

Abstract
Background
Genetic susceptibility in lung cancer risk has long been recognized but remains ill defined, as does the role of tobacco smoke exposure and chronic obstructive pulmonary disease (COPD).
Methods
Using a dual case-control design, we tested whether alpha1-antitrypsin deficiency (α1ATD) carriers are predisposed to a higher risk of lung cancer, adjusting for the effects of tobacco smoke exposure and COPD. A total of 1856 patients with incident lung cancer were included in the study; 1585 community residents served as controls. A second control group was composed of 902 full siblings of the patients. We first modeled 1585 case-control pairs without the α1ATD variable using multiple logistic regression analysis and then modeled the α1 ATD allele type in the presence of other known risk factors of lung cancer.
Results
We found a significantly increased lung cancer risk among α1ATD carriers from 2 parallel case-control comparisons: when patients were compared with unrelated controls, α1ATD carriers had a 70% higher risk of developing lung cancer than noncarriers (odds ratio, 1.7; 95% confidence interval, 1.2–2.4). In a further comparison of patients with their cancer-free siblings, we found a 2-fold increased lung cancer risk in α1ATD carriers (95% confidence interval, 1.4–2.7). Stratified analysis by tumor histologic subtypes showed a significant increase for adenocarcinoma and squamous cell carcinoma among α1ATD carriers.
Conclusion
Our results suggest that α1ATD carriers are at a 70% to 100% increased risk of lung cancer and may account for 11% to 12% of the patients with lung cancer in our study.
A history of chronic obstructive pulmonary disease (COPD) has been reported to increase lung cancer risk,1 and COPD aggregates with lung cancer within the same families. Chronic obstructive pulmonary disease is characterized by airway obstruction, which is not fully reversible and usually results from emphysema and/or chronic bronchitis.2,3 Since 1977, numerous studies have shown COPD to be an independent risk factor for lung cancer.1,4–8 However, it remains unclear whether emphysema, chronic bronchitis, or both predispose an individual to an elevated lung cancer risk at the same magnitude. In addition, the biologic mechanism linking COPD and lung cancer development is largely unknown.
Alpha1-antitrypsin deficiency (α1ATD) is one of the most common genetic disorders affecting the US population, especially European descendents, and can lead to early age onset of emphysema among homozygous individuals.9 The level of alpha1-antitrypsin (α1AT) is inherited as a Mendelian codominant trait that is determined by the α1AT gene (or Pi1 locus) on chromosome 14q32.1.10 The Pi1 locus is highly polymorphic, with more than 70 variants reported.10 Each variant is designated by a letter corresponding to the migration of the α1AT protein in an isoelectric focusing assay, the standard clinical diagnostic test for more than 20 years.11 The most common wild type is M (including subtypes M1, M2, and M3), with most normal individuals being homozygous for this allele (designated as MM or PiMM with the subtypes, eg, M1M1). Of the variants that lead to a deficiency of α1 AT, only Z and S alleles are common. Uncommon deficient alleles include I, MMalton, MPittsburgh, null, and other rare alleles.10 Alpha-1 antitrypsin deficiency carriers (ie, heterozygous individuals) do not normally have severe α1ATD-related diseases, and most of them are not aware of their carrier status. These carriers may be more vulnerable to carcinogen-containing tobacco smoke than noncarriers, especially when their α1AT levels are compromised under physiologic stress or they have subclinical lung tissue damage.9 The goals of this study were to evaluate 3 risk factors (α1ATD, COPD, and cigarette smoking) together and their interplay in lung cancer development and to estimate their relative contributions to lung cancer risk in the general population.
METHODS
STUDY SUBJECTS
Patients With Lung Cancer
All study subjects were enrolled using our rapid patient identification and enrollment procedure approved by the Mayo Clinic institutional review board.12,13 Briefly, newly diagnosed cases of lung cancer are identified by a daily electronic pathology reporting system. Once identified, study consents are obtained from the patients for enrollment, their medical records are abstracted, and interviews are conducted. The overall participation and blood sample donation rates were 87% and 73%, respectively.
Unrelated Controls
We selected community residents who were identified by having had a general medical examination and a leftover blood sample from routine clinical tests.14,15 Excluded were individuals who had been diagnosed with any cancer (except nonmelanoma skin cancer) or major organ failure (eg, heart, brain, lung, kidney, or liver) on or prior to this visit. An invitation letter was mailed out to potential study participants who were found to be the best potential controls matched to patients on age, sex, and race/ethnicity. If a subject declined to participate, the same procedure was repeated for the next best potential control. A self-administered questionnaire with the same questions as obtained from patients with lung cancer was completed by the controls. Two to 6 months were required to obtain a matched control for each enrolled patient. Of the respondents, 78% gave permission to use their blood sample, of whom 84% completed and returned the questionnaire; 11% declined to participate; and 1% were deceased or could not be located. Ninety-five percent of these study subjects were white, representing a US Midwestern population in and surrounding Minnesota.
Full Sibling Controls
We recruited full siblings through the index cases of lung cancer probands. All full siblings who were free of cancer, as reported by the probands as well as the siblings themselves, and who donated a blood sample were included in the study. Sibling participation rates could not be accurately estimated because family members were invited into the study through the probands, based on our approved protocol. Even though all living full siblings were eligible to participate, probands decided which family members to contact; no disclosure was provided to the study.
DATA COLLECTION
Complete family history information regarding cancer (including lung cancer and other cancer sites), other lung disease history (including emphysema, chronic bronchitis, COPD, tuberculosis, asthma, allergy, asbestosis, silicosis, and other), and cigarette smoking history for each relative was obtained from all study subjects via a combination of a structured subject interview, self-administered questionnaire, and medical records.13–16 Cigar or pipe smokers were excluded from the never smokers. Never smokers were defined as having smoked fewer than 100 cigarettes during their lifetimes, and former smokers were defined as having quit smoking for 6 months or more before the lung cancer diagnosis for patients or the date of blood draw for controls. Tobacco history information included current and/or previous use, duration, the average number of cigarettes smoked per day, the number of years since the patient quit smoking, and passive smoking or environmental tobacco smoking (ETS).17 In this analysis, ETS exposure was modeled as a dichotomized covariate (yes vs no). Light smokers were those who smoked for fewer than 20 pack-years; moderate smokers, between 20 and 40 pack-years; and heavy smokers, more than 40 pack-years.
For 12% of the patients and none of the controls, we obtained study information from the next of kin, primarily a spouse; most of these patients had advanced-stage disease and died within 1 year after diagnosis. A careful examination of tobacco smoke exposure (including ETS) and family history data, obtained from medical records or interviews, showed almost identical results between patient and proxy interview groups (Table 1). As expected, patients who could not be directly interviewed were more likely to have advanced-stage disease and die within 1 year after diagnosis. We chose to include the proxy information to maintain a reasonable proportion of cases of advanced-stage disease.
Table 1.
Patient Characteristics by Method of Interviewa
| Variable and Information Source | From Patient | From Proxy |
|---|---|---|
| Information From Medical Records | ||
| Sex ratio, men to women | 1.1:1 | 2.8:1 |
| Age, mean (SD), y | 63.8 (11.3) | 65.9 (12.3) |
| Race/ethnicity | ||
| White | 94 | 97 |
| Alaskan/Native American | 4 | 1 |
| African American | 1 | 1 |
| Other | 1 | 1 |
| Tobacco smoking history | ||
| Never smoker | 18 | 16 |
| Former smoker | 47 | 47 |
| Current smoker | 35 | 37 |
| Tumor histologic findings | ||
| Adenocarcinoma | 39 | 35 |
| Squamous cell carcinoma | 22 | 22 |
| Other NSCLC | 29 | 29 |
| Small cell carcinoma | 10 | 14 |
| Survival after diagnosis | ||
| Died within 1 year after diagnosis | 37 | 70 |
| Median survival, mo | 19.5 | 9.1 |
| Disease stage | ||
| Early | 60 | 30 |
| Late | 40 | 70 |
| History of COPD | 38 | 39 |
|
| ||
| Information From Interview | ||
| Family history of lung cancer | 28 | 28 |
| First-degree relative | 19 | 20 |
| Second-degree relative | 10 | 9 |
| Tobacco smoke exposure | ||
| Pack-year data missing | 2 | 2 |
| ETS data missing | 3 | 0 |
| ETS and pack-year data missing | 24 | 24 |
| History of COPD | 23 | 21 |
Abbreviations: COPD, chronic obstructive pulmonary disease; ETS, environmental tobacco smoking; NSCLC, non–small cell lung cancer.
Unless otherwise indicated, data are reported as percentage of subjects.
The medical records of each patient were reviewed, and the following information was abstracted: demographic characteristics; vital status (alive or dead); education; history of tobacco exposure and alcohol use; lung cancer histologic findings, staging, anatomic location, and treatment; and other medical conditions. The history of COPD was determined based on explicit diagnosis recorded in the medical history; most patients but a minority of controls underwent pulmonary function tests to support this diagnosis.
α1AT ALLELE TYPING
Isoelectric focusing assay, performed in the Mayo Clinic Protein and Immunopathology Laboratory, was used to type α1AT alleles (Figure 1).19 The samples were tested in the order of their assigned blind identification number. The apparent differing genotyping rate across study groups was owing to a natural termination of the grant funding. The typed subjects were compared with the total subjects among patients, unrelated controls, or sibling controls, as appropriate, indicating that typed subjects represented the total subjects in major clinical and demographic characteristics.
Figure 1.

Isoelectric focusing test for alpha1-antitrypsin allele types. A, Selected Pi variants as revealed by isoelectric focusing (polyacrylamide gel, anode at the top and cathode at the bottom) (nomenclature of allele names from Cox et al18). B, Selected Pi variants observed in human plasma using isoelectric focusing (polyacrylamide gel, anode is at the top) from Mayo Clinic Immunopathology Protein Laboratory.
STATISTICAL ANALYSIS
We tested the Hardy-Weinberg equilibrium in 2 ways: (1) all alleles and (2) dichotomizing normal vs deficient alleles collectively.20 For the unrelated controls, the α1ATD allele distribution did not depart from the Hardy-Weinberg equilibrium either tested using all observed alleles (P>.99) or using the dichotomized normal vs deficient alleles (P=.16).
UNRELATED CONTROL ANALYSIS
Standard contingency table statistics were used to test whether the α1ATD high-risk genotype frequency differed between patients and controls. The McNemar test for the equality of proportions in matched samples was used to test for an association between α1ATD and lung cancer, measured by odds ratios (ORs). Conditional logistic regression models for the matched set21 were used to test for the effects of history of chronic lung diseases (emphysema only, chronic bronchitis only, and both), tobacco smoke exposure, and high-risk genotype on the association with lung cancer risk. These models used case or control status as an outcome variable, and high-risk genotype, existing chronic lung diseases, history of tobacco smoke exposure, and hypothesis-driven 2-way interactions of these variables, as well as other known confounding variables (eg, family history) as predictor variables. Variation in the association between lung cancer risk and high-risk genotype by histologic subtype, smoking history, or family history of cancer was tested by stratified case-control analysis.
UNAFFECTED SIBLING CONTROL ANALYSIS
The generalized estimating equations approach was used to test for an association between the α1ATD gene and lung cancer (proc GENMOD in SAS, version 8.2; SAS Institute, Cary, North Carolina)22 for its ability to accommodate any sibship size.23
ATTRIBUTABLE RISK ESTIMATION
We estimated the expected population attributable risk (EAR) in percentage that was modeled for each risk factor according to the expected population frequency of that risk factor and the adjusted OR based on our multivariable regression models.23
RESULTS
During the 6-year study period (1997–2003), we enrolled 1856 patients with lung cancer, of whom 1585 were matched with unrelated controls and 1053 had full sibling controls. The α1AT allele types were tested in 1443 patients, 797 unrelated controls, and 902 full siblings in the order the samples were received. Table 2 lists the basic demographic characteristics of the patients and controls. For these variables, patients with lung cancer who were tested for α1AT allele type were very similar to those who were not (no statistically significant difference, P=.31-.96); likewise, α1AT-typed controls were very similar to all controls enrolled in our study.
Table 2.
Basic Description of Study Participants
| Study Group | Age, Mean (SD), y | Participants, % | α1ATD Carriers, % |
|---|---|---|---|
| Patients With Lung Cancer | |||
| Enrolled (n=1856) | 65 (11) | 100 | NA |
| Men | 987 | 53.2 | NA |
| Women | 869 | 46.8 | NA |
| α1AT tested (n=1443) | 64 (11) | 100 | 13.4a |
| Men | 765 | 53.0 | 13.5a |
| Women | 677 | 46.9 | 13.3a |
|
| |||
| Unrelated Controls | |||
| Matched (n=1585) | 64 (13) | 100 | NA |
| Men | 768 | 48.5 | NA |
| Women | 817 | 51.5 | NA |
| α1AT tested (n=797) | 66 (15) | 100 | 7.8 |
| Men | 381 | 47.8 | 7.4 |
| Women | 416 | 52.2 | 8.2 |
|
| |||
| Sibling Controls | |||
| Enrolled (n=1053) | 61 (12) | 100 | NA |
| Men | 629 | 59.7 | NA |
| Women | 424 | 40.3 | NA |
| α1AT tested (n=902) | 61 (12) | 100 | 9.9 |
| Men | 543 | 60.2 | 9.9 |
| Women | 359 | 39.8 | 9.8 |
Abbreviations: α1ATD, alpha1-antitrypsin deficiency; α1AT, alpha1- antitrypsin; NA, not applicable.
P<.01 comparing patients with respective controls.
COMPARISON BETWEEN PATIENTS AND UNRELATED CONTROLS
We applied multiple logistic regression models to examine the effects of tobacco exposure history, COPD, and α1ATD carrier state on lung cancer risk. First, we modeled 1585 case-control pairs (Table 3 [model 1]) without the α1ATD variable. This model demonstrated the relative importance of COPD as a risk factor and different levels of tobacco smoke exposure, including ETS alone and light, moderate, and heavy smokers. Individuals with COPD had a greater than 6-fold higher lung cancer risk, and smokers were at a 2- to 9-fold higher risk than never smokers, depending on smoking intensity. Although ETS did not significantly increase lung cancer risk (OR, 1.2), a trend test of a 5-level cigarette smoking exposure intensity (never smokers with no ETS, never smokers with ETS, and light, moderate, and heavy smokers) was significant (P<.01).
Table 3.
Comparison of Patients and Unrelated Controls by Multiple Logistic Regression Analysis
| Estimated Odds Ratio (95% CI)
|
||
|---|---|---|
| Risk Factor | Model 1a | Model 2b |
| α1ATD carrier | 1.7 (1.2–2.4)d | |
| All COPD | 6.4 (4.8–8.3)d | 3.9 (3.0–5.1)d |
| Chronic bronchitis only | 4.8 (3.0–7.7) | 4.4 (2.6–7.4) |
| Emphysema only | 5.9 (3.5–10.1) | 5.2 (2.7–10.2) |
| Chronic bronchitis and emphysema | 6.6 (2.8–15.3) | 3.2 (1.3–8.2) |
| Smoke exposure | 3.2 (2.6–3.9)d | 4.0 (3.2–4.9)d |
| ETS vs never smokerc | 1.2 (0.8–1.9) | 2.0 (1.3–3.0) |
| Light vs never smoker | 1.7 (1.2–2.6) | 3.4 (2.2–5.2) |
| Moderate vs never smoker | 4.6 (3.0–7.0) | 7.2 (4.7–11.2) |
| Heavy vs never smoker | 9.4 (6.1–14.5) | 18.3 (11.8–28.4) |
Abbreviations: α1ATD, alpha1-antitrypsin deficiency; CI, confidence interval; COPD, chronic obstructive pulmonary disease; ETS, environmental tobacco smoking.
Model 1 is based on 1585 subject pairs matched by sex and age; Pi1 locus was not tested in all instances.
Model 2 adjusted for sex, age, and unspecified COPD.
Never smokers without ETS exposure.
P≤.001.
We then built a model including the α1ATD allele types (Table 3 [model 2]). The overall α1ATD carrier rate among 1443 patients with lung cancer was 13.4%. The carrier rate in 797 unrelated controls was 7.8% (P<.001). The uneven proportion genotyped (78%, 50%, and 75% of patients, unrelated controls, and sibling controls, respectively) was driven by the natural process of subject enrollment. No significant difference between tested and untested groups was found.
The α1ATD alleles included mainly S (6.7%), Z (3.2%), and other rare ones (I, P, V, and null type making up collectively 2.6% of the allele type distribution in all study participants). Individuals carrying 1 or 2 deficient alleles were all considered as high-risk allele carriers and more than 90% were heterozygous (including SZ) or carriers. These carriers were at a 70% higher risk of developing lung cancer than the noncarriers. Among nonsmokers, ETS was associated with a 2-fold increase in lung cancer independent of α1ATD carrier status. Compared with model 1, the risk for chronic bronchitis or emphysema was similar, whereas the risks associated with the different levels of smoking intensity were approximately doubled.
To further test whether the association between lung cancer risk and α1ATD carrier status varied by history of tobacco smoke exposure and/or COPD, histologic type of lung cancer, and family history of lung cancer and/or other malignancies, we conducted stratified analyses on our case-unrelated control data set.
SMOKING HISTORY
Among never smokers, we found that α1ATD allele carriers were at a 2.2-fold risk for lung cancer compared with noncarriers after adjusting for age, sex, and history of unspecified COPD. In addition, the increased risk for never smokers with ETS was 2.0-fold (95% confidence interval [CI], 1.3–3.0). Figure 2 depicts the risk estimates for lung cancer (OR) stratified by smoking intensity. Among moderate to heavy smokers (≥20 pack-years), the α1ATD allele–associated lung cancer risk was estimated at 2.3-fold, whereas among light smokers (<20 pack-years), the α1ATD allele–associated lung cancer risk was 2.0-fold (not significant [95% CI, 0.9–4.3]). Having a history of COPD increases lung cancer risk significantly for all 3 groups of smokers (from 2.5- to 5.9-fold), with the largest effect on never smokers (OR, 5.9; 95% CI, 2.7–12.8).
Figure 2.
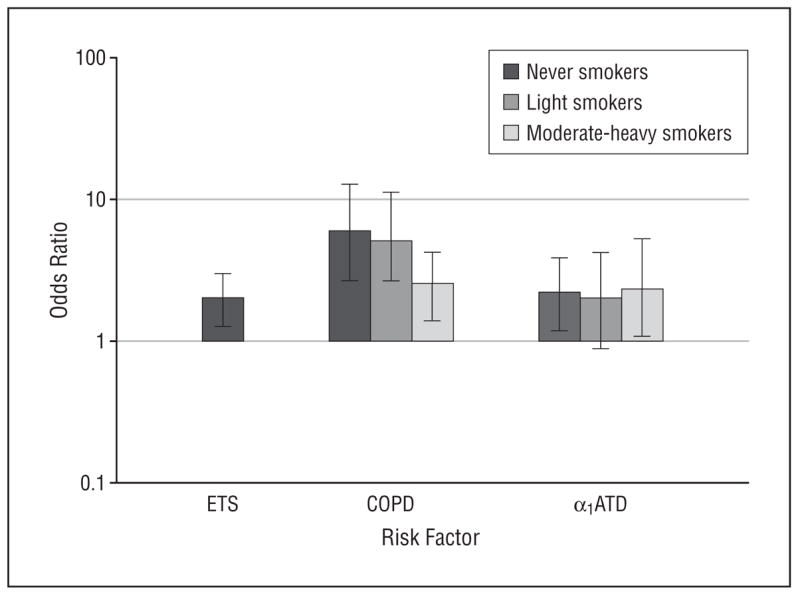
Adjusted odds ratio of risk factors by smoking intensity. α1ATD indicates alpha1-antitrypsin deficiency; COPD, chronic obstructive pulmonary disease; and ETS, environmental tobacco smoke. Error bars indicate 95% confidence intervals.
TUMOR HISTOLOGIC FEATURES
Stratified analysis by selected histologic groups showed varying odds ratios of specific lung cancer risk associated with α1ATD allele carriers (Figure 3), from not significant in small cell lung cancer (OR, 1.4; 95% CI, 0.6–3.1) to significant in adenocarcinoma, particularly bronchioalveolar carcinoma (OR, 2.0; 95% CI, 1.1–3.8), and also significant in squamous cell carcinoma (OR, 2.5; 95% CI, 1.2–5.3). While the effect of COPD on various types of lung cancer was relatively constant with the exception of small cell carcinoma, the effect of smoking history was dramatically different across histologic groups (OR, 2.0–12.1), with the strongest effect on squamous cell carcinoma (OR, 12.1; 95% CI, 4.9–30.2).
Figure 3.

Adjusted odds ratios (95% confidence intervals) of risk factors by cell types of lung cancer. α1ATD indicates alpha1-antitrypsin deficiency; COPD, chronic obstructive pulmonary disease; AD, adenocarcinoma; BAC, bronchioalveolar carcinoma; SQCLC, squamous cell lung cancer; and SCLC, small cell lung cancer.
FAMILY HISTORY OF CANCER
Patients with a family history of lung cancer or other cancers in their first-degree relatives had a similar α1ATD carrier rate to those without such a family history, all significantly higher than the controls. This finding suggests that increased lung cancer risk among α1ATD carriers is independent of a family history of cancer.
COMPARISON BETWEEN PATIENTS AND UNAFFECTED SIBLINGS
Consistent with the results of patient–unrelated-control comparisons, α1ATD allele carriers were at a 2-fold higher lung cancer risk than noncarriers (OR, 2.2; 95% CI, 1.6–2.9) in univariate analysis, and COPD was a very strong risk factor for lung cancer (OR, 5.7; 95% CI, 4.4–7.3). When adding COPD to the model that included α1ATD and the other covariates (Table 4), we found that the odds ratio for an increased lung cancer risk in α1ATD carriers was 2.0 (95% CI, 1.4–2.7), supporting an independent role of α1ATD state in lung cancer development. Noted is the lack of difference or trend among the 3 odds ratios for COPD subgroups (chronic bronchitis only, emphysema only, and both); this could reflect the low occurrence of COPD in unaffected siblings and therefore the unstable risk estimates.
Table 4.
Comparison of Patients and Sibling Controlsa
| Risk Factor | Odds Ratio (95% CI) |
|---|---|
| α1ATD | 2.0 (1.4–2.7) |
| Unspecified COPD | 8.1 (4.0–16.5) |
| Chronic bronchitis only | 5.6 (3.4–9.3) |
| Emphysema only | 3.3 (2.0–5.3) |
| Chronic bronchitis and emphysema | 4.2 (2.1–8.8) |
| Ever smoker | 3.0 (2.2–4.0) |
Abbreviations: α1ATD, alpha1-antitrypsin deficiency; CI, confidence interval; COPD chronic obstructive pulmonary disease.
Model adjusted for sex and age.
POPULATION ATTRIBUTABLE RISK ESTIMATION
We estimated the EAR based on our multivariable models. The EAR for α1ATD carriers, COPD, and ETS among never smokers was 12%, 10%, and 41%, respectively. Among heavy smokers, the EAR for α1ATD carriers was 11%, and for COPD it was 12%, adjusting for pack-years of cigarettes smoked. Therefore, the α1ATD allele type might explain 11% to 12% of lung cancer occurrence in our population, which was predominantly from the US Midwest, represented by the majority of our study subjects.
COMMENT
Our results demonstrated that the α1ATD allele can double an individual’s lung cancer risk and also confirmed that COPD is an independent risk factor for lung cancer, with an EAR of 10% to 12%. An EAR of 16% for COPD in lung cancer was reported in 19927 after controlling for smoking; the somewhat lower EAR found in our study could be explained by the inclusion of α1AT allele types in the risk models. An alternative explanation would be that the 1992 study estimated the attributable risk associated with a larger grouping of previous lung diseases including chronic bronchitis, emphysema, tuberculosis, and asthma. The inclusion of these additional lung diseases may also have accounted for a higher EAR in that study. In contrast, tuberculosis and asthma were not significant contributors in the present study. Importantly, our results support the hypothesis that the excess risk of lung cancer among patients with COPD may be a result of lung tissue damage (emphysema), chronic infection or inflammation (chronic bronchitis), or both.
A report from the World Health Organization24 disclosed that α1ATD is seriously underdiagnosed. There are at least 10 million Americans and 120 million people worldwide who are α1ATD carriers.25 Our results indicate a potentially significant impact of α1ATD on lung cancer risk and potential benefit for identifying and protecting these individuals who may be vulnerable to carcinogens. Even though the proportion of α1ATD carriers in the population is less than 10%, the relative and attributable lung cancer risk is suggested to be more than 10%, which is among the highest for major gene effects on the risk of a common cancer.
A recent report showed that over 12 million people in the US population have been clinically diagnosed with COPD, and this only accounted for half of all who have the disease.26 Chronic obstructive pulmonary disease is an indolent process that can remain subclinical because the lungs have a tremendous reserve that is not called on, even during mild exertion; therefore, many persons with COPD may not be diagnosed until the disease is advanced.
A limitation to our study is the potential underdiagnosis of COPD, which should be based on symptom complex, clinical signs, and the most reliable test of all, laboratory evaluation of fixed airway obstruction by a pulmonary function test.2 Emphysema can be diagnosed by adequate radiographic or pathologic evaluation and is defined as abnormal permanent enlargement of the air spaces distal to the terminal bronchioles accompanied by destruction of their walls without obvious fibrosis.2
Another potential limitation is the possibility of population stratification in gene-disease association due to heterogeneous ancestral background of the study subjects. One method of overcoming this limitation is by a case-control study that matches ethnicity at subject enrollment or adjusts for ethnicity in analysis. However, owing to the diversity of the ancestral background of US whites, it is a serious challenge to combine 4 lines of ethnicity (or country of origin) from both sets of grandparents into meaningful groups.13,16 In our study, we identified 25 distinct ethnic groups, which makes 254 or 390 625 possible combinations to consider for 4 grandparents. Moreover, because the α1ATD allele frequency varies significantly within many European countries, for example with a north to south gradient,9 the finest classification of ethnicity by country of origin, if possible at all, may not resolve the issue of population admixture. In the present study, 95% of our patients were white, as were 92% of controls. Because of the small proportion of the nonwhites (that included Native American, Asian Indian, Asian, African American, and Hispanic ethnic backgrounds), our study results were not changed by either dropping them from the analysis or adjusting the potential effect of ethnicity as a covariate in the analytic models.
Sibling controls are a valid way to avoid population stratification. We applied the generalized estimating equations model that uses all the information provided from the multiple siblings per proband while properly accounting for the correlation between observations within a sibship.27 A potential confounder is that younger unaffected siblings might become patients in time, but age was adjusted for along with other factors. Another potential drawback is that sibling controls and the patients are overmatched for both their genes and early life environment. Siblings in general had shared more common environmental risk factors with the patients than had the unrelated controls, which may partly explain the relative lower ORs for smoking and COPD when using siblings compared with using unrelated controls.28,29
A third limitation is the use of community-based controls to match the clinically based patients.14,15 Nonetheless, the advantages of this design are time- and cost-effectiveness, assuming a reasonable participation rate.14–16 By using this unrelated population control group, we were able to test the hypothesis that patients with lung cancer are more likely to be α1ATD carriers, and we could also provide an accurate estimate of the expected exposure rate (ie, α1ATD carrier rate) in a reference population of Olmsted County, Minnesota.
In summary, our findings demonstrate a paradigm in lung cancer etiology research and risk assessment that incorporates clinical and genetic markers for lung damage into a gene-environment interaction. A logical next step is to further investigate whether the excess risk of lung cancer among patients with COPD is a result of lung tissue damage, airway obstruction, or both; such studies will require accurate assessment of these phenotypes beyond current clinical diagnosis. Lung cancer development is a complicated multistage process involving many factors. Carcinogens or harmful substances in tobacco smoke not only cause damage to the DNA of cells but also stimulate inflammatory response in the lung. Meanwhile, the host has various mechanisms to inactivate harmful substances from the smoke, remove or repair damaged DNA, and adapt the stimulants by structural changes in the lung tissue. The intricate interplay among these factors underlies the mechanisms of lung cancer development. This knowledge may prove to be useful in further understanding the pathologic mechanisms of lung cancer development and in refining lung cancer risk assessment.
Acknowledgments
Funding/Support: This study was supported by grants NIH-CA 77118, NIH-CA 80127, and NIH-CA 84354 from the National Institutes of Health (Dr Yang).
Footnotes
Financial Disclosure: None reported.
Additional Contributions: Noralane M. Lindor, MD, Timothy G. Lesnick, MS, and Kimberly A. Wentzlaff, MS, contributed at various stages to this work, specifically during patient identification and enrollment. Susan Ernst, BS, provided technical assistance with the manuscript.
Author Contributions: Dr Yang had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Yang, Krowka, Bamlet, Midthun, Marks, and de Andrade. Acquisition of data: Yang, Sun, Aubry, Thibodeau, and Katzmann. Analysis and interpretation of data: Yang, Sun, Krowka, Bamlet, Wampfler, Allen, Marks, and de Andrade. Drafting of the manuscript: Yang, Sun, Krowka, Aubry, Bamlet, and Midthun. Critical revision of the manuscript for important intellectual content: Yang, Sun, Krowka, Bamlet, Wampfler, Thibodeau, Katzmann, Allen, Marks, and de Andrade. Statistical analysis: Bamlet, Wampfler, and de Andrade. Obtained funding: Yang. Administrative, technical, and material support: Sun, Krowka, Thibodeau, Katzmann, and Midthun. Study supervision: Yang and Allen.
References
- 1.Wu AH, Fontham ETH, Reynolds P, et al. Previous lung disease and risk of lung cancer among lifetime nonsmoking women in the United States. Am J Epidemiol. 1995;141(11):1023–1032. doi: 10.1093/oxfordjournals.aje.a117366. [DOI] [PubMed] [Google Scholar]
- 2.American Thoracic Society. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma: this official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am Rev Respir Dis. 1987;136(1):225–244. doi: 10.1164/ajrccm/136.1.225. [DOI] [PubMed] [Google Scholar]
- 3.Gómez FP, Rodriguez-Roisin R. Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines for chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2002;8(2):81–86. doi: 10.1097/00063198-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Samet JM, Humble CG, Pathak DR. Personal and family history of respiratory disease and lung cancer risk. Am Rev Respir Dis. 1986;134(3):466–470. doi: 10.1164/arrd.1986.134.3.466. [DOI] [PubMed] [Google Scholar]
- 5.Skillrud DM, Offord KP, Miller RD. Higher risk of lung cancer in chronic obstructive pulmonary disease. Ann Intern Med. 1986;105(4):503–507. doi: 10.7326/0003-4819-105-4-503. [DOI] [PubMed] [Google Scholar]
- 6.Cohen BH, Graves CG, Levy DA, et al. A common familial component in lung cancer and chronic obstructive pulmonary disease. Lancet. 1977;2(8037):523–526. doi: 10.1016/s0140-6736(77)90663-8. [DOI] [PubMed] [Google Scholar]
- 7.Alavanja MCR, Brownson RC, Boice JD, Hock E. Preexisting lung disease and lung cancer among nonsmoking women. Am J Epidemiol. 1992;136(6):623–632. doi: 10.1093/oxfordjournals.aje.a116542. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz AG, Yang P, Swanson GM. Familial risk of lung cancer among non-smokers and their relatives. Am J Epidemiol. 1996;144(6):554–562. doi: 10.1093/oxfordjournals.aje.a008965. [DOI] [PubMed] [Google Scholar]
- 9.Sun Z, Yang P. Neutrophil elastase and alpha-1 antitrypsin: the role of imbalance in cancer development and progression: a review. Lancet Oncol. 2004;5(3):182–190. doi: 10.1016/S1470-2045(04)01414-7. [DOI] [PubMed] [Google Scholar]
- 10.Cox DW. Alpha-1 antitrypsin deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 7. New York, NY: McGraw-Hill; 1995. pp. 4125–4158. [Google Scholar]
- 11.Kueppers F. Determination of alpha-1-antitrypsin phenotypes by isoelectric focusing on polyacrylamide gels. J Lab Clin Med. 1976;88(1):151–155. [PubMed] [Google Scholar]
- 12.Yang P, Allen MS, Aubry MC, et al. Clinical features of 5,628 primary lung cancer patients: experience at Mayo Clinic from 1997–2003. Chest. 2005;128(1):452–462. doi: 10.1378/chest.128.1.452. [DOI] [PubMed] [Google Scholar]
- 13.Yang P, Wentzlaff KA, Katzmann JA, et al. Alpha1-antitrypsin deficiency allele carriers in lung cancer patients. Cancer Epidemiol Biomarkers Prev. 1999;8(5):461–465. [PubMed] [Google Scholar]
- 14.Taniguchi K, Yang P, Jett J, et al. Polymorphisms in the promoter region of the neutrophil elastase gene are associated with lung cancer development. Clin Cancer Res. 2002;8(4):1115–1120. [PubMed] [Google Scholar]
- 15.Yang P, Kollmeyer TM, Buckner K, Bamlet W, Ballman KV, Jenkins RB. Polymorphisms in GLTSCR1 and ERCC2 are associated with the development of oligodendrogliomas. Cancer. 2005;103(11):2363–2372. doi: 10.1002/cncr.21028. [DOI] [PubMed] [Google Scholar]
- 16.Yang P, Bamlet W, Ebbert JO, et al. Alpha-1 antitrypsin and neutrophil elastase imbalance and lung cancer risk. Chest. 2005;128(1):445–452. doi: 10.1378/chest.128.1.445. [DOI] [PubMed] [Google Scholar]
- 17.de Andrade M, Ebbert JO, Wampfler JA, et al. Environmental tobacco smoke exposure in women with lung cancer. Lung Cancer. 2004;43(2):127–134. doi: 10.1016/j.lungcan.2003.08.025. [DOI] [PubMed] [Google Scholar]
- 18.Cox DW, Johnson AM, Fagerhol MK. Report of nomenclature meeting for alpha 1-antitrypsin, INSERM, Rouen/Bois-Guillaume-1978. Hum Genet. 1980;53(3):429–433. doi: 10.1007/BF00287070. [DOI] [PubMed] [Google Scholar]
- 19.Tietz NW. Clinical Guide to Laboratory Tests. Philadelphia, PA: WB Saunders Co; 1990. [Google Scholar]
- 20.Guo SW, Thompson ET. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48(2):361–372. [PubMed] [Google Scholar]
- 21.Hosmer DW, Lemeshow S. Applied Logistic Regression. New York, NY: John Wiley & Sons; 1989. [Google Scholar]
- 22.Ramon C, Freund RJ, Specotr PC. SAS System for Linear Models. Cary, NC: SAS Institute Inc; 1991. [Google Scholar]
- 23.Bruzzi P, Green SB, Byar DP, Brinton LA, Schairer C. Estimating the population attributable risk for multiple risk factors using case-control data. Am J Epidemiol. 1985;122(5):904–914. doi: 10.1093/oxfordjournals.aje.a114174. [DOI] [PubMed] [Google Scholar]
- 24.World Health Organization. Alpha1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397–415. [PMC free article] [PubMed] [Google Scholar]
- 25.de Serres FJ. Alpha-1 antitrypsin deficiency is not a rare disease but a disease rarely diagnosed. Environ Health Perspect. 2003;111(16):1851–1854. doi: 10.1289/ehp.6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.National Institutes of Health, National Heart, Lung, and Blood Institute, US Department of Health and Human Services. Data Fact Sheet: Chronic Obstructive Pulmonary Disease. Bethesda, MD: National Institutes of Health; 2003. pp. 1–6. NIH publication 03–5229. [Google Scholar]
- 27.Lipsitz SR, Fitzmaurice GM, Orav EJ, Laird NM. Performance of generalized estimating equations in practical situations. Biometrics. 1994;50(1):270–278. [PubMed] [Google Scholar]
- 28.Witte JS, Gauderman WJ, Thomas DC. Asymptotic bias and efficiency in case-control studies of candidate genes and gene-environment interactions: basic family designs. Am J Epidemiol. 1999;149(8):693–705. doi: 10.1093/oxfordjournals.aje.a009877. [DOI] [PubMed] [Google Scholar]
- 29.Schaid DJ, Buetow K, Weeks DE, et al. Discovery of cancer susceptibility genes: study designs, analytic approaches, and trends in technology. J Natl Cancer Inst Monogr. 1999;(26):1–16. doi: 10.1093/oxfordjournals.jncimonographs.a024219. [DOI] [PubMed] [Google Scholar]