

Abstract
Molecules that govern the formation, integrity and function of the hippocampus remain an important area of investigation. Here we show that absence of the proneuropeptide processing enzyme, carboxypeptidase E (CPE) in CPE knock-out (KO) mice had a profound effect on memory, synaptic physiology and the cytoarchitecture of the hippocampus in these animals. Adult CPE-KO mice displayed deficits in memory consolidation as revealed by the water-maze, object preference and social transmission of food preference tests. These mice also showed no evoked long term-potentiation. Additionally, CPE-KO mice at 4 weeks of age and older, but not at 3 weeks of age, exhibited marked degeneration specifically of the pyramidal neurons in the hippocampal CA3 region which normally expresses high levels of CPE. Immunohistochemistry revealed that the neuronal marker, NeuN, was reduced, while the glial marker, GFAP, was increased, characteristic of gliosis in the CA3 area of CPE-KO mice. Calbindin staining indicated early termination of the mossy fibers before reaching the CA1 region in these mice. Thus, absence of CPE leads to degeneration of the CA3 neurons and perturbation of the cytoarchitecture of the hippocampus. Ex vivo studies showed that overexpression of CPE in cultured hippocampal neurons protected them against H2O2 oxidative-stress induced cell death. These findings taken together indicate that CPE is essential for the survival of adult hippocampal CA3 neurons to maintain normal cognitive function.
Keywords: learning, CA3 neurons, neuroprotection, neuronal survival, long term potentiation
INTRODUCTION
Carboxypeptidase E (CPE) was first identified as an exopeptidase that is expressed primarily in neuronal and endocrine tissues and plays a role in pro-neuropeptide and prohormone processing (Fricker, 1988). The soluble form of CPE cleaves C-terminally extended basic amino acids from neuropeptide intermediates derived from the proform to yield the bioactive neuropeptides (Hook and Loh, 1984). The membrane-bound form of CPE serves as a sorting receptor for some proneuropeptides and proBDNF to target them into the regulated secretory pathway (Cool et al., 1997; Lou et al., 2005). Hence, cortical and hippocampal neurons from the CPE-KO mouse showed a lack of processing of proBDNF and absence of activity-dependent release of BDNF (Lou et al., 2005). More recently, the cytoplasmic tail of the transmembrane form of CPE was shown to be a key molecule in the anchoring of ACTH vesicles to the microtubule-based transport system for post-Golgi delivery of the vesicles to the release site (Park et al., 2008). Thus it would be expected that lack of CPE would have a major impact on the function of the endocrine and nervous systems in the CPE-KO mice since the enzyme is required for the processing and secretion of proneuropetides. Indeed, these CPE-KO mice exhibit not only endocrinological deficits such as diabetes, infertility and obesity (Cawley et al., 2004), but also neurological deficits including poor muscle tone (Cawley et al., 2004) and diminished glutamate-transmission mediated b-wave in their retinograms (Zhu et al., 2005). It has also been shown that lack of CPE in a C. elegans mutant resulted in a defect in acetylcholine neurotransmission at the neuromuscular junction (Jacob and Kaplan, 2003). Interestingly, it has also been reported that after transient global ischemia, there is a greater and more sustained increase in CPE expression in the CA3 region of the hippocampus which correlated with survival of those neurons, in contrast to neurons of the CA1 region which only showed a transient increase of CPE expression and were more vulnerable (Jin et al., 2001). Thus, CPE appears to play multiple roles in the nervous system, including activity-dependent BDNF secretion, neurotransmission, and possibly maintenance of hippocampal neuron survival after global ischemia. Nevertheless, CPE-KO animals survive and neurological deficits may be subtle perhaps due to partial compensatory mechanisms. However, close examination of CPE-KO mice with respect to behavior, electrophysiology and brain morphology to characterize deficits that are severe and not compensated in these animals will likely identify important and unique functions of CPE in the central nervous system.
In this study, we have investigated hippocampal function in CPE-KO mice, since activity-dependent secretion of BDNF, which is important in hippocampal-based memory and learning (Egan et al., 2003) is diminished in neurons of these mice; and therefore this BDNF-rich region of the brain is likely to be affected. We evaluated potential deficits in hippocampal function in CPE-KO mice using several different behavioral tests that rely upon an intact hippocampus (Kogan et al., 2000; Logue et al., 1997). As well, we performed electrophysiological analyses to examine hippocampal long-term potentiation (LTP) following tetanic stimulation in these mice. Finally we examined the cytoarchitecture of the hippocampus in adult CPE-KO mice. Collectively, our findings indicate that CPE plays a critical role in hippocampal neuron survival and memory in adult mice.
MATERIALS AND METHODS
Animals
Two colonies of WT and CPE-KO mice were used; one housed at NIH and the other at Duke University Medical Center. At both facilities, animals were housed in groups of 2–5 under a 14:10 hr light-dark cycle in a temperature- (22°C) and humidity- (45%) controlled room and provided food and water ad libitum. All experiments were conducted with approved protocols and accordingly with NIH guidelines for the care and use of laboratory animals.
Behavioral studies
Social transmission of food preference (STFP)
This test was run at Duke University and the procedure has been described previously (Kogan et al., 2000; Rodriguiz, 2006; Rodriguiz et al., 2004). Mice (8–12 weeks old) housed in groups of 3–4 animals were segregated by genotype and sex for several weeks before testing. Flavored diets (vanilla or chocolate) were prepared by mixing 50 g ground rodent diet with 50 ml water and 10 ml of a vanilla- or chocolate-flavored soy-based drink (Ensure®; Abbott Laboratories). On the day before testing, all mice were tail marked and food was removed with water available ad libitum. Following 18 hrs of food deprivation, one mouse (i.e., the demonstrator) was removed from the home-cage and placed into a clean 20 × 20 × 30 cm acrylic chamber with a single 4 cm (diameter) bowl containing 10 g of flavored diet. The choice of flavor was randomized across mouse cages and genotypes. After eating for 30 min, the demonstrator was returned to the home-cage where interactions between the demonstrator and other mice (i.e., testers) were observed for 20 min. Demonstrator mice were then removed from the home-cage for the remainder of the study and the tester mice were placed into a clean mouse cage and presented with two bowls: one flavored with vanilla and the other with chocolate diet positioned at opposite corners of the cage. Tests were conducted at 20 min for one-half the mice or at 24 hrs for the remainder. Each tester was allowed to eat for 30 min and consumption (weight) of the novel and familiar foods was recorded. In an additional study, tester animals were examined both at 20 min and 24 hrs using the same paradigm, except the demonstrator animal remained in the home-cage with the tester. All bowls were weighed before and after each test and any spillage was recovered and noted. Preference for a given diet was determined by calculating the difference between the two flavored diets consumed relative to the total amount consumed. Positive scores indicated preference for the diet fed the demonstrator mouse, whereas negative scores denoted preferences for the novel or unfamiliar diet. Scores approaching zero indicated no preference for either diet.
Object recognition
Testing was conducted at Duke University as outlined (Rodriguiz, 2006). Prior to testing, a small group of mice (8–12 weeks of age) was examined with a series of objects to exclude innate object preferences or neophobia. To further exclude any potential object bias, one-half of the animals from each genotype was initially exposed to one pair of identical objects (2 × 3 × 1 cm in size; Legos), while the remainder was presented with another pair of identical objects of similar size but differing in color-pattern and shape. The two pairs of objects constituted the “familiar” and “novel” objects for the test. At pre-exposure, two identical objects were placed into opposite corners of the arena (20 × 20 × 30 cm) and affixed to the floor with double-sided tape. Parenthetically, the inside walls of the arena were homogenous without any cues to provide spatial orientation during the exposure phase and testing. Mice were allowed to interact with the objects for 30 min. Animals were returned to the home-cage for 20 min and then placed into the arena for 30 min with one familiar and a novel object. Twenty-four hrs later, mice were returned to the arena and were exposed to the same familiar and novel object for 5 min. All behaviors were videotaped and coded by trained observers with the Noldus Observer (Noldus Information Technologies) who were blind to the genotype of the animal and test interval. Scored behaviors included latency to first contact an object, time spent with an object, and the number of physical contacts (sniffing, climbing or sitting) with an object. Recognition memory scores were calculated over the 5 min of testing by subtracting the time spent with the novel from time spent with the familiar object, and dividing this difference by the time spent with both objects. Positive scores signified recognition of the familiar object, whereas negative scores indicated preferences for the novel object. Scores approaching “zero” denoted recognition of neither object.
Morris water maze
This test is often used as an index of hippocampal spatial learning and memory (Morris, 1984) and was run at the NIH. WT and CPE-KO mice were compared at 10–13 wks of age before these animals became obese. The test was administered in the following order: visible platform training (3 days), hidden platform training (5 days). Testing was performed in a circular pool, 1 m in diameter, filled with water made opaque with the addition of nontoxic white paint. Videotracking was conducted with a video camera and navigational parameters were analyzed subsequently with the Videomex Water Maze Program (Columbus Instruments). Prior to testing, mice were placed on the submerged platform and acclimatized to the apparatus. For visible platform testing, the submerged platform had a flag on it and mice were given 4 trials/day over 3 days where the position of the platform was moved to a different quadrant each day. For hidden platform testing, animals were placed into the pool, facing the wall of the pool, in a new quadrant on each of the successive 4 trials/day. The hidden platform remained in the same position for all these trials. Mice were allowed a maximum of 60s to locate the platform and were left on the platform for 30 s before being removed. If the platform was not reached in 60s, the mouse was guided to the platform and allowed to sit on it for 30 sec. An average latency of 10 seconds or less to locate the platform across the block of 4 consecutive trials each day was the criterion for learning.
Statistical analyses
Data analyses were performed with the Statistical Package for the Social Sciences (SPSS), Version 11.0 or Statview (Abacus Concepts). The data are presented as means and standard errors of the means. For the STFP test, univariate ANOVA was used to analyze preference at 20 min and 24 hrs in the absence and presence of the demonstrator. A repeated-measures ANOVA (RMANOVA) was used for the object recognition tests where the animals were examined at 20 min and 24 hrs. Aposteriori analyses were performed using Bonferroni corrected pair-wise comparisons. For the Morris water maze, the data were analyzed with RMANOVA and contrasts between groups were performed with Fisher’s pair-wise least-significant differences. In all cases, p < 0.05 was considered significant.
Electrophysiological studies
Preparation of hippocampal slices
Hippocampal slices were obtained from 7–9 week-old WT and CPE-KO mice. Animals were anesthetized with halothane, decapitated, and brains quickly removed and placed in ice-cold solution containing (in mM): 125 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaHPO4, 1.5 MgSO4, 1.5 CaCl2, and 10 glucose that was saturated with 95% O2 and 5% CO2 (pH 7.4). Hippocampal slices were cut at 350 µm thickness in coronal plane with a vibratome (Leica VT 1000S) and placed into a slice-holding chamber filled with artificial cerebral spinal fluid (aCSF) containing (in mM): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaHPO4, 1.5 MgSO4, 1.5 CaCl2, and 10 glucose that was saturated with 95% O2 and 5% CO2 (pH 7.4).
Electrophysiological recording
Following ~60 min incubation at 22°C, individual slices were transferred to a submerged slice chamber mounted on the stage of an upright microscope (Nikon Eclipse E600FN) and perfused with oxygenated aCSF at 35 °C at a rate of 2–3 ml/min. Recording electrodes were pulled from borosilicated glass (WPI) and filled with aCSF. The field excitatory postsynaptic potential (fEPSP) was evoked by a bipolar stimulation electrode (FHC) placed in the Schaffer collateral pathway. The fEPSPs were recorded every 1 min from the striatum radiatum of the hippocampal CA1 region by half maximal intensity of stimulation. After 10–20 min recording to establish a stable baseline fEPSP, tetanic stimulation was applied to generate LTP. The tetanic stimulation protocol consisted of two trains of stimuli (1 sec each, at 100 Hz) with 3 sec intervals. Both tetanic and test stimulations were generated by a MASTER-8 stimulator (AMPI) and delivered through a stimuli isolation unit (Iso-Flex; AMPI). Signals were recorded with an Axopatch 1D (Axon Instrument) and sampled at 10 KHz. Data were collected using Clampex 9.2 (Axon Instrument) through an analog-to-digital converter (Digidata 1322A; Axon Instrument) and analyzed with Clampfit 9.2 software (Axon Instrument).
Histology and immunohistochemistry studies
Embedding and sectioning
The brains of 12 CPE-KO mice and 12 WT littermates (2 mice of each genotype and age) were dissected from perfused mice and embedded (24 mouse brains per block) in a gelatin matrix using MultiBrain™ Technology (NeuroScience Associates).
Immunohistochemistry
For CPE and calbindin immunohistochemistry, free-floating sections were used. All incubation solutions from the blocking-serum step onwards used PBS with 0.1%Triton X-100 and all rinses were with PBS. After treatment with 5% hydrogen peroxide for 30 min and blocking with goat serum, sections were incubated with a primary antibody: rabbit anti-calbindin (1:1500 dilution; Chemicon) or rabbit anti-CPE antibody (1:1500 dilution; GeneTex) overnight at 22°C. The Vectastain ABC kit; (Vector) was used to visualize the reaction. For NeuN, GFAP and Nf-160 staining, brains of CPE-KO and WT perfused mice were dissected, placed into the fixative solution containing 4% paraformaldehyde in 1X PBS and kept in 4% parafomaldehyde and 10% sucrose at 4°C overnight. Coronal frozen sections were cut at 50 µm, and incubated with primary antibodies in 1X PBS, 0.3% Triton X-100, and 5% normal goat serum overnight at 4°C. The primary antibodies used were: anti-GFAP polyclonal rabbit (1:1000 dilution; Chemicon), anti-NeuN mouse monoclonal (1:200 dilution; Chemicon) and anti-Nf-160 mouse monoclonal antibodies (1:500 dilution; Sigma-Aldrich). After washing with 1X PBS-0.3% Triton-X100, sections were incubated with secondary antibodies conjugated with Alexa 488 and Alexa 568 (1:200, Molecular Probe) and counterstained with 1µg/ml DPAI (Molecular Probes).
Nissl staining
Fifty µm sections were mounted onto gelatinized slides, dehydrated through an alcohol series, defatted in a chloroform/ether/alcohol solution, and stained in 0.05% thionine/0.08M acetate buffer (pH 4.5).
Hippocampal neuronal survival assay
E18 rat primary hippocampal neurons (Gene Therapy Systems) were dissociated and seeded into chamber slides coated with polylysine. The culture was incubated with 10% (FBS) and 90% Dulbecco's modified Eagle's (DME) medium. After 4–5 days the culture medium was changed to Neurobasal medium (Gibco) supplied with 2% B27 supplement (Gibco) and 10 mM cytosin-arabinosid (AraC). The culture was infected (MOI 100) with adenovirus expressing β-galactosidase (LacZ) or CPE for 48 hours. After this time, the cells were treated with 50 µM H202 for 24 hrs to induce cell death (Numakawa et al., 2007). Subsequently, the hippocampal cultures were fixed in 4% paraformaldehyde and stained with 1 mM DAPI (Sigma-Aldrich). Fluorescent images taken using a confocal microscope (LSM 510, Carl Zeiss) were processed using Confocal Assistant, Adobe Photoshop 6.0 and Image J software. Cell death was calculated using the formula: % Cell Death = 100 × [(condensed-nucleus) / (intact nucleus + condensed nucleus)]. Data are presented as means ± SEM. One-way ANOVA together with a post-hoc test were used for statistical analysis.
RESULTS
CPE-KO mice display deficits in learning and memory
To evaluate non-spatial memory processes that require an intact hippocampus in WT and CPE-KO mice, we conducted a test of social olfactory learning and memory, the social transmission of food preference (STFP) test (Kogan et al., 2000). Mice were studied under two different conditions. In one, animals were examined at 20 min and at 24 hrs where the demonstrator mouse had been removed from the home-cage following the initial training episode. In a second condition, an additional set of mice was evaluated where the demonstrator was housed continuously in the home-cage with the tester animals from training through testing at 24 hrs. Under both conditions, WT animals showed a preference for the familiar diet at 20 min and at 24 hrs, regardless of presence of the demonstrator (Fig. 1A). By comparison, CPE-KO animals failed to display a preference at 20 min or at 24 hrs when the demonstrator mouse was absent before testing. However, the CPE-KO mice exhibited a preference for the familiar diet when the demonstrator remained in the home-cage. Despite genotypic differences for preference, WT and CPE-KO mice interacted for similar amounts of time with the demonstrator in the home-cage (data not shown), consumed similar amounts of food in all tests (Fig. 1B), and their latencies to contact the familiar and novel diets at 20 min or 24 hrs were also similar (data not shown). Together, these results show that CPE-KO animals are impaired in olfactory social learning and memory at 20 min and at 24 hr; however, if allowed continuous interaction with the demonstrator in the home-cage, CPE-KO mice show normal recognition memory at 24 hrs. The results from the STFP test indicated that the presence of the demonstrator and/or scent of the flavored diet on the demonstrator animal may strengthen the preference for the familiar diet at 24 hrs.
Figure 1.
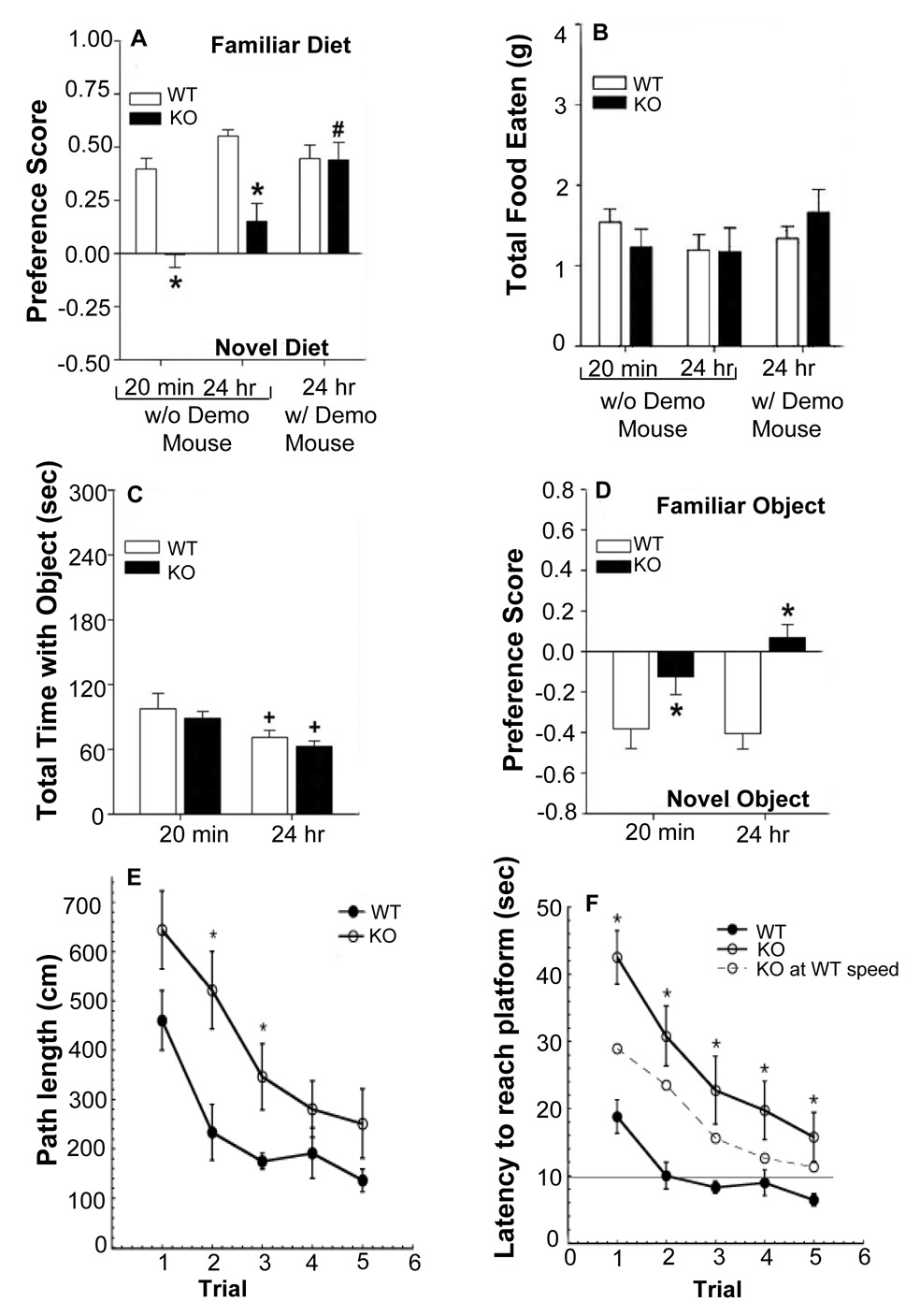
CPE-KO mice show impaired learning and memory in the social transmission of food preference and object recognition tests. (A) Preference scores for the familiar- and novel-flavored diets by WT and CPE-KO mice at 20 min and 24 hrs without the demonstrator present and at 24 hrs with the demonstrator mouse present immediately before testing. ANOVA found the main effects of genotype [F1,151 = 36.466, p < 0.001], test [F2,51 = 12.517, p < 0.001] and the test by genotype interaction [F2,51 = 8.581, p < 0.001] to be significant. Bonferroni corrected pair-wise comparisons confirmed that without the demonstrator present, CPE-KO mice had reduced preferences for the familiar diet relative to WT controls at 20 min and at 24 hrs (ps < 0.001); however, the presence of the demonstrator immediately before testing at 24 hrs was sufficient to improve memory of CPE-KO mice to levels of the WT littermates (p = 0.846). WT controls preferred the familiar diet at 20 min or at 24 hrs, regardless of presence of the demonstrator (ps > 0.229). CPE-KO mice showed no differences in preferences at 20 min and 24 hrs if the demonstrator was not present (p = 0.208); however, housing tester CPE-KO animals with the demonstrator resulted in a preference for the familiar flavored diet at 24 hrs compared to the two previous conditions without the demonstrator (ps < 0.001). (B) Total amount of food eaten by WT and CPE-KO mice at 20 min and 24 hrs under both testing conditions: with or without demonstrator mice present. ANOVA for the within subjects comparison failed to detect significant differences for test or for the test by genotype interaction; no effect of genotype for the between-subjects comparison was observed. For the STFP test, 9–11 mice/genotype/test condition was used. *p < 0.05, WT versus CPE-KO mice; #p < 0.05, with versus without the demonstrator present at 24 hrs. (C) Total contact time with the familiar and novel objects in an object recognition test. RMANOVA revealed significant main effects of test-time [F1,16 = 3.843, p < 0.051]; the test-time by genotype interaction was not significant. Bonferroni planned comparisons showed at 24 hrs, WT and CPE-KO mice spent less time investigating objects than at the 20 min test (ps < 0.044). (D) Preference for the novel object at 20 min and at 24 hr was clearly demonstrated by the WT, but not by the CPE-KO animals. RMANOVA within-subjects test failed to find test-time or a test-time by genotype interaction to be significant; however, the between-subjects effects for genotype were highly significant [F1,16 = 43.844, p < 0.001]. Bonferroni corrected pair-wise comparisons confirmed that at both test times, WT mice exhibited a marked preference for the novel object, whereas mutants showed no preference for either object (ps < 0.010). (E) Path lengths for the hidden-platform for WT and CPE-KO mice in the Morris water maze. Path lengths during the hidden-platform test were decreased for WT and CPE-KO mice. However, path lengths were longer for mutants on the first 3 days of testing. (F) Latency to reach the hidden platform in WT and CPE-KO mice. Both WT and CPE-KO mice show decreased latencies to reach the platform. This calculation demonstrates that the slower swim speed of the CPE-KO mice made only a small contribution to their swim latencies. *p < 0.05, compared with WT mice.
WT and CPE-KO mice were examined in another test that assesses hippocampal function, the object recognition memory test. Mice were exposed to two identical objects and then reexamined 20 min and 24 hrs later in a 2- choice test where the familiar and novel object were presented. Although all mice interacted with the objects longer at 20 min than at 24 hr, no genotypic differences were discerned in total object interaction time (Fig. 1C). Nonetheless, at the 20 min test, WT controls showed a marked preference for the novel object and this preference was maintained at 24 hrs (Fig. 1D). By contrast, CPE-KO mice failed to show a preference for either object at either time (Fig. 1D). These results confirm the STFP findings that an absence of external cues between the 20 min and 24 hr tests renders the CPE-KO animals deficient in hippocampal-based declarative memory.
Performance of WT and CPE-KO mice was also assessed in the Morris water maze, a test of spatial and/or place learning (Morris, 1984). Over the 3 days of visible platform testing, no genotypic differences in performance were observed. In the hidden platform version of the test, WT and CPE-KO mice showed decreased path lengths (Fig. 1E) and reduced latencies to reach the platform over trials (Fig. 1F). However, CPE-KO mice took significantly longer paths searching for the platform than WT mice on the first 3 trial days (Fig. 1E) and had significantly longer latencies to reach the platform than WT mice on all test days (Fig. 1F) where they did not reach the criterion of 10 seconds on any trial day. These data indicate that CPE-KO mice have significant impairment in learning the task. Although swim speed was significantly slower in CPE-KO mice (CPE-KO: 18.1 ± 1.2 cm/sec versus WT: 22.5 ± 0.7 cm/sec, p < 0.05), the significantly longer path lengths taken by CPE-KO mice while searching for the platform contributed to the longer latencies of these mice to reach the platform, indicating poor memory of their location. The calculated latency for CPE-KO mice to reach the platform if they swam as fast as the WT mice (Fig. 1F – CPE-KO latency at WT speed) revealed that, even at this faster swim speed the CPE-KO mice had longer latencies than WT mice and did not reach the criterion of 10 seconds. There were no sex differences in performance of either group. Collectively, the STFP, object preference, and Morris water maze tests show that the CPE-KO mice are deficient in these tasks relative to their WT controls.
Tetanic stimulation fails to induce LTP in hippocampus of CPE-KO mice
To evaluate hippocampal electrophysiological function, we first examined basal synaptic transmission in the Schaffer collateral - CA1 pathway from hippocampal slices of WT and CPE-KO mice (age 7–9 weeks) and constructed stimulation-response relationships for fEPSP. The negative-going fEPSP, following presynaptic fiber volley (PFV), was generated by a given stimulus and an example trace is shown in Figure 2A. The slope of fEPSP (Fig. 2B) can be seen to be steeper for WT (p < 0.001, unpaired student t-test) than for CPE-KO mice (N = 7 slices from 4 mice/group, 1.00 ± 0.10 mV/ms in WT and 0.33 ± 0.04 mV/ms in CPE-KO mice at 300 µA stimulation intensity), indicating that the CPE-KO mice have weaker synaptic strength.
Figure 2.
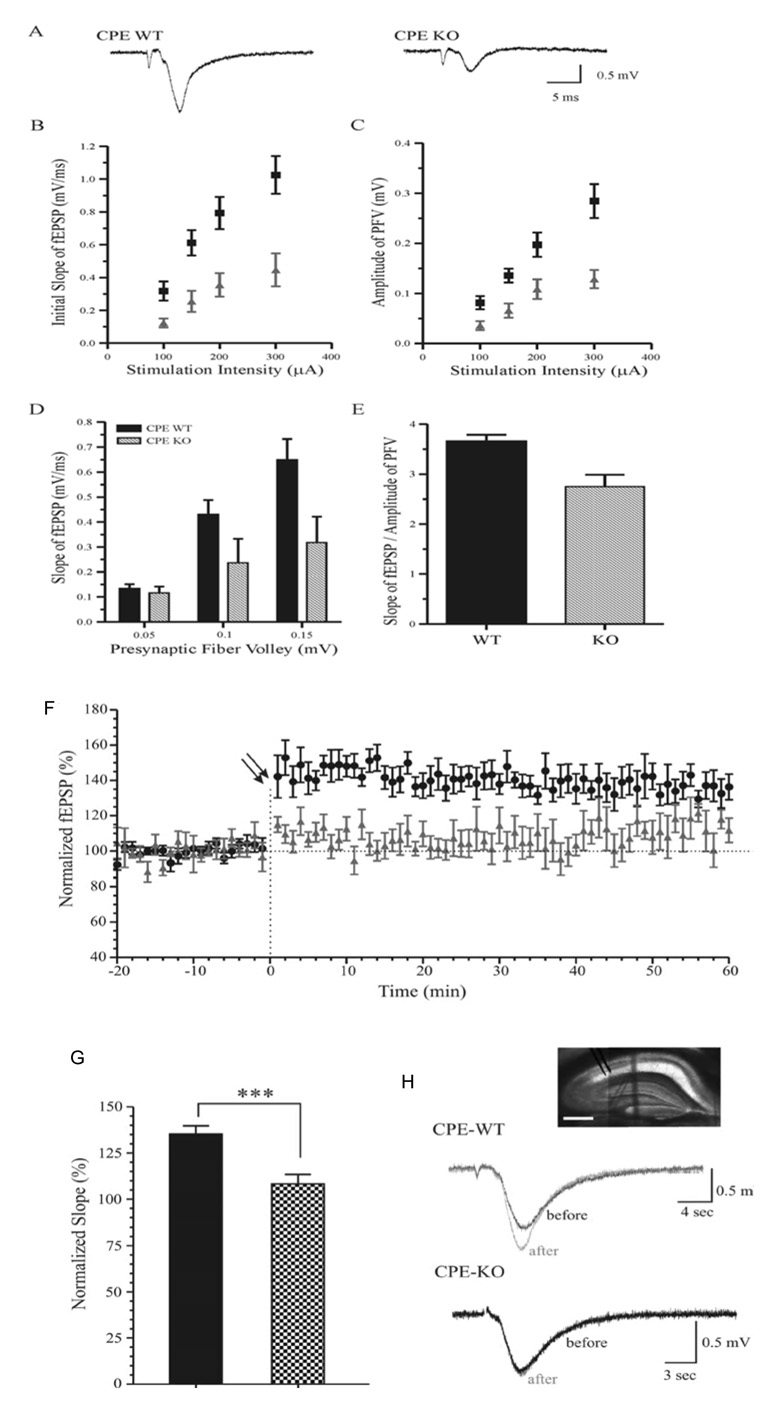
Basal synaptic transmission in the Schaffer collateral - CA1 pathway of hippocampal slices from WT and CPE-KO mice. (A) Representative traces of fEPSP in WT and CPE-KO mice are evoked by the same stimulation intensity. (B–C) Slope of the initial fEPSP (B) and the amplitude of presynaptic fiber volley or PFV (C) are plotted against the stimulus intensity for WT (■) and CPE KO mice (▲). N = 7 slices from 4 mice/group. (D) The initial slopes of fEPSP are plotted against the amplitude of PFV for both WT and CPE-KO mice. N = 5 slices from 4 mice/group. (E) To measure synaptic strength, the ratio of fEPSP slope to the amplitude of PFV over a range of stimulus intensities was calculated. Synaptic strength in CPE-KO mice was smaller than that in WT mice (N = 55, among 11 slices from 4 WT mice; N = 50, among 14 slices from 4 CPE-KO mice; p < 0.001, unpaired student t-test). (F) Hippocampal LTP from WT and CPE-KO mice. Percentage (%) increase in the initial slope of fEPSP was recorded following tetanic stimulation in slices from WT (●) and CPE-KO mice (▲). Each point represents the mean ± SEM (n = 8 slices from 4 mice/group; p < 0.001, two-way ANOVA). Double arrows indicate onset of tetanic stimulation. (G) The percentage increase of the slope of fEPSP at 60 min after the tetanic stimulation in slices from WT (black bar) and the CPE KO mice (p < 0.001; unpaired student t-test, *** p < 0.001). (H) Representative fEPSP in WT and CPE-KO mice obtained immediately before and 60 min after the tetanic stimulation. Inset, photo of hippocampal slices in WT mice. Scale bar = 0.5 mm.
To determine whether changes in the response of presynaptic terminals contribute to the decrease in synaptic transmission, the amplitudes of PFV were plotted against the stimulus intensity (Fig. 2C). Slices from CPE-KO mice showed significantly (p < 0.005, unpaired student t-test) smaller amplitude of the PFV in response to the same intensity of stimulation compared to those from WT mice (N = 8 slices from 4 mice/group; 0.28 ± 0.03 mV in WT and 0.12 ± 0.01 mV in CPE-KO at 300 µA stimulation intensity). Since the size of PFV is proportional to the number of presynaptic firing neurons recruited, these results imply that either a smaller number of presynaptic neurons were involved in the firing of action potential in the CPE-KO mice or the intrinsic properties of presynaptic neurons in the CPE-KO mice were changed resulting in less effective action potentials. For further analysis of synaptic strength, we plotted the amplitude of PFV against the slope of fEPSP (Fig. 2D). With the same amplitude of presynaptic activation, a steeper slope of the fEPSP was obtained in WT slices compared to that in CPE-KO slices. In the whole range of stimuli, the ratio of fEPSP slope to the amplitude of PFV was significantly smaller in the CPE-KO (p < 0.001, unpaired student t-test) than that in WT mice (Fig. 2E; N = 55, among 11 slices from 4 WT mice; N = 50, among 14 slices from 4 CPE-KO mice). This result shows that the basic synaptic strength in CPE-KO mice is reduced compared to that in WT mice.
Next, we examined whether there is an alteration of synaptic plasticity in CPE-KO mice by comparing the percentage of LTP induced by tetanic stimulation. In WT mice, LTP at 60 min was 135 ± 5 % of baseline (n = 8 slices from 4 mice; Fig. 2F–H). However, in CPE-KO mice the tetanic stimulation failed to induce detectable LTP (n = 8 slices from 4 mice; Fig. 2F–H). Taken together, these data indicate that basic synaptic transmission and synaptic plasticity is perturbed in the hippocampus of CPE-KO mice.
Neuronal degeneration is evident in CA3 hippocampus of CPE-KO mice
The effect of the lack of expression of CPE on the cytoarchitecture of the hippocampus was examined in the CPE-KO mouse. Six, 14 and 30 week-old animals were perfused and the brains were removed, processed, sectioned, and stained with thionine (Nissl). Hippocampal sections were also analyzed by immunohistochemistry for the expression of CPE. Immunostaining of CPE was evident in the CA1 and CA3 regions of the hippocampus from 14 week old WT mice, with the most intense staining in the CA3 region (Fig. 3A). As expected, CPE immunoreactivity was absent in CPE-KO hippocampus (Fig. 3B). Light microscopic analysis of Nissl-stained sections showed most strikingly that the pyramidal cell bodies in the CA3 area were specifically missing in CPE-KO hippocampus (Fig. 3C), in contrast to WT mice which showed typical normal morphology (Fig. 3D). Six and 30 week-old CPE-KO animals had the same abnormal hippocampal morphology as 14 week-old CPE-KO mice (data not shown). These observations suggest that lack of CPE leads to specific loss of neurons in the hippocampal CA3 region in CPE-KO mice. Immunostaining with anti-NeuN antibodies further demonstrated characteristic patterns of neurodegeneration in the hippocampal CA3 region of CPE-KO mice (Fig. 4A, B). There was a profound decrease in the number of neurons in the pyramidal cell layer (NeuN, green) in the hippocampal CA3 area in CPE-KO mice (Fig. 4B), compared to WT littermates (Fig. 4A). Concomitantly, glial cells were generated in the periphery of the CA3 region (Fig.4D), as indicated by the increase in the number of GFAP positive cells (red) (Fig. 4C, D). Disappearance of pyramidal cells (DAPI blue) in the CA3 hippocampus of CPE-KO mice was also evident. Additionally, neurofilaments staining with NF160 indicate premature termination of the mossy fibers (white arrows).
Figure 3.
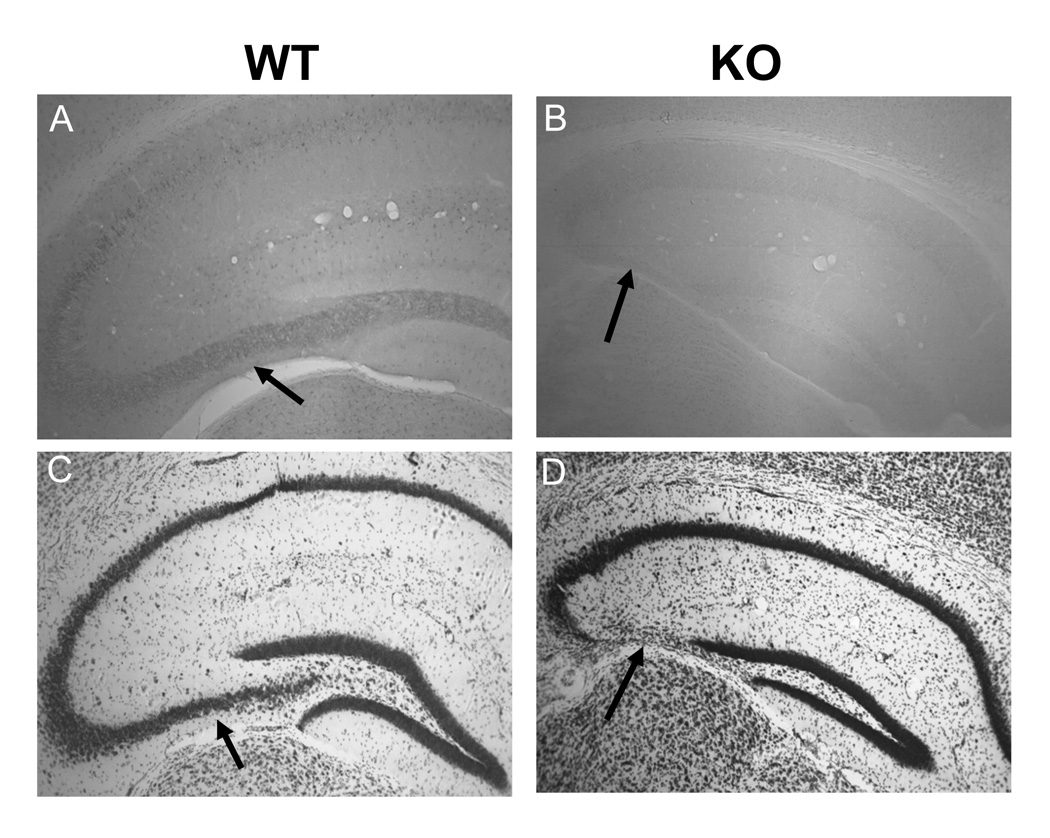
Cytoarchitecture of the hippocampus of 14 week-old WT and CPE-KO mice. (A) CPE immunohistochemistry showing intense staining (arrow) in the CA3 region as well as in pyramidal cell layer of the CA1 region in WT mice, whereas the staining is absent in CPE-KO mice (B). Nissl staining of coronal anterior hippocampal sections from WT (C) and CPE-KO mice (D). Note the complete lack of CA3 region in CPE-KO mice (arrow). (n=2)
Figure 4.
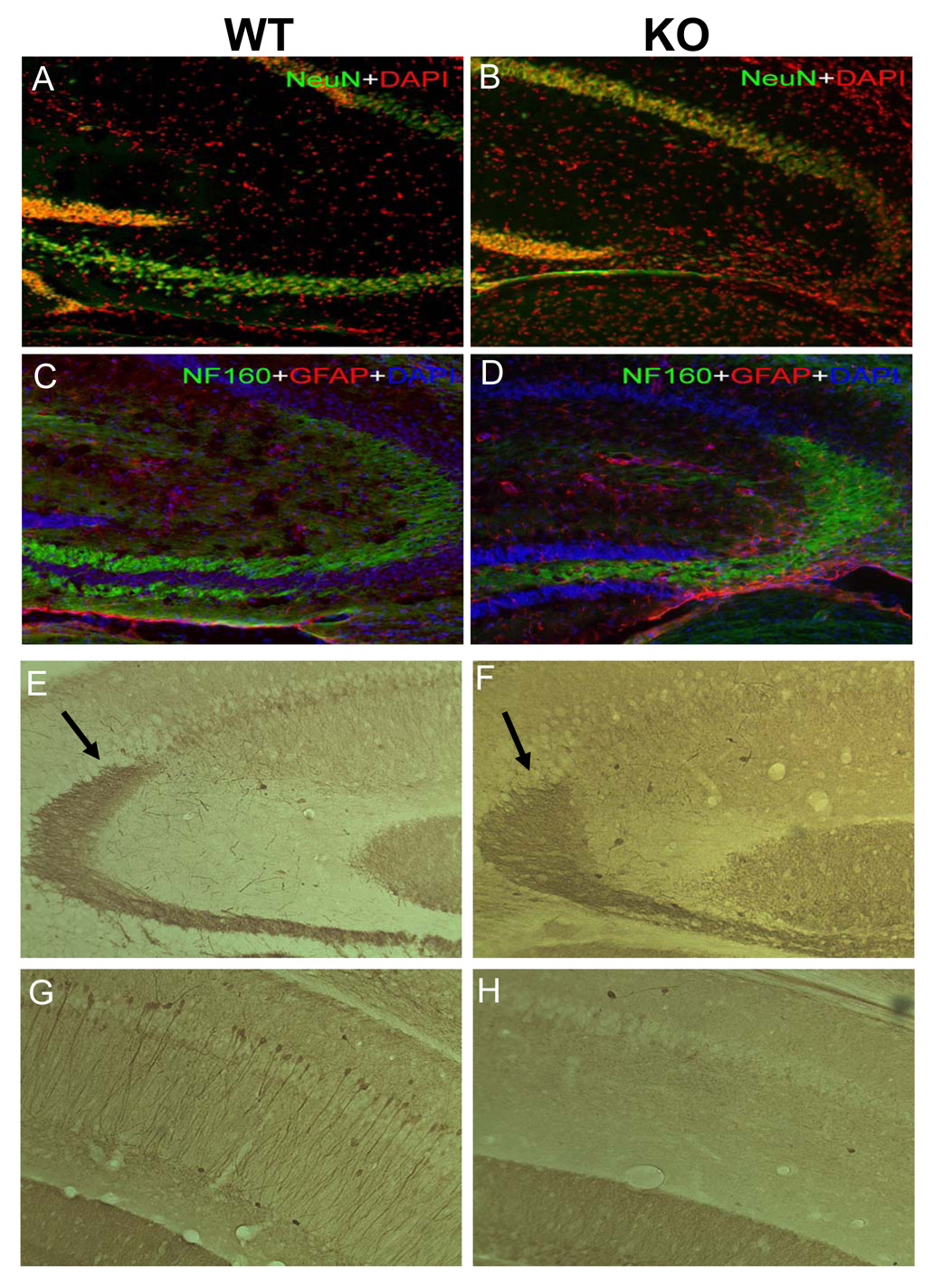
Immunohistochemistry of the hippocampus of CPE-KO mice. (A) NeuN immunoreactivity and DAPI staining in WT mice. (B) NeuN immunoreactivity and DAPI staining showing characteristic patterns of neurodegeneration in the hippocampus of CPE-KO mice. A decrease in the number of neurons in pyramidal cell layer (NeuN, green), was evident in the CA3 hippocampus in CPE-KO mice as compared to WT littermates. (D) More GFAP positive cells (red) in the hippocampus of CPE-KO mice are evident as compared to WT controls (C). Note disappearance of pyramidal cells (DAPI blue) and premature ending of the neuronal fibers (NF160 green see white arrows), in the CA3 region of CPE-KO mice. (E, F) Calbindin immunoreactivity showing that mossy fibers in hippocampus of CPE-KO mice have an abnormal course and end abruptly (black arrow). (G, H) Absence of labeling of soma and dendrites of pyramidal cells in the stratum pyramidale and stratum radiatum and less intense band of axons in the stratum lacunosum-moleculare in the CA1 region of CPE-KO mice. (n=2)
To further define the morphological abnormalities in the hippocampus of CPE-KO mice, sections were immunoassayed for calbindin (Fig. 4E, F, G, and H). Parenthetically, calbindin D-28k is a marker that can be utilized to selectively visualize certain neurons and pathways in the CNS. Although the cytoarchitecture of the molecular and granule cell layers of the dentate gyrus was normal, the mossy fibers in the hippocampus of CPE-KO mice were disorganized, from where they left the hilar zone of the dentate gyrus and throughout the CA3 region. Furthermore, the mossy fibers were observed to terminate before reaching the CA1 region (Fig. 4F). Thus, in contrast to WT mice (Fig. 4G), the CA1 region of CPE-KO mice showed a lack of dense staining of the stratum pyramidale and the apical dendritic arborizations extending into the stratum radiatum (Fig. 4H).
To determine if the defect in the CA3 region of the hippocampus of CPE-KO mice was a developmental problem, we analyzed embryonic and post-natal (PN) brains by Nissl stain and found them to be apparently normal up to PN21 (data not shown). Further analysis demonstrated that while normal morphology was seen at 3 weeks of age (Fig. 5B), significant loss of neurons in the CA3 region was observed at 4 weeks of age (Fig. 5D) indicating that rapid degeneration occurred between 3–4 weeks of age in these animals (Fig. 5C, D). These results indicate that CPE is required for the survival of CA3 neurons after 3 weeks of age and furthermore suggests a hitherto unknown specific role of CPE in neuroprotection.
Figure 5.
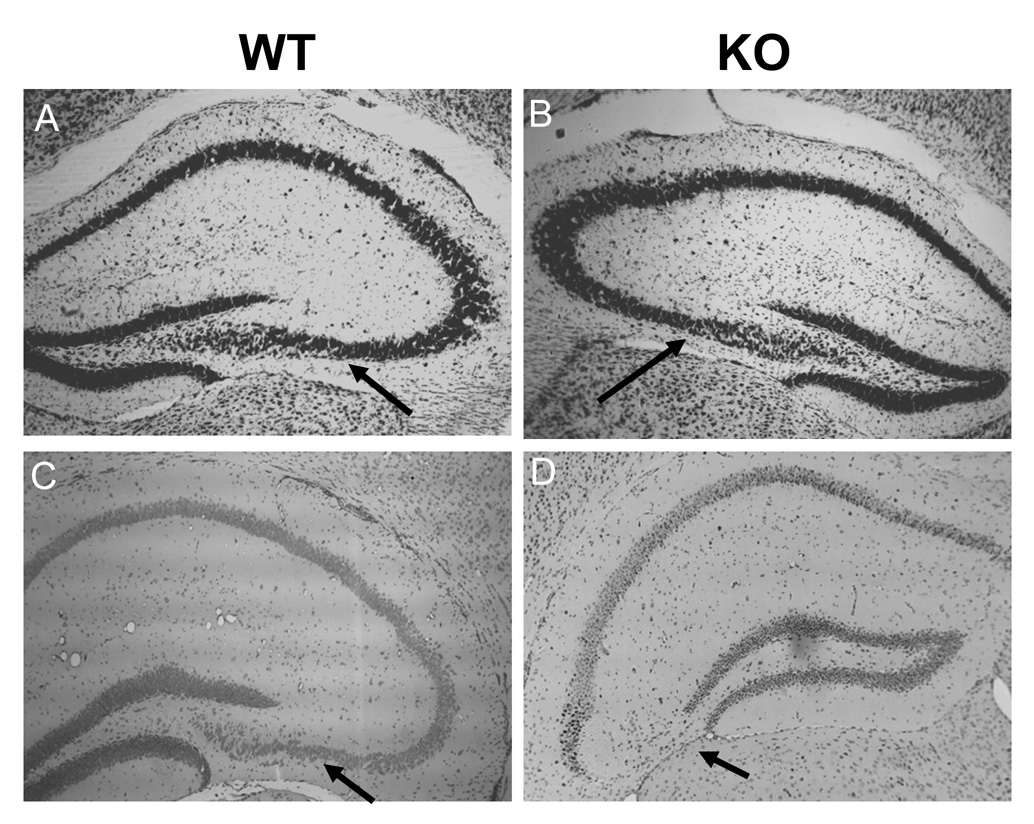
Cytoarchitecture of the hippocampus of 3 and 4 week-old WT and CPE-KO mice. Nissl staining of coronal anterior hippocampal sections from WT and CPE-KO of 3 week old (A, B), and 4 week old mice (C, D). Note that the cytoarchitecture of the hippocampus is indistiguihable between WT and CPE-KO mice at 3 weeks of age, but the CA3 region has completely degenerated by 4 weeks of age in CPE-KO mice (D). (n=2)
CPE protects oxidative stress-induced neuronal cell death in hippocampal neurons
To further assess the ability of CPE to protect neurons against cell death, ex vivo studies on hippocampal neurons in culture were conducted using the model of induced oxidative stress (Numakawa et al., 2007). Rat hippocampal cultures were infected with adenovirus encoding encoding β-galactosidase (LacZ) or CPE. Subsequently, cultures were exposed to 50 µM H2O2 as an oxidative stress to induce neuronal cell death. Twenty-four hrs later, cells were stained with DAPI and observed by confocal microscopy and analyzed (Fig. 6A). In control culture, the percentage of basal cell death was 10.8 ± 1.9%, while with H2O2 treatment, the percentage of cell death, rose to 28.3 ± 4.3%, indicating that oxidative stress promoted apoptosis in the hippocampal culture (p < 0.05). In contrast, the percentage of cell death in the H2O2-treated CPE infected cultures (18.9 ± 3.6%) was not significantly different from basal cell death (16.7 ± 3.2%). Thus, oxidative stress-induced cell death of hippocampal neurons was completely protected by overexpression of exogenous CPE (Fig. 6B).
Figure 6.
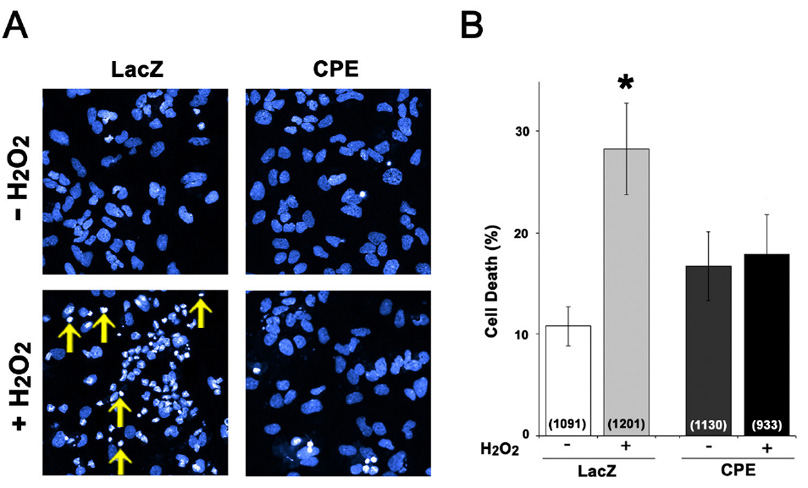
Overexpression of CPE protects hippocampal neurons from apoptosis. (A) Rat hippocampal neuronal cultures were infected with LacZ (control) or CPE adenovirus and treated with or without H2O2 to induce apoptosis. One day after cell death induction, cultures were stained with DAPI. Yellow arrows illustrate the pattern of DAPI staining in the nuclei of a group of apoptotic cells after H2O2 treatment. (B) Bar graphs show the % of cells that exhibited cell death per total cell population. The numbers in parenthesis within the bar show the number of cells for each condition. H2O2 treatment induced apoptosis in the LacZ-expressing culture (p < 0.05), while no effect on cell death was observed in CPE-transduced neurons. n = 6 independent wells of cells for each group.
DISCUSSION
In this study, we have examined the effect of the absence of CPE on hippocampus-based behavior, electrophysiological function and neuronal survival in the hippocampus of CPE-KO mice. We found that mice lacking CPE exhibited specific neuronal cell death in the CA3 region of the hippocampus which occurred between three to four weeks of age and persisted from age four weeks onwards. Interestingly, this pattern of degeneration is characteristic of the morphology seen in the hippocampus of seizure induced mice; i.e. the CA3 region also degenerates specifically (Murphy et al., 2007). However, we have not found any evidence of seizures thus far in the CPE-KO mice.
Immunostaining revealed that the major loss of CPE-rich neurons (Fig. 4, NeuN staining) in this region of the CPE-KO hippocampus was accompanied by proliferation of GFAP-positive glial cells. Moreover, calbindin staining revealed that the mossy fibers, which normally course through the CA3 to the CA1 regions, terminated prematurely and abruptly prior to reaching the CA1 region. These morphological findings mice suggest that CPE plays a critical role in maintaining the levels of specific hippocampal neurons in adult brain which in turn preserves the proper structure and function of the hippocampus. While several correlative studies have suggested a role for CPE in neuronal survival under ischemic conditions (Jin et al., 2001; Zhou et al., 2004), our results presented here are the first to demonstrate a specific connection between CPE and neuronal survival in vivo. In addition to the CPE-KO mouse data, our ex vivo studies of rat hippocampal neurons provide supporting evidence for this connection. We demonstrated that hippocampal neurons infected with adenovirus overexpressing WT CPE were protected against oxidative stress-induced cell death, in contrast to neurons infected with the control Lac-Z adenovirus. Taken together, these data provide strong evidence that CPE might be involved in neuroprotection as a mode of neuronal survival.
As mentioned above, several studies have observed a correlation between CPE expression and neuronal survival in the ischemic brain. Previously, Jin et al., (Jin et al., 2001) found that following global ischemia, there was a sustained increase in CPE mRNA and protein in the hippocampal CA3 neurons which survived such injury. In contrast, CA1 neurons which showed only a transient rise in CPE were more vulnerable to ischemia. Additionally, Zhou et al reported (Zhou et al., 2004) an exacerbation in ischemic injury in the cortex of the CPEfat/fat mutant mice, a mouse strain that lacks an active CPE protease (Naggert et al., 1995), after an otherwise non-injurious focal ischemia compared to normal mice. Furthermore, studies using an ischemia tolerance model showed an elevated level of CPE protein expression in cortical neurons that survived ischemic injury (A. Zhou, unpublished data). These findings suggest that CPE plays a role in neuroprotection under oxidative stress conditions in vivo similar to the ex vivo oxidative stress model we used here (Fig. 6).
The neurodegeneration of the CA3 neurons in CPE-KO mice resulted in structural abnormalities of the hippocampus which had a significant impact on function, especially since the CA3 region receives sensory information from the mossy fibers of granule cells in the dentate gyrus and from projections of cells in the entorhinal cortex along the perforant path (Tamura et al., 2006). As well, CA3 pyramidal cells form recurrent collaterals with other CA3 neurons to mediate NMDA receptor-dependent changes in synaptic strength (Walker et al., 2007). Hence, not surprisingly, electrophysiological recordings showed that synaptic strength was weaker and that tetanic stimulation was unable to induce LTP in the hippocampus of these CPE-KO mice (Fig. 2). Since hippocampal LTP is necessary for learning and memory processes (Miyamoto, 2006), the absence of LTP with tetanic stimulation would most likely be responsible for the deficiency in the hippocampus-based learning and memory processes (Eichenbaum, 2001; Morris, 1984; Rampon et al., 2000; Rodriguiz, 2006) observed in the CPE-KO mice (Fig. 1).
In the object recognition test, CPE-KO mice were impaired when tested at 20 min and at 24 hrs. Similarly, in the STFP test, CPE-KO mice were deficient when evaluated at 20 min and at 24 hrs, if the demonstrator animal was removed from the home-cage. However, when the demonstrator was continually present, retention of the task at 24 hrs was similar to that of the WT controls. As the CPE-KO mice were deficient also at 20 min, the continual presence of the demonstrator may serve as a constant reminder cue for selection of the familiar diet at 24 hrs. A similar relationship was observed in the Morris water maze. Although the CPE-KO animals were not impaired on the visible-platform, signifying that their sensory and motor functions were intact, they were deficient on the hidden-platform version of the test. When path length was considered, CPE-KO mice appeared to acquire the task more slowly than WT controls. Following repeated exposures to the maze (i.e., more trials) performance of CPE-KO animals becomes similar to that of their WT littermates. Hence, the STFP and water maze data indicate that CPE-KO mice may be slow learners and that repetition of the learning experience is sufficient to place the information into long-term memory. Together these results show that CPE-KO mice are impaired in hippocampus-based memory, however, this debility is not fixed since reminder cues, or increased training can overcome the deficiency.
What is the molecular mechanism underlying the function of CPE in either promoting neuronal survival or inhibiting neuronal cell death? One likely possibility is that CPE is required for the processing, sorting and packaging of a growth factor or an anti-apoptotic protein into the regulated secretory pathway for release in hippocampal neurons. A candidate protein could be BDNF which is involved in promoting neuronal survival (Huang and Reichardt, 2003). Since its sorting to the regulated secretory pathway is dependent upon CPE and hence activity-dependent secretion of BDNF is defective in the CPE-KO mice (Lou et al., 2005), this could contribute to the degeneration of the CA3 hippocampal neurons observed in our study. However, this relationship may not be so simple since, Korte et al. did not find any obvious anatomical or morphological differences between hippocampi from wild-type and BDNF-KO mice between the ages P12–P67 (Korte et al., 1995). Hence, lack of BDNF alone cannot account for the specific neurodegeneration of the CA3 region in the hippocampus of the adult CPE-KO mouse. Thus, identification of other putative neuropeptides involved in this process would be very important in helping our understanding of the structure and maintenance of the hippocampus. The fact that the CA3 region of the hippocampus is highly enriched in CPE and that survival of CA3 neurons after global ischemia is specific and coupled with a sustained increase in expression of CPE argues for another possible mechanism in which a more direct role of CPE itself in supporting survival or inhibiting degeneration of these neurons exists.
In summary, this study has uncovered a novel player: CPE, which now has a new and essential role in preventing neuronal cell death in the CA3 hippocampus so as to maintain normal cytoarchitecture for optimal electrophysiological and cognitive function in the adult brain. Additionally, enhanced CPE expression appears to be important in protecting hippocampal neurons from oxidative stress-induced apoptosis. Future investigations into the mechanism of action of CPE in this respect are necessary and could lead to identification of therapeutic targets for development of pharmaceutical agents for neuroprotection of the adult brain.
ACKNOWLEDGEMENTS
We thank Drs. Leslie Ungerleider, (NIMH) and Barry Hoffer (NIDA) for critical reading of the manuscript. Special thanks to Lindsey Phillips (Duke Univ.) who assisted with the STFP and object recognition tests, and Liping Du and Jiechun Zhou (Duke Univ.) for genotyping and husbandry of the animals used in behavioral testing and their assistance in decoding the videotape for the STFP and object recognition tests.
This research was supported by NIH contract 263-MD-516126 (W.C.W.), NIH grant# RO1NS049470 (Z-G. X.), NIH grant # RO1NS46560-01A2 (A.Z.) and by the Intramural Research Program of the National Institute of Child Health and Human Development, National Institutes of Health, USA.
REFERENCES
- Cawley NX, Zhou J, Hill JM, Abebe D, Romboz S, Yanik T, Rodriguiz RM, Wetsel WC, Loh YP. The carboxypeptidase E knockout mouse exhibits endocrinological and behavioral deficits. Endocrinology. 2004;145(12):5807–5819. doi: 10.1210/en.2004-0847. [DOI] [PubMed] [Google Scholar]
- Cool DR, Normant E, Shen F, Chen HC, Pannell L, Zhang Y, Loh YP. Carboxypeptidase E is a regulated secretory pathway sorting receptor: genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell. 1997;88(1):73–83. doi: 10.1016/s0092-8674(00)81860-7. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. The hippocampus and declarative memory: cognitive mechanisms and neural codes. Behav Brain Res. 2001;127(1–2):199–207. doi: 10.1016/s0166-4328(01)00365-5. [DOI] [PubMed] [Google Scholar]
- Fricker LD. Carboxypeptidase E. Annu Rev Physiol. 1988;50:309–321. doi: 10.1146/annurev.ph.50.030188.001521. [DOI] [PubMed] [Google Scholar]
- Hook VY, Loh YP. Carboxypeptidase B-like converting enzyme activity in secretory granules of rat pituitary. Proc Natl Acad Sci U S A. 1984;81(9):2776–2780. doi: 10.1073/pnas.81.9.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jacob TC, Kaplan JM. The EGL-21 carboxypeptidase E facilitates acetylcholine release at Caenorhabditis elegans neuromuscular junctions. J Neurosci. 2003;23(6):2122–2130. doi: 10.1523/JNEUROSCI.23-06-02122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Graham SH, Nagayama T, Goldsmith PC, Greenberg DA, Zhou A, Simon RP. Altered expression of the neuropeptide-processing enzyme carboxypeptidase E in the rat brain after global ischemia. J Cereb Blood Flow Metab. 2001;21(12):1422–1429. doi: 10.1097/00004647-200112000-00006. [DOI] [PubMed] [Google Scholar]
- Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10(1):47–56. doi: 10.1002/(SICI)1098-1063(2000)10:1<47::AID-HIPO5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92(19):8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111(1):104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Lou H, Kim SK, Zaitsev E, Snell CR, Lu B, Loh YP. Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase e. Neuron. 2005;45(2):245–255. doi: 10.1016/j.neuron.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100(5):433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11(1):47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Murphy B, Dunleavy M, Shinoda S, Schindler C, Meller R, Bellver-Estelles C, Hatazaki S, Dicker P, Yamamoto A, Koegel I, et al. Bcl-w protects hippocampus during experimental status epilepticus. Am J Pathol. 2007;171(4):1258–1268. doi: 10.2353/ajpath.2007.070269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naggert JK, Fricker LD, Varlamov O, Nishina PM, Rouille Y, Steiner DF, Carroll RJ, Paigen BJ, Leiter EH. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet. 1995;10(2):135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- Numakawa Y, Matsumoto T, Yokomaku D, Taguchi T, Niki E, Hatanaka H, Kunugi H, Numakawa T. 17beta-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of mitogen-activated protein kinase and in the accumulation of intracellular calcium. Endocrinology. 2007;148(2):627–637. doi: 10.1210/en.2006-1210. [DOI] [PubMed] [Google Scholar]
- Park JJ, Cawley NX, Loh YP. Carboxypeptidase e cytoplasmic tail-driven vesicle transport is key for activity-dependent secretion of Peptide hormones. Mol Endocrinol. 2008;22(4):989–1005. doi: 10.1210/me.2007-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ. Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci. 2000;3(3):238–244. doi: 10.1038/72945. [DOI] [PubMed] [Google Scholar]
- Rodriguiz RM, Wetsel WC. Assessments of cognitive deficits in mutant mice. In: Levin ED, Buccafusco JJ, editors. Animal Models of Cognitive Impairment. Boca Raton, FL: CRC Press; 2006. pp. 223–282. 223–282. [PubMed] [Google Scholar]
- Rodriguiz RM, Chu R, Caron MG, Wetsel WC. Aberrant responses in social interaction of dopamine transporter knockout mice. Behav Brain Res. 2004;148(1–2):185–198. doi: 10.1016/s0166-4328(03)00187-6. [DOI] [PubMed] [Google Scholar]
- Tamura M, Koyama R, Ikegaya Y, Matsuki N, Yamada MK. K252a, an inhibitor of Trk, disturbs pathfinding of hippocampal mossy fibers. Neuroreport. 2006;17(5):481–486. doi: 10.1097/01.wnr.0000208997.23448.ea. [DOI] [PubMed] [Google Scholar]
- Walker CD, Long H, Williams S, Richard D. Long-lasting effects of elevated neonatal leptin on rat hippocampal function, synaptic proteins and NMDA receptor subunits. J Neurosci Res. 2007;85(4):816–828. doi: 10.1002/jnr.21173. [DOI] [PubMed] [Google Scholar]
- Zhou A, Minami M, Zhu X, Bae S, Minthorne J, Lan J, Xiong ZG, Simon RP. Altered biosynthesis of neuropeptide processing enzyme carboxypeptidase E after brain ischemia: molecular mechanism and implication. J Cereb Blood Flow Metab. 2004;24(6):612–622. doi: 10.1097/01.WCB.0000118959.03453.17. [DOI] [PubMed] [Google Scholar]
- Zhu X, Wu K, Rife L, Cawley NX, Brown B, Adams T, Teofilo K, Lillo C, Williams DS, Loh YP, et al. Carboxypeptidase E is required for normal synaptic transmission from photoreceptors to the inner retina. J Neurochem. 2005;95(5):1351–1362. doi: 10.1111/j.1471-4159.2005.03460.x. [DOI] [PubMed] [Google Scholar]