophys. 2003;410:89–95. doi: 10.1016/s0003-9861(02)00656-2. [DOI] [PubMed] [Google Scholar]
- 62.Aniruddha S, Vidwans TFU, Hewett JA, Hewett SJ. Differential modulation of prostaglandin H synthase-2 by nitric oxide-related species in intact cells. Biochemistry. 2001;40:11533–11542. doi: 10.1021/bi0108960. [DOI] [PubMed] [Google Scholar]
- 63.Naughton P, Foresti R, Bains SK, Hoque M, Green CJ, Motterlini R. Induction of heme oxygenase 1 by nitrosative stress. J Biol Chem. 2002;277:40666–40674. doi: 10.1074/jbc.M203863200. [DOI] [PubMed] [Google Scholar]
- 64.Costa G, Labadia A, Triguero D, Jimenez E, Garcia-Pascual A. Nitrergic relaxation in urethral smooth muscle: involvement of potassium channels and alternative redox forms of NO. Naunyn-Schmied Arch Pharmacol. 2001;364:516–523. doi: 10.1007/s002100100480. [DOI] [PubMed] [Google Scholar]
- 65.DeMaster EG, Redfern B, Nagasawa HT. Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 66.Huang Y. Hydroxylamine-induced relaxation inhibited by K+ channel blockers in rat aortic rings. Eur J Biochem. 1998;349:53–60. doi: 10.1016/s0014-2999(98)00178-2. [DOI] [PubMed] [Google Scholar]
- 67.Booth BP, Tabrizi-Fard MA, Fung HL. Calcitonin gene-related peptide-dependent vascular relaxation of rat aorta - an additional mechanism for nitroglycerin. Biochem Pharmacol. 2000;59:1603–1609. doi: 10.1016/s0006-2952(00)00290-2. [DOI] [PubMed] [Google Scholar]
- 68.Zhang R, Pinson A, Samuni A. Both hydroxylamine and nitroxide protect cardiomyocytes from oxidative stress. Free Radic Biol Med. 1998;24:66–75. doi: 10.1016/s0891-5849(97)00165-2. [DOI] [PubMed] [Google Scholar]